

The Marine-Derived Macrolactone Mandelalide A Is an Indirect Activator of AMPK

 , ,
, ,  ,
,  and
and 
Abstract
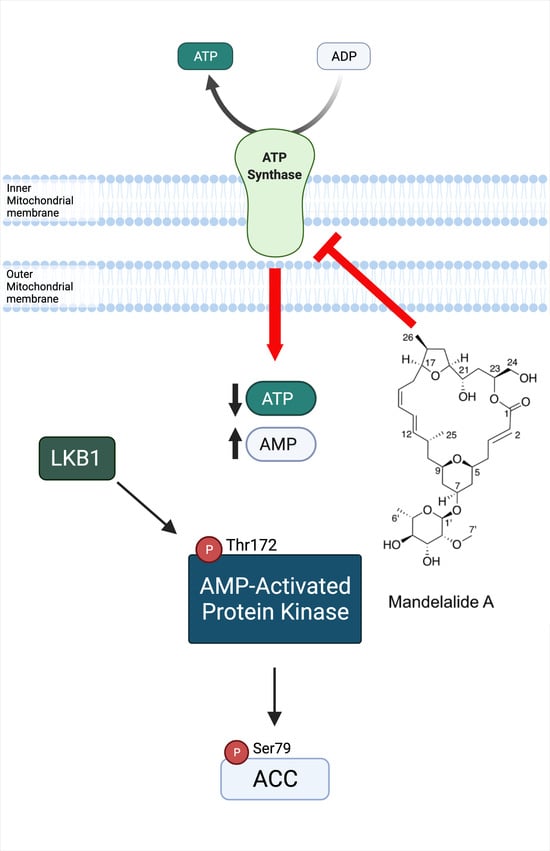
:
1. Introduction
2. Results
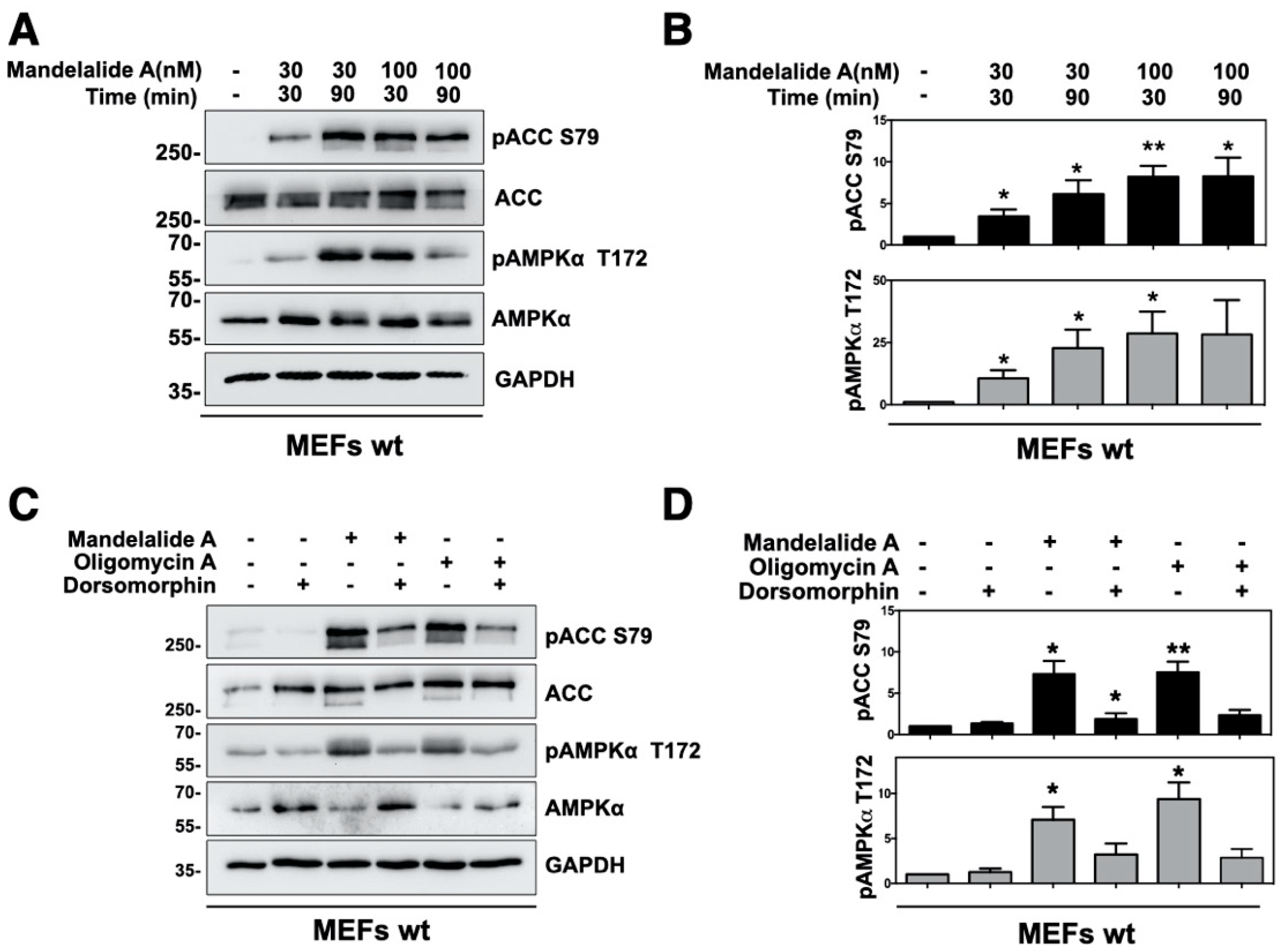
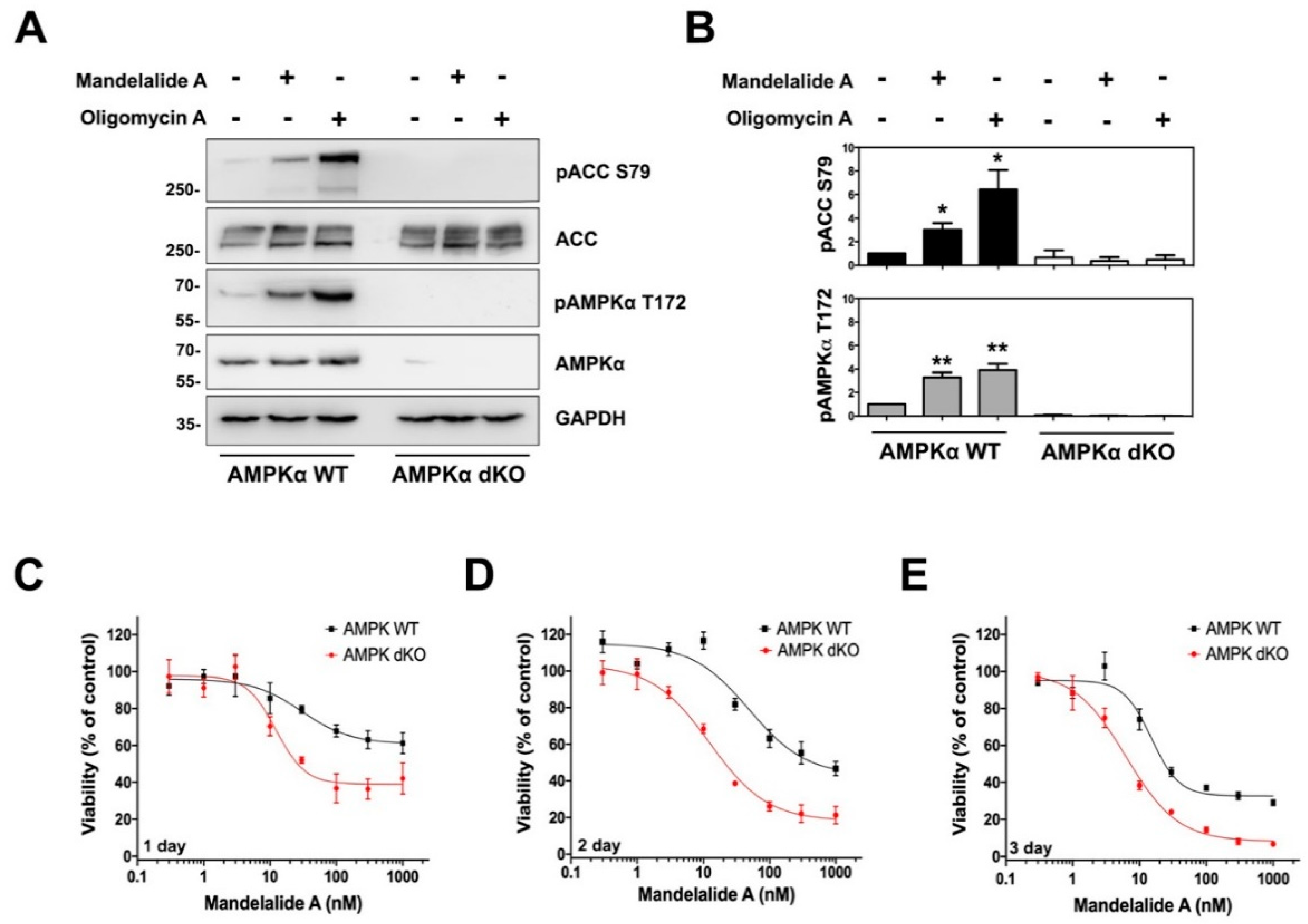
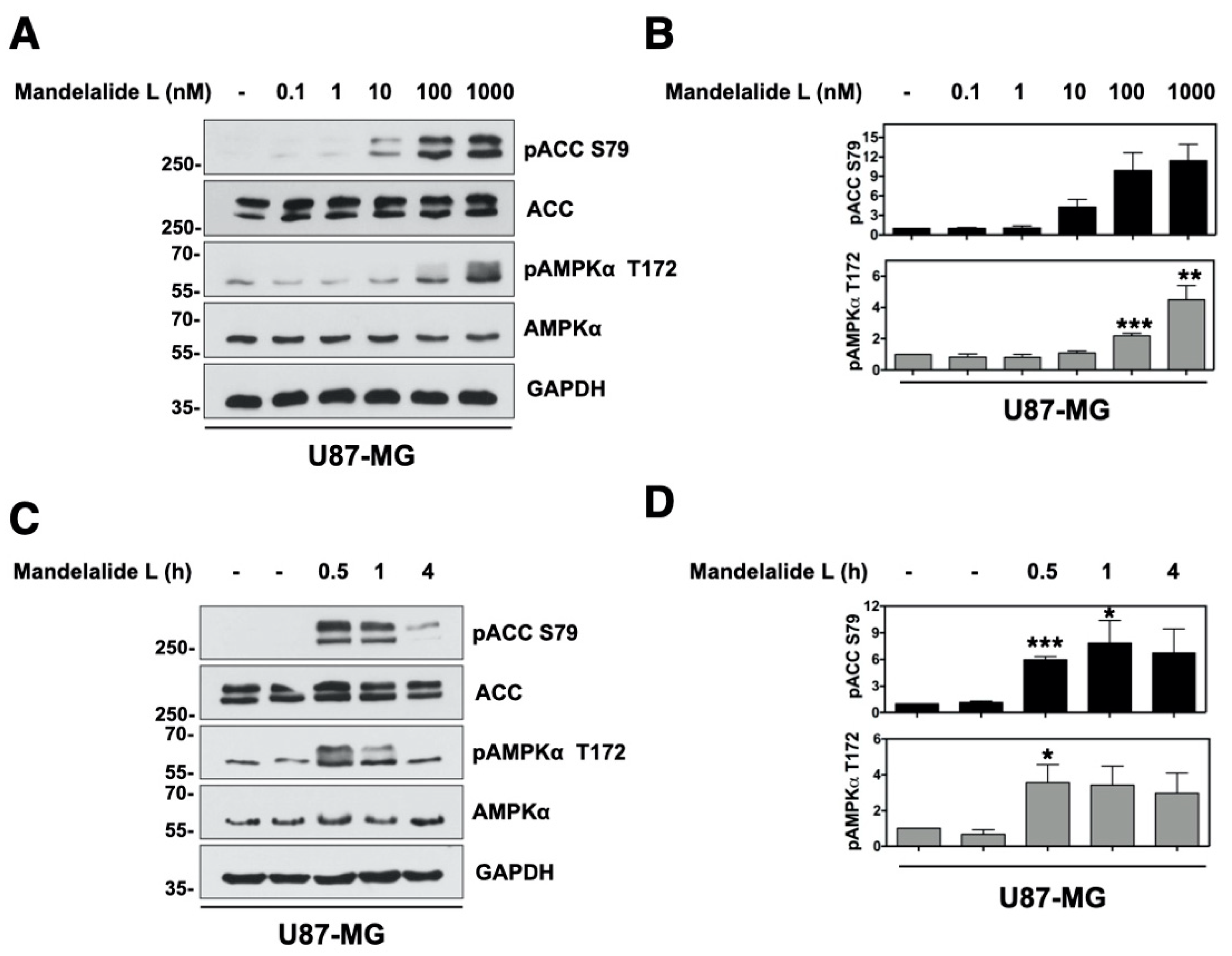
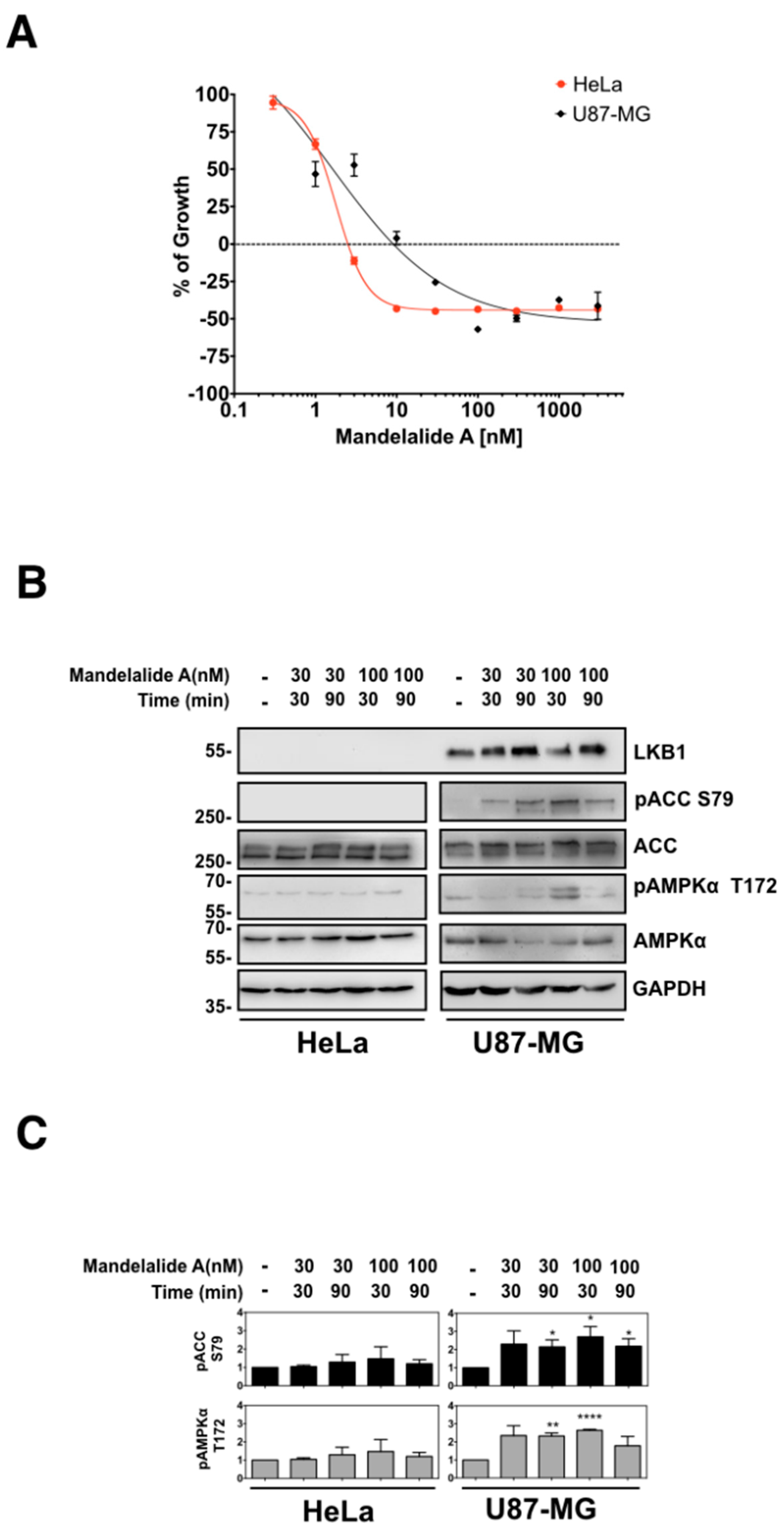
2.1. Mandelalide A Is an Indirect Activator of AMPK
2.2. AMPKα Confers a Survival Advantage against Mandelalide A-Induced Cytotoxicity
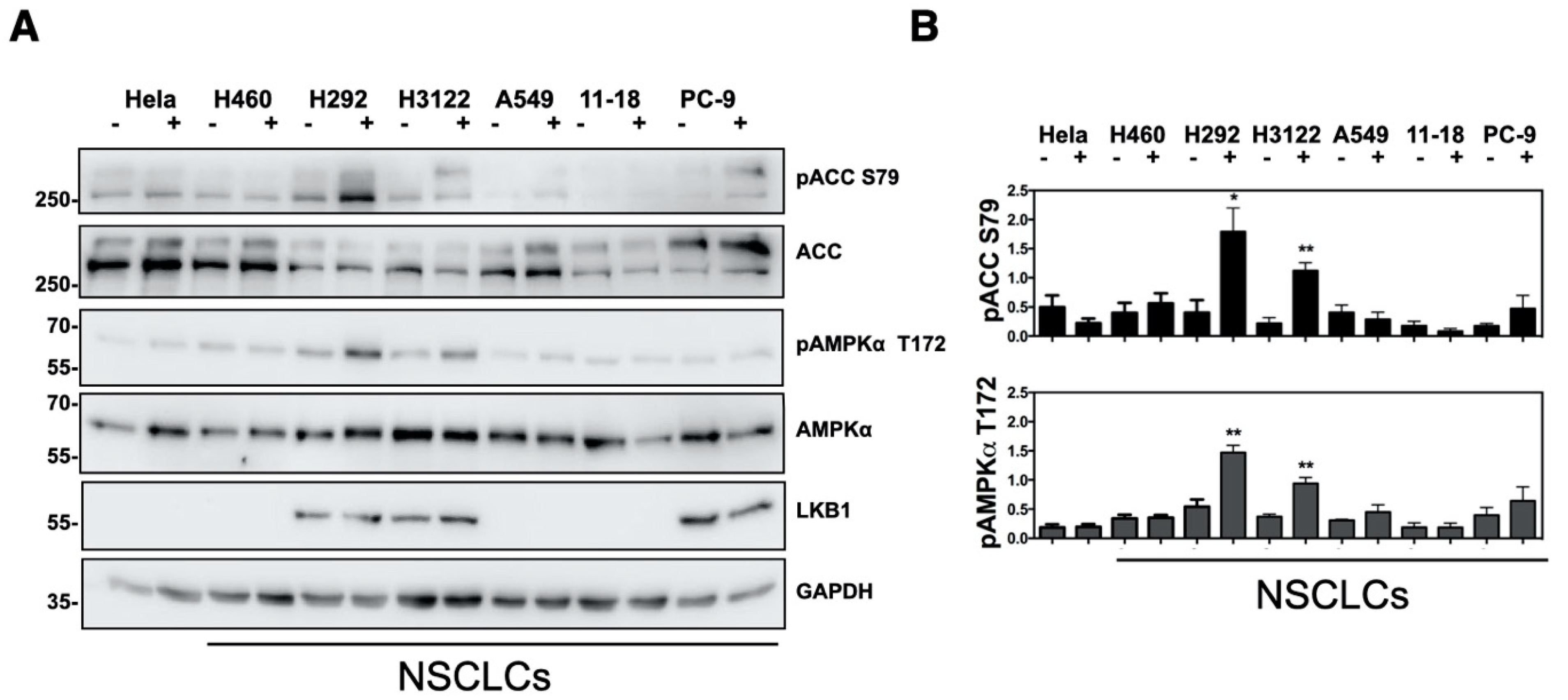
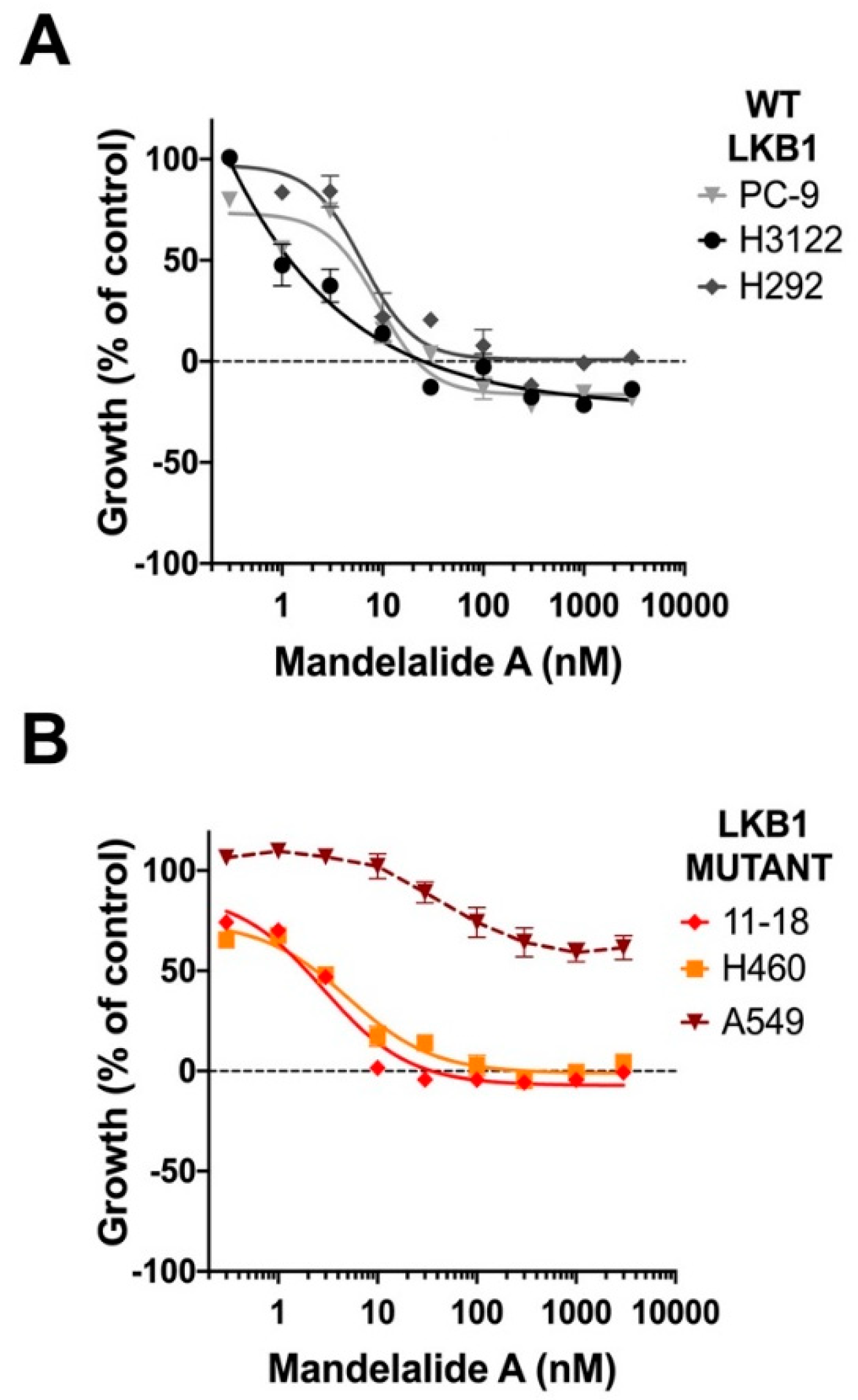
2.3. Analysis of Mandelalide A-Induced Changes in AMPK Status and NSCLC Cell Growth as a Function of LKB1 Expression
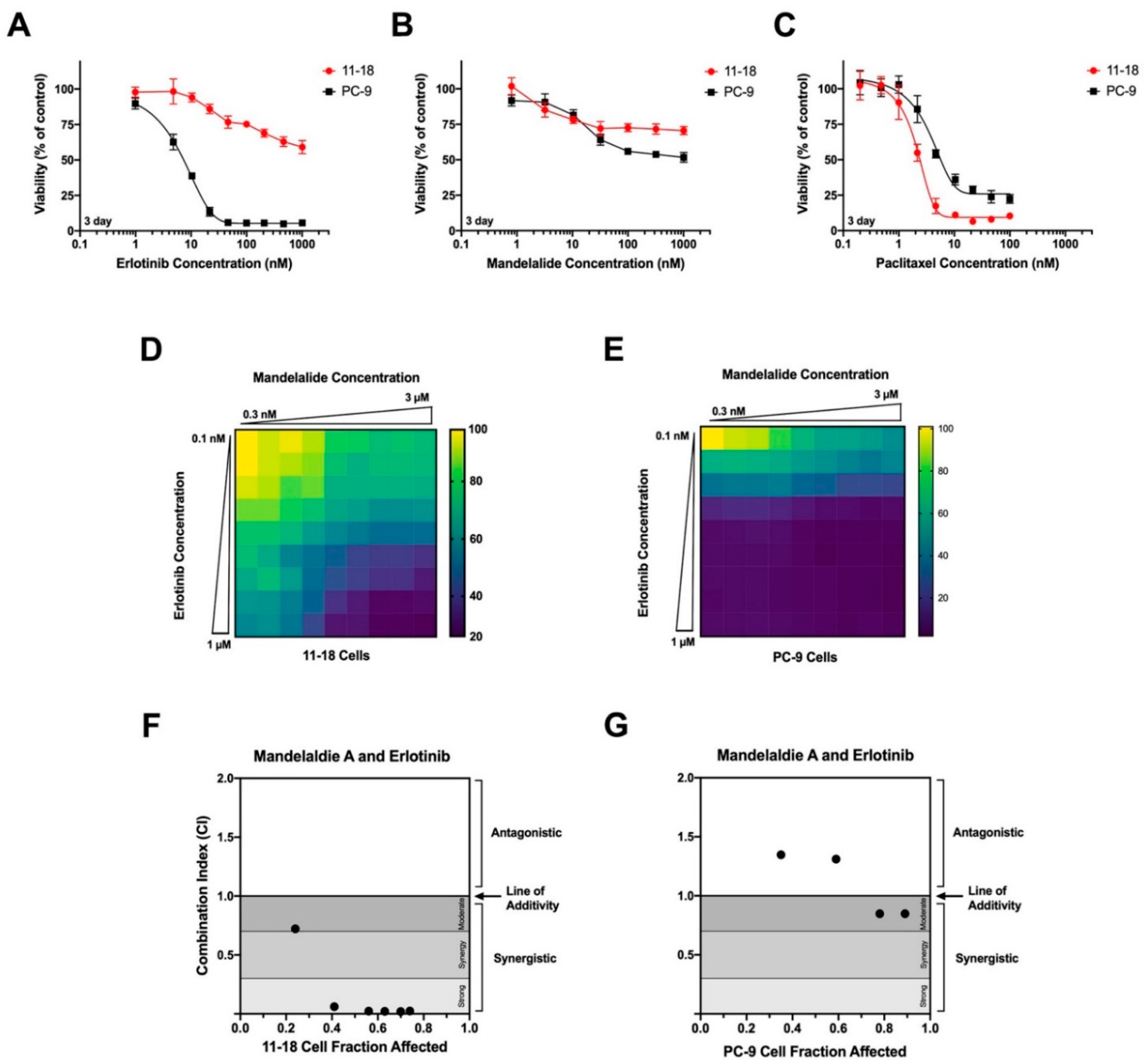
2.4. Analysis of Mandelalide-Induced Cytotoxicity in EGFR Mutant NSCLC Cells Alone and in Combination with Erlotinib
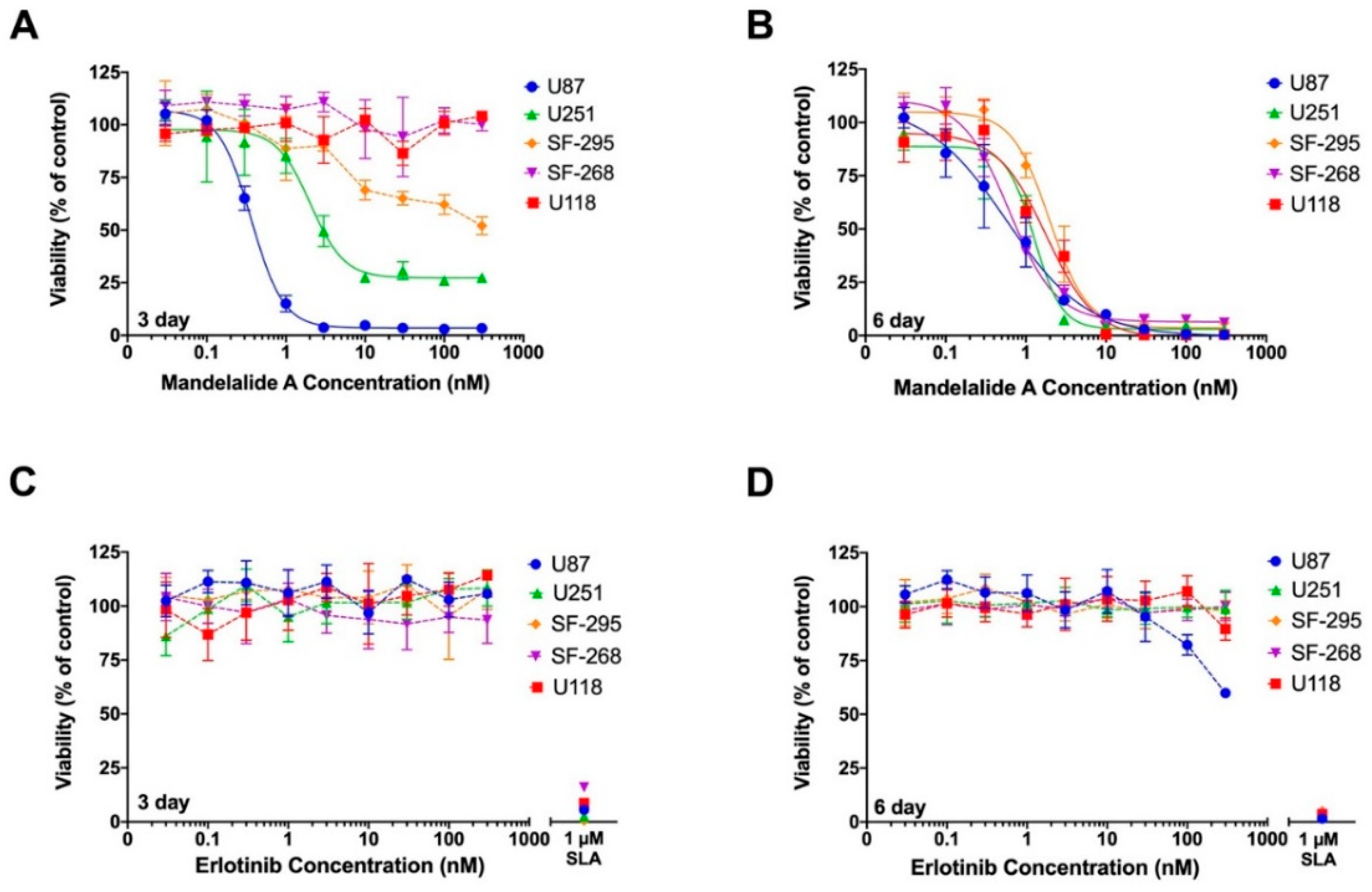
2.5. Mandelalide A-Induced Loss of ATP Is Lethal to Cultured Human Glioblastoma Cells
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Mammalian Cell Culture
4.3. Cell Lysis and Immunoblot Analysis
4.4. Cell Viability Assay
4.5. Drug Combination Assays
4.6. Data Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

References
- Sikorska, J.; Hau, A.M.; Anklin, C.; Parker-Nance, S.; Davies-Coleman, M.T.; Ishmael, J.E.; McPhail, K.L. Mandelalides A–D, cytotoxic macrolides from a new Lissoclinum species of South African tunicate. J. Org. Chem. 2012, 77, 6066–6075. [Google Scholar] [CrossRef] [PubMed]
- Lei, H.; Yan, J.; Yu, J.; Liu, Y.; Wang, Z.; Xu, Z.; Ye, T. Total synthesis and stereochemical reassignment of mandelalide A. Angew. Chem. Int. Ed. Engl. 2014, 53, 6533–6537. [Google Scholar] [CrossRef] [PubMed]
- Nazari, M.; Serrill, J.D.; Sikorska, J.; Ye, T.; Ishmael, J.E.; McPhail, K.L. Discovery of Mandelalide E and Determinants of Cytotoxicity for the Mandelalide Series. Org. Lett. 2016, 18, 1374–1377. [Google Scholar] [CrossRef]
- Nazari, M.; Serrill, J.D.; Wan, X.; Nguyen, M.H.; Anklin, C.; Gallegos, D.A.; Smith, A.B., III; Ishmael, J.E.; McPhail, K.L. New Mandelalides Expand a Macrolide Series of Mitochondrial Inhibitors. J. Med. Chem. 2017, 60, 7850–7862. [Google Scholar] [CrossRef]
- Lopera, J.; Miller, I.J.; McPhail, K.L.; Kwan, J.C. Increased Biosynthetic Gene Dosage in a Genome-Reduced Defensive Bacterial Symbiont. Msystems 2017, 2, e00096-17. [Google Scholar] [CrossRef] [PubMed]
- Willwacher, J.; Furstner, A. Catalysis-based total synthesis of putative mandelalide A. Angew. Chem. Int. Ed. Engl. 2014, 53, 4217–4221. [Google Scholar] [CrossRef]
- Brutsch, T.M.; Bucher, P.; Altmann, K.H. Total Synthesis and Biological Assessment of Mandelalide A. Chemistry 2016, 22, 1292–1300. [Google Scholar] [CrossRef]
- Veerasamy, N.; Ghosh, A.; Li, J.; Watanabe, K.; Serrill, J.D.; Ishmael, J.E.; McPhail, K.L.; Carter, R.G. Enantioselective Total Synthesis of Mandelalide A and Isomandelalide A: Discovery of a Cytotoxic Ring-Expanded Isomer. J. Am. Chem. Soc. 2016, 138, 770–773. [Google Scholar] [CrossRef]
- Nguyen, M.H.; Imanishi, M.; Kurogi, T.; Smith, A.B., III. Total Synthesis of (-)-Mandelalide A Exploiting Anion Relay Chemistry (ARC): Identification of a Type II ARC/CuCN Cross-Coupling Protocol. J. Am. Chem. Soc. 2016, 138, 3675–3678. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O.; Wind, F.; Negelein, E. The Metabolism of Tumors in the Body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef]
- Palm, W.; Thompson, C.B. Nutrient acquisition strategies of mammalian cells. Nature 2017, 546, 234–242. [Google Scholar] [CrossRef]
- Grasmann, G.; Mondal, A.; Leithner, K. Flexibility and Adaptation of Cancer Cells in a Heterogenous Metabolic Microenvironment. Int. J. Mol. Sci. 2021, 22, 1476. [Google Scholar] [CrossRef]
- Ashton, T.M.; McKenna, W.G.; Kunz-Schughart, L.A.; Higgins, G.S. Oxidative Phosphorylation as an Emerging Target in Cancer Therapy. Clin. Cancer Res. 2018, 24, 2482–2490. [Google Scholar] [CrossRef]
- Wang, T.; Ma, F.; Qian, H.L. Defueling the cancer: ATP synthase as an emerging target in cancer therapy. Mol. Ther. Oncolytics 2021, 23, 82–95. [Google Scholar] [CrossRef]
- Sica, V.; Bravo-San Pedro, J.M.; Stoll, G.; Kroemer, G. Oxidative phosphorylation as a potential therapeutic target for cancer therapy. Int. J. Cancer 2020, 146, 10–17. [Google Scholar] [CrossRef]
- Shelton, P.M.M.; Kapoor, T.M. A wrench in the motor. Nat. Chem. Biol. 2022, 18, 355–356. [Google Scholar] [CrossRef] [PubMed]
- Patel, B.A.; D’Amico, T.L.; Blagg, B.S.J. Natural products and other inhibitors of F1FO ATP synthase. Eur. J. Med. Chem. 2020, 207, 112779. [Google Scholar] [CrossRef] [PubMed]
- Lardy, H.A.; Johnson, D.; Mc, M.W. Antibiotics as tools for metabolic studies. I. A survey of toxic antibiotics in respiratory, phosphorylative and glycolytic systems. Arch. Biochem. Biophys. 1958, 78, 587–597. [Google Scholar] [CrossRef]
- Kim, J.W.; Adachi, H.; Shin-Ya, K.; Hayakawa, Y.; Seto, H. Apoptolidin, a new apoptosis inducer in transformed cells from Nocardiopsis sp. J. Antibiot. 1997, 50, 628–630. [Google Scholar] [CrossRef] [PubMed]
- Serrill, J.D.; Tan, M.; Fotso, S.; Sikorska, J.; Kasanah, N.; Hau, A.M.; McPhail, K.L.; Santosa, D.A.; Zabriskie, T.M.; Mahmud, T.; et al. Apoptolidins A and C activate AMPK in metabolically sensitive cell types and are mechanistically distinct from oligomycin A. Biochem. Pharmacol. 2015, 93, 251–265. [Google Scholar] [CrossRef]
- Murakami, R.; Tomikawa, T.; Shin-Ya, K.; Shinozaki, J.; Kajiura, T.; Kinoshita, T.; Miyajima, A.; Seto, H.; Hayakawa, Y. Ammocidin, a new apoptosis inducer in Ras-dependent cells from Saccharothrix sp. I. Production, isolation and biological activity. J. Antibiot. 2001, 54, 710–713. [Google Scholar] [CrossRef]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef]
- Ji, H.; Ramsey, M.R.; Hayes, D.N.; Fan, C.; McNamara, K.; Kozlowski, P.; Torrice, C.; Wu, M.C.; Shimamura, T.; Perera, S.A.; et al. LKB1 modulates lung cancer differentiation and metastasis. Nature 2007, 448, 807–810. [Google Scholar] [CrossRef]
- Hawley, S.A.; Boudeau, J.; Reid, J.L.; Mustard, K.J.; Udd, L.; Makela, T.P.; Alessi, D.R.; Hardie, D.G. Complexes between the LKB1 tumor suppressor, STRAD alpha/beta and MO25 alpha/beta are upstream kinases in the AMP-activated protein kinase cascade. J. Biol. 2003, 2, 28. [Google Scholar] [CrossRef]
- Harrison, P.T.; Vyse, S.; Huang, P.H. Rare epidermal growth factor receptor (EGFR) mutations in non-small cell lung cancer. Semin. Cancer Biol. 2020, 61, 167–179. [Google Scholar] [CrossRef]
- Bi, J.; Chowdhry, S.; Wu, S.; Zhang, W.; Masui, K.; Mischel, P.S. Altered cellular metabolism in gliomas—An emerging landscape of actionable co-dependency targets. Nat. Rev. Cancer 2020, 20, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G. AMPK: A target for drugs and natural products with effects on both diabetes and cancer. Diabetes 2013, 62, 2164–2172. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, G.R.; Carling, D. AMP-activated protein kinase: The current landscape for drug development. Nat. Rev. Drug Discov. 2019, 18, 527–551. [Google Scholar] [CrossRef]
- Hingst, J.R.; Kjobsted, R.; Birk, J.B.; Jorgensen, N.O.; Larsen, M.R.; Kido, K.; Larsen, J.K.; Kjeldsen, S.A.S.; Fentz, J.; Frosig, C.; et al. Inducible deletion of skeletal muscle AMPKalpha reveals that AMPK is required for nucleotide balance but dispensable for muscle glucose uptake and fat oxidation during exercise. Mol. Metab. 2020, 40, 101028. [Google Scholar] [CrossRef] [PubMed]
- Huet, C.; Boudaba, N.; Guigas, B.; Viollet, B.; Foretz, M. Glucose availability but not changes in pancreatic hormones sensitizes hepatic AMPK activity during nutritional transition in rodents. J. Biol. Chem. 2020, 295, 5836–5849. [Google Scholar] [CrossRef]
- Hardie, D.G. Regulation of AMP-activated protein kinase by natural and synthetic activator. Acta Pharm. Sin. B 2016, 6, 1–19. [Google Scholar] [CrossRef]
- Garcia, D.; Shaw, R.J. AMPK: Mechanisms of Cellular Energy Sensing and Restoration of Metabolic Balance. Mol. Cell 2017, 66, 789–800. [Google Scholar] [CrossRef] [PubMed]
- Woods, A.; Johnstone, S.R.; Dickerson, K.; Leiper, F.C.; Fryer, L.G.; Neumann, D.; Schlattner, U.; Wallimann, T.; Carlson, M.; Carling, D. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr. Biol. 2003, 13, 2004–2008. [Google Scholar] [CrossRef]
- Shaw, R.J.; Kosmatka, M.; Bardeesy, N.; Hurley, R.L.; Witters, L.A.; DePinho, R.A.; Cantley, L.C. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc. Natl. Acad. Sci. USA 2004, 101, 3329–3335. [Google Scholar] [CrossRef]
- Hemminki, A.; Markie, D.; Tomlinson, I.; Avizienyte, E.; Roth, S.; Loukola, A.; Bignell, G.; Warren, W.; Aminoff, M.; Hoglund, P.; et al. A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature 1998, 391, 184–187. [Google Scholar] [CrossRef]
- Jenne, D.E.; Reimann, H.; Nezu, J.; Friedel, W.; Loff, S.; Jeschke, R.; Muller, O.; Back, W.; Zimmer, M. Peutz-Jeghers syndrome is caused by mutations in a novel serine threonine kinase. Nat. Genet. 1998, 18, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Shackelford, D.B.; Shaw, R.J. The LKB1-AMPK pathway: Metabolism and growth control in tumour suppression. Nat. Rev. Cancer 2009, 9, 563–575. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.; Huang, L.L.; Chen, J.H.; Wu, J.; Xu, Q. The emerging treatment landscape of targeted therapy in non-small-cell lung cancer. Signal Transduct. Target. Ther. 2019, 4, 61. [Google Scholar] [CrossRef] [PubMed]
- De Rosa, V.; Iommelli, F.; Monti, M.; Fonti, R.; Votta, G.; Stoppelli, M.P.; Del Vecchio, S. Reversal of Warburg Effect and Reactivation of Oxidative Phosphorylation by Differential Inhibition of EGFR Signaling Pathways in Non-Small Cell Lung Cancer. Clin. Cancer Res. 2015, 21, 5110–5120. [Google Scholar] [CrossRef]
- Kang, J.H.; Lee, S.H.; Lee, J.S.; Nam, B.; Seong, T.W.; Son, J.; Jang, H.; Hong, K.M.; Lee, C.; Kim, S.Y. Aldehyde dehydrogenase inhibition combined with phenformin treatment reversed NSCLC through ATP depletion. Oncotarget 2016, 7, 49397–49410. [Google Scholar] [CrossRef]
- Kim, S.; Im, J.H.; Kim, W.K.; Choi, Y.J.; Lee, J.Y.; Kim, S.K.; Kim, S.J.; Kwon, S.W.; Kang, K.W. Enhanced Sensitivity of Nonsmall Cell Lung Cancer with Acquired Resistance to Epidermal Growth Factor Receptor-Tyrosine Kinase Inhibitors to Phenformin: The Roles of a Metabolic Shift to Oxidative Phosphorylation and Redox Balance. Oxid. Med. Cell Longev. 2021, 2021, 5428364. [Google Scholar] [CrossRef]
- Chen, N.; Zhou, Y.S.; Wang, L.C.; Huang, J.B. Advances in metforminbased metabolic therapy for nonsmall cell lung cancer (Review). Oncol. Rep. 2022, 47, 55. [Google Scholar] [CrossRef]
- Shackelford, D.B.; Abt, E.; Gerken, L.; Vasquez, D.S.; Seki, A.; Leblanc, M.; Wei, L.; Fishbein, M.C.; Czernin, J.; Mischel, P.S.; et al. LKB1 inactivation dictates therapeutic response of non-small cell lung cancer to the metabolism drug phenformin. Cancer Cell 2013, 23, 143–158. [Google Scholar] [CrossRef]
- Reisman, B.J.; Guo, H.; Ramsey, H.E.; Wright, M.T.; Reinfeld, B.I.; Ferrell, P.B.; Sulikowski, G.A.; Rathmell, W.K.; Savona, M.R.; Plate, L.; et al. Apoptolidin family glycomacrolides target leukemia through inhibition of ATP synthase. Nat. Chem. Biol. 2022, 18, 360–367. [Google Scholar] [CrossRef]
- Symersky, J.; Osowski, D.; Walters, D.E.; Mueller, D.M. Oligomycin frames a common drug-binding site in the ATP synthase. Proc. Natl. Acad. Sci. USA 2012, 109, 13961–13965. [Google Scholar] [CrossRef]
- Kalender, A.; Selvaraj, A.; Kim, S.Y.; Gulati, P.; Brule, S.; Viollet, B.; Kemp, B.E.; Bardeesy, N.; Dennis, P.; Schlager, J.J.; et al. Metformin, independent of AMPK, inhibits mTORC1 in a rag GTPase-dependent manner. Cell Metab. 2010, 11, 390–401. [Google Scholar] [CrossRef]
- Ben Sahra, I.; Tanti, J.F.; Bost, F. The combination of metformin and 2 deoxyglucose inhibits autophagy and induces AMPK-dependent apoptosis in prostate cancer cells. Autophagy 2010, 6, 670–671. [Google Scholar] [CrossRef] [PubMed]
- Ben Sahra, I.; Laurent, K.; Giuliano, S.; Larbret, F.; Ponzio, G.; Gounon, P.; Le Marchand-Brustel, Y.; Giorgetti-Peraldi, S.; Cormont, M.; Bertolotto, C.; et al. Targeting cancer cell metabolism: The combination of metformin and 2-deoxyglucose induces p53-dependent apoptosis in prostate cancer cells. Cancer Res. 2010, 70, 2465–2475. [Google Scholar] [CrossRef]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef]
- Shi, Y.; Lim, S.K.; Liang, Q.; Iyer, S.V.; Wang, H.Y.; Wang, Z.; Xie, X.; Sun, D.; Chen, Y.J.; Tabar, V.; et al. Gboxin is an oxidative phosphorylation inhibitor that targets glioblastoma. Nature 2019, 567, 341–346. [Google Scholar] [CrossRef]
- Sighel, D.; Notarangelo, M.; Aibara, S.; Re, A.; Ricci, G.; Guida, M.; Soldano, A.; Adami, V.; Ambrosini, C.; Broso, F.; et al. Inhibition of mitochondrial translation suppresses glioblastoma stem cell growth. Cell Rep. 2021, 35, 109024. [Google Scholar] [CrossRef]
- Rios, M.; Foretz, M.; Viollet, B.; Prieto, A.; Fraga, M.; Costoya, J.A.; Senaris, R. AMPK activation by oncogenesis is required to maintain cancer cell proliferation in astrocytic tumors. Cancer Res. 2013, 73, 2628–2638. [Google Scholar] [CrossRef]
- Chhipa, R.R.; Fan, Q.; Anderson, J.; Muraleedharan, R.; Huang, Y.; Ciraolo, G.; Chen, X.; Waclaw, R.; Chow, L.M.; Khuchua, Z.; et al. AMP kinase promotes glioblastoma bioenergetics and tumour growth. Nat. Cell. Biol. 2018, 20, 823–835. [Google Scholar] [CrossRef]
- Nguyen, M.H.; Imanishi, M.; Kurogi, T.; Wan, X.; Ishmael, J.E.; McPhail, K.L.; Smith, A.B., III. Synthetic Access to the Mandelalide Family of Macrolides: Development of an Anion Relay Chemistry Strategy. J. Org. Chem. 2018, 83, 4287–4306. [Google Scholar] [CrossRef]
- Laderoute, K.R.; Amin, K.; Calaoagan, J.M.; Knapp, M.; Le, T.; Orduna, J.; Foretz, M.; Viollet, B. 5′-AMP-activated protein kinase (AMPK) is induced by low-oxygen and glucose deprivation conditions found in solid-tumor microenvironments. Mol. Cell Biol. 2006, 26, 5336–5347. [Google Scholar] [CrossRef]
- Chou, T.C.; Talalay, P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv. Enzyme Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef]
- Chou, T.C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Molinski, T.F.; Dalisay, D.S.; Lievens, S.L.; Saludes, J.P. Drug development from marine natural products. Nat. Rev. Drug Discov. 2009, 8, 69–85. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Relative IC50 (nM) ± SEM | |||
|---|---|---|---|
| 24 h | 48 h | 72 h | |
| Wild-type | 28.2 ± 6.3 | 37.1 ± 0.1 | 33.0 ± 4.9 |
| AMPKα-null | 9.3 ± 1.3 | 13.5 ± 1.4 ** | 10.6 ± 1.9 * |
| Cell Line | LKB1 Status | EGFR Status | GI50 (nM) ± SEM | TGI (nM) ± SEM |
|---|---|---|---|---|
| 11-18 | Loss | L848R | 2.0 ± 0.4 | 174 ± 26 |
| H460 | Loss | WT | 4.3 ± 0.6 | 50.2 ± 8.9 |
| A549 | Loss | WT | ~300 | >3000 |
| H3122 | WT | WT | 4.7 ± 0.6 | 45.8 ± 13 |
| H292 | WT | WT | 3.4 ± 0.2 | 51.5 ± 12 |
| PC-9 | WT | E746- A750 del | 3.6 ± 0.7 | 133 ± 46 |
| Relative IC50 (nM) ± SEM | |||
|---|---|---|---|
| Mandelalide A | Erlotinib | Paclitaxel | |
| 11-18 | ~100 1 | ~100 1 | 2.94 ± 0.80 |
| PC-9 | ~100 1 | 5.24 ± 2.35 | 5.20 ± 2.13 |
| Relative IC50 (nM) ± SEM | ||||
|---|---|---|---|---|
| Cell Line | Mandelalide A 3 Day | Mandelalide A 6 Day | Erlotinib 3 Day | Erlotinib 6 Day |
| U87-MG | 0.38 ± 0.01 | 0.85 ± 0.11 | >300 | ~100 1 |
| U251 | 1.72 ± 0.22 | 1.21 ± 0.26 | >300 | >300 |
| SF-295 | >10 1 | 1.26 ± 0.23 | >300 | >300 |
| SF-268 | >300 | 1.07 ± 0.26 | >300 | >300 |
| U118-MG | >300 | 1.21 ± 0.19 | >300 | >300 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mattos, D.R.; Wan, X.; Serrill, J.D.; Nguyen, M.H.; Humphreys, I.R.; Viollet, B.; Smith, A.B., III; McPhail, K.L.; Ishmael, J.E. The Marine-Derived Macrolactone Mandelalide A Is an Indirect Activator of AMPK. Mar. Drugs 2022, 20, 418. https://doi.org/10.3390/md20070418
Mattos DR, Wan X, Serrill JD, Nguyen MH, Humphreys IR, Viollet B, Smith AB III, McPhail KL, Ishmael JE. The Marine-Derived Macrolactone Mandelalide A Is an Indirect Activator of AMPK. Marine Drugs. 2022; 20(7):418. https://doi.org/10.3390/md20070418
Chicago/Turabian StyleMattos, Daphne R., Xuemei Wan, Jeffrey D. Serrill, Minh H. Nguyen, Ian R. Humphreys, Benoit Viollet, Amos B. Smith, III, Kerry L. McPhail, and Jane E. Ishmael. 2022. "The Marine-Derived Macrolactone Mandelalide A Is an Indirect Activator of AMPK" Marine Drugs 20, no. 7: 418. https://doi.org/10.3390/md20070418
APA StyleMattos, D. R., Wan, X., Serrill, J. D., Nguyen, M. H., Humphreys, I. R., Viollet, B., Smith, A. B., III, McPhail, K. L., & Ishmael, J. E. (2022). The Marine-Derived Macrolactone Mandelalide A Is an Indirect Activator of AMPK. Marine Drugs, 20(7), 418. https://doi.org/10.3390/md20070418