
Connection of Isolated Stereoclusters by Combining 13C-RCSA, RDC, and J-Based Configurational Analyses and Structural Revision of a Tetraprenyltoluquinol Chromane Meroterpenoid from Sargassum muticum

 , , ,
, , ,  and
and 
Abstract
:1. Introduction
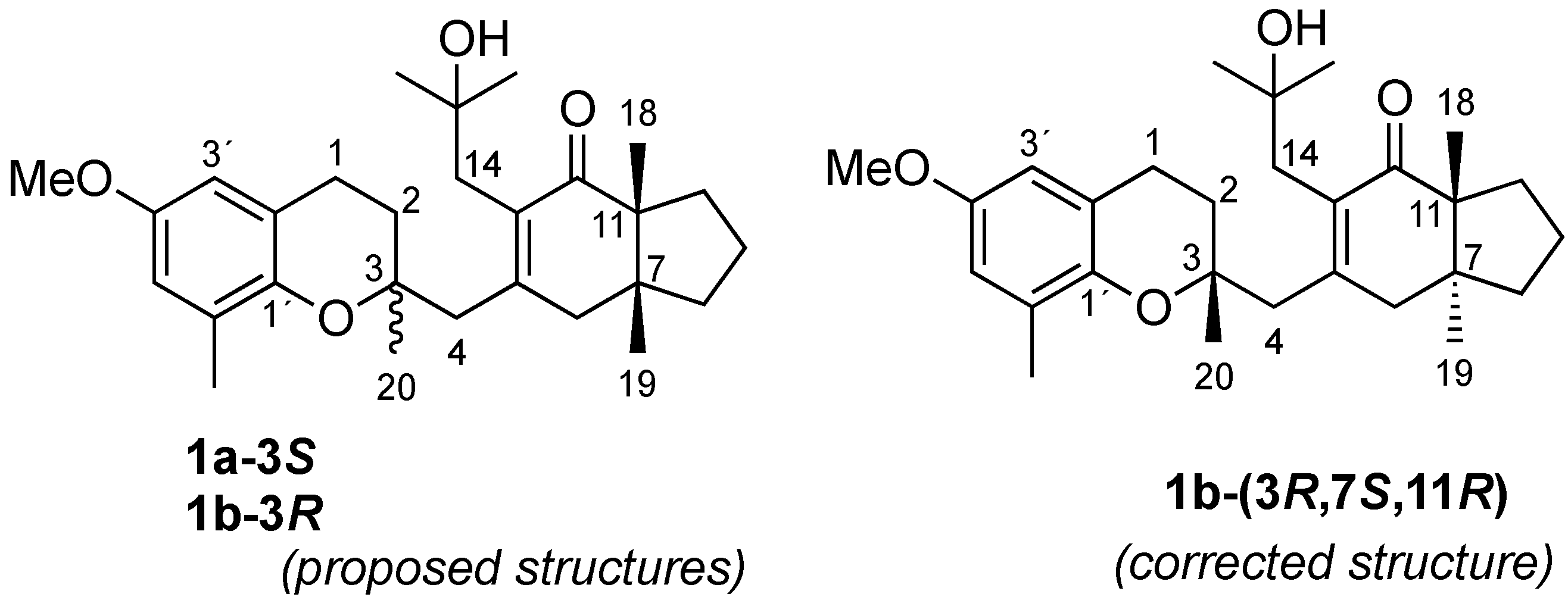
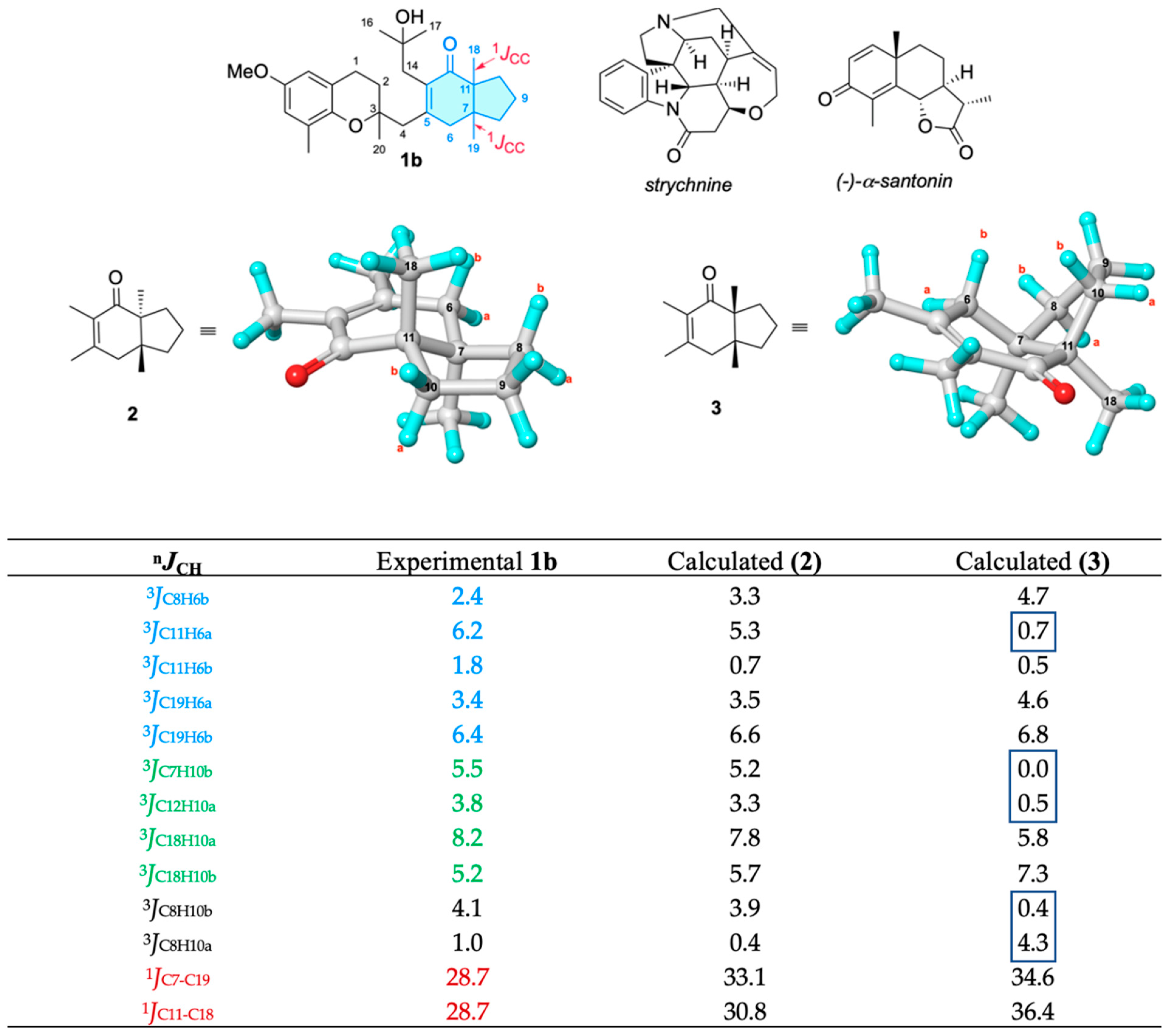
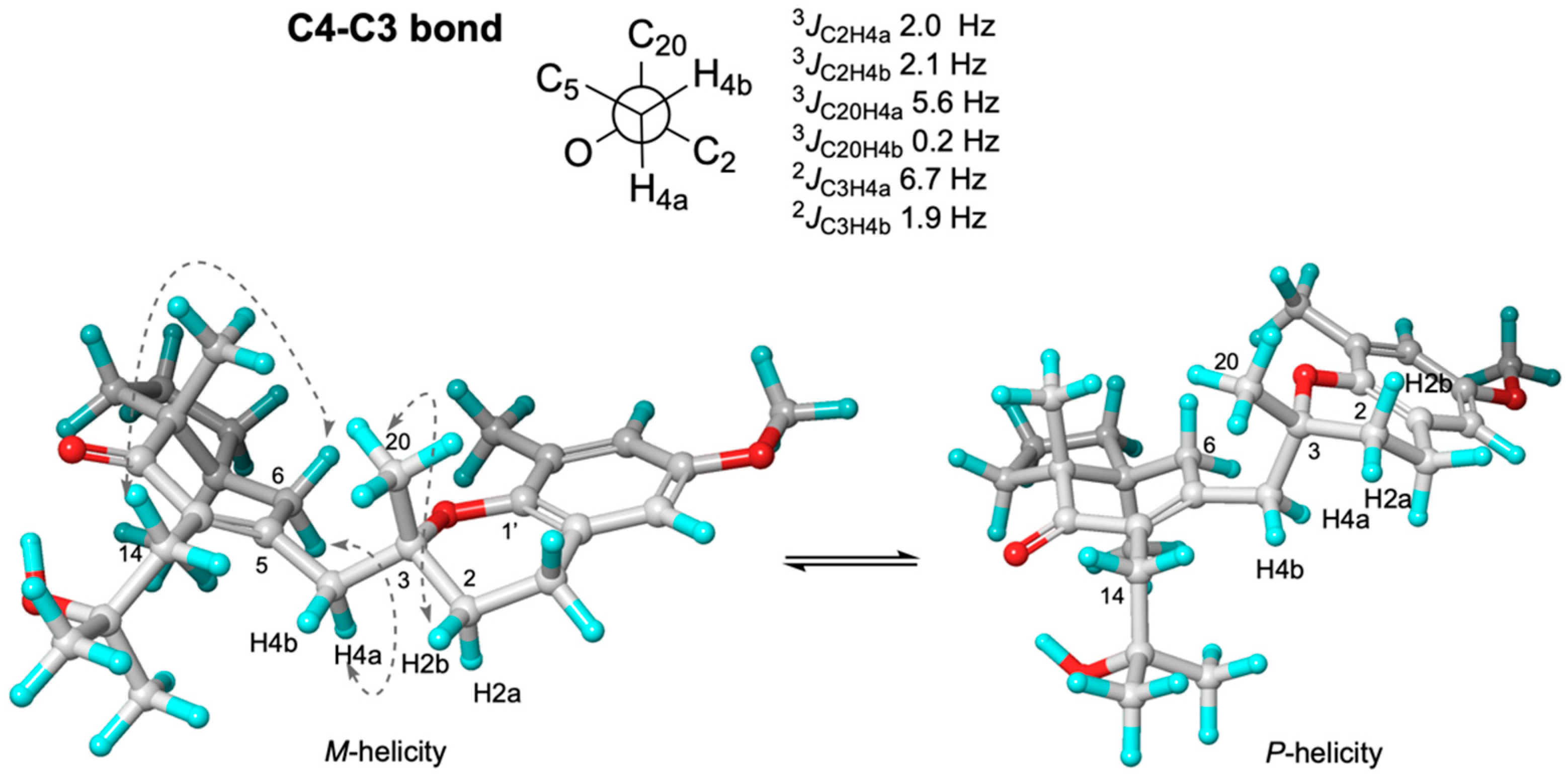
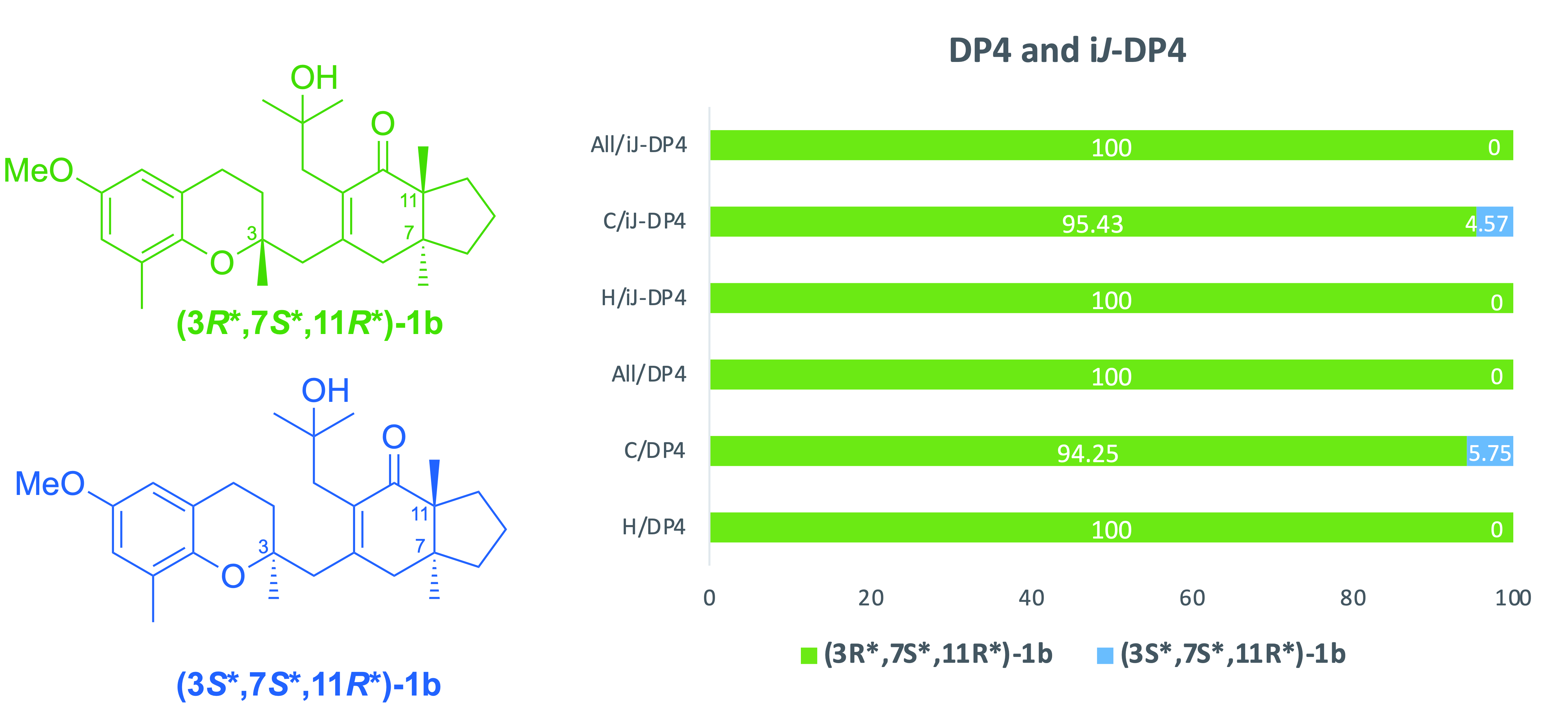
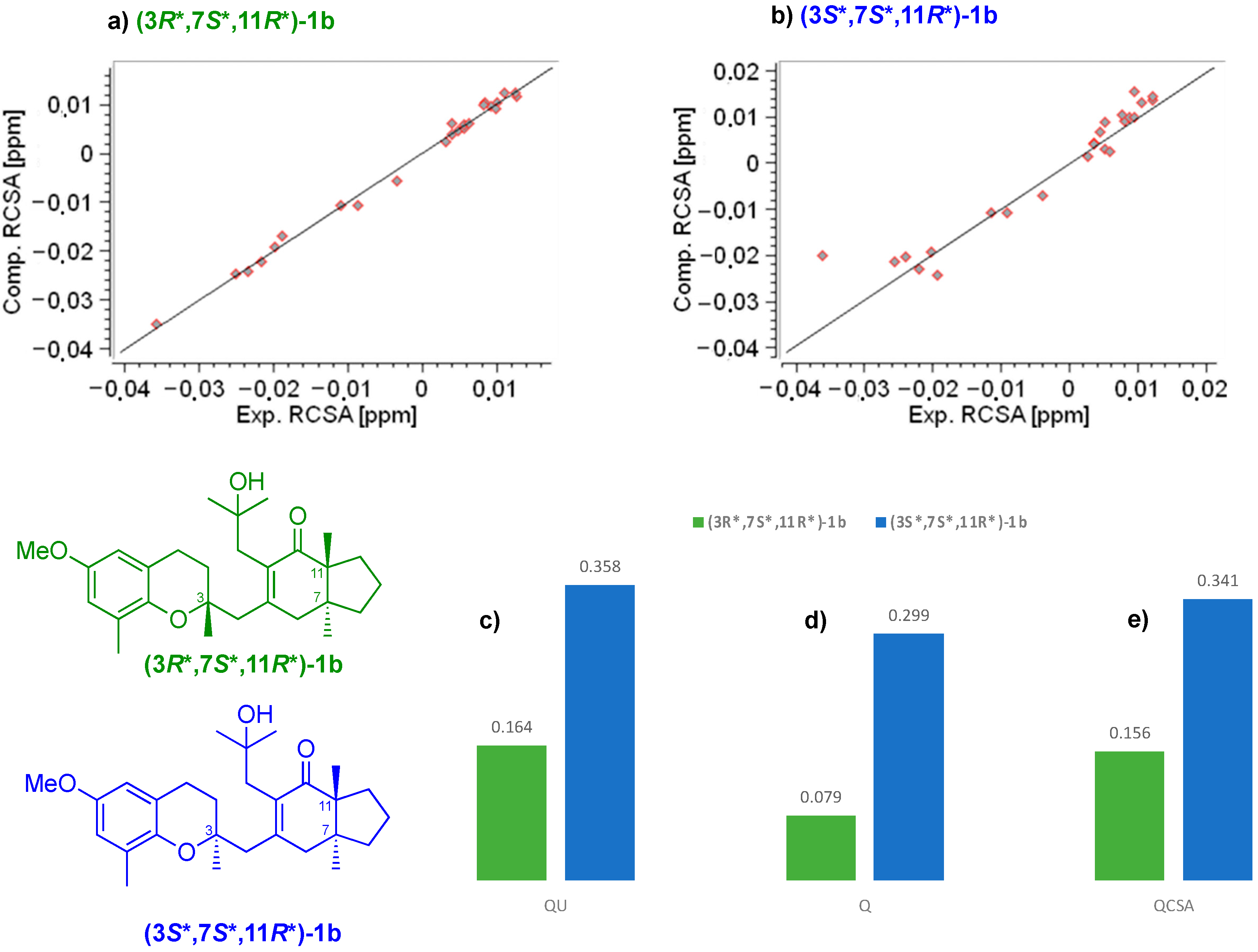
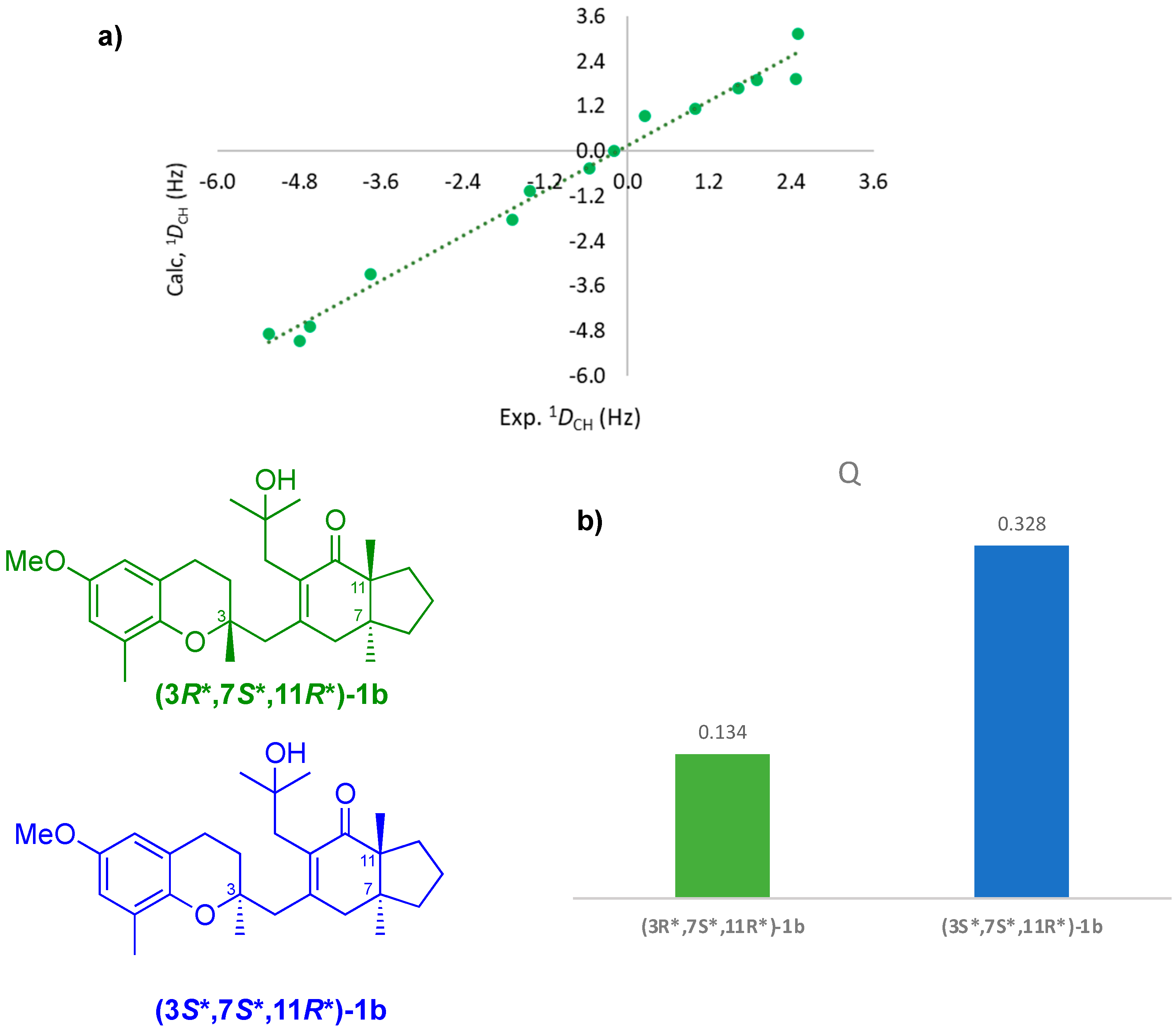
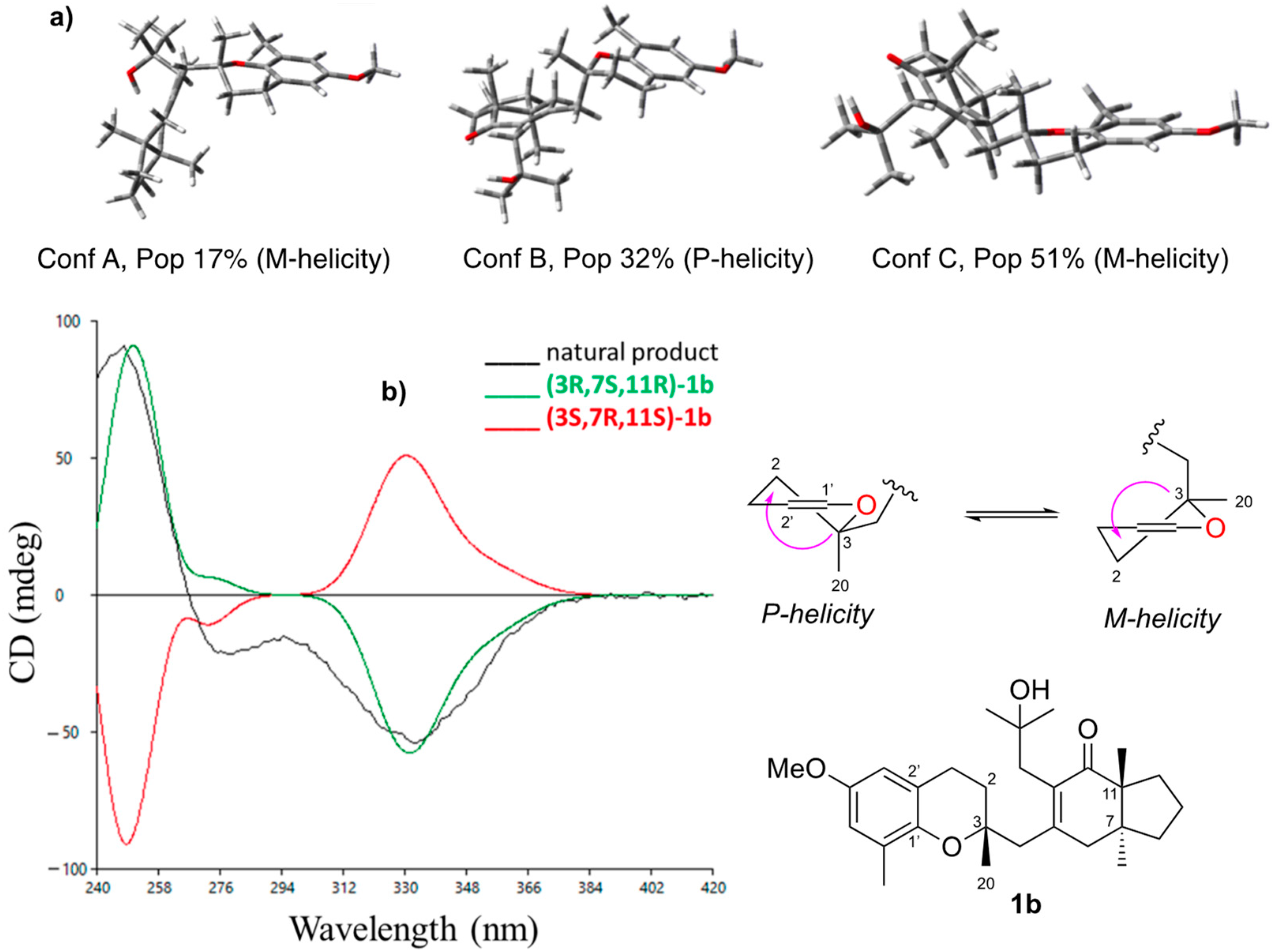
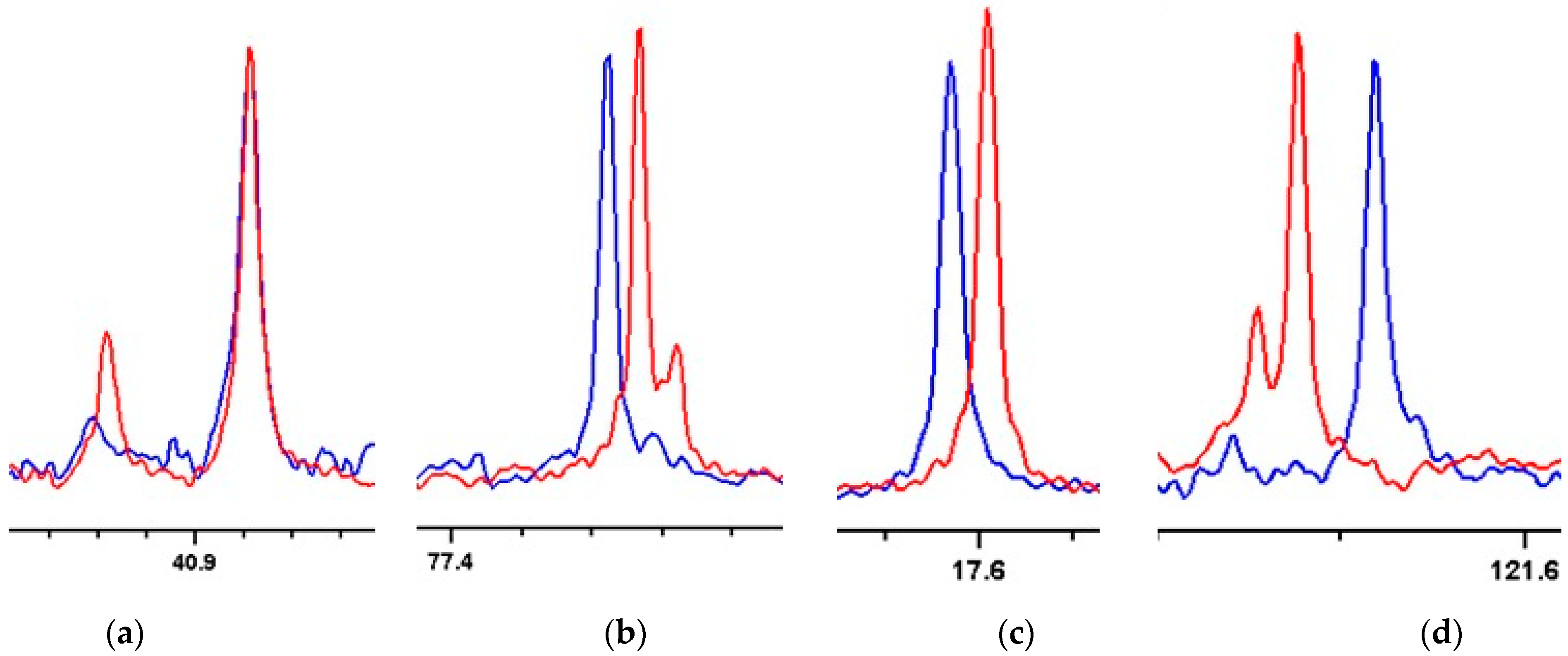
2. Results and Discussion
3. Materials and Methods
3.1. General
3.2. Data Processing
3.3. Raw Material
3.4. Extraction and Isolation
Purification of 1b
3.5. Computational Section
J-Coupling Calculation
3.6. 13C-RCSA Measurements
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Norton, T.A. The Growth and Development of Sargassum Muticum (Yendo) Fensholt. J. Exp. Mar. Biol. Ecol. 1977, 26, 41–53. [Google Scholar] [CrossRef]
- Critchley, A.T. Sargassum Muticum: A Taxonomic History Including World-Wide and Western Pacific Distributions. J. Mar. Biol. Assoc. U. K. 1983, 63, 617–625. [Google Scholar] [CrossRef]
- Davison, D.M. Sargassum Muticum in Scotland, 2008: A Review of Information, Issues and Implications; Commissioned Report No. 324; Scottish Natural Heritage: Perth, Scotland, 2009. [Google Scholar]
- Balboa, E.; Conde, E.; Constenla, A.; Falqué, E.; Domínguez, H. Sensory Evaluation and Oxidative Stability of a Suncream Formulated with Thermal Spring Waters from Ourense (NW Spain) and Sargassum Muticum Extracts. Cosmetics 2017, 4, 19. [Google Scholar] [CrossRef]
- Park, S.Y.; Seo, I.S.; Lee, S.J.; Lee, S.P. Study on the Health Benefits of Brown Algae Sargassum Muticum in Volunteers. J. Food Nutr. Res. 2015, 3, 126–130. [Google Scholar] [CrossRef] [Green Version]
- Heo, S.J.; Jeon, Y.J. Protective Effect of Fucoxanthin Isolated from Sargassum Siliquastrum on UV-B Induced Cell Damage. J. Photochem. Photobiol. B Biol. 2009, 95, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Nazir, M.; Saleem, M.; Tousif, M.I.; Anwar, M.A.; Surup, F.; Ali, I.; Wang, D.; Mamadalieva, N.Z.; Alshammari, E.; Ashour, M.L.; et al. Meroterpenoids: A Comprehensive Update Insight on Structural Diversity and Biology. Biomolecules 2021, 11, 957. [Google Scholar] [CrossRef]
- Kim, J.A.; Ahn, B.N.; Kong, C.S.; Kim, S.K. The Chromene Sargachromanol e Inhibits Ultraviolet A-Induced Ageing of Skin in Human Dermal Fibroblasts. Br. J. Dermatol. 2013, 168, 968–976. [Google Scholar] [CrossRef]
- Kang, H.S.; Kim, J.P. New Chromene Derivatives with Radical Scavenging Activities from the Brown Alga Sargassum Siliquastrum. J. Chem. Res. 2017, 41, 116–119. [Google Scholar] [CrossRef]
- Balboa, E.M.; Li, Y.-X.; Ahn, B.-N.; Eom, S.-H.; Domínguez, H.; Jiménez, C.; Rodríguez, J. Photodamage Attenuation Effect by a Tetraprenyltoluquinol Chromane Meroterpenoid Isolated from Sargassum Muticum. J. Photochem. Photobiol. B: Biol. 2015, 148, 51–58. [Google Scholar] [CrossRef]
- Ferdous, U.T.; Yusof, Z.N.B. Algal Terpenoids: A Potential Source of Antioxidants for Cancer Therapy. In Terpenes and Terpenoids; Perveen, S., Al-Taweel, A.M., Eds.; IntechOpen: Rijeka, Croatia, 2021. [Google Scholar]
- Fisch, K.M.; Böhm, V.; Wright, A.D.; König, G.M. Antioxidative Meroterpenoids from the Brown Alga Cystoseira Crinita. J. Nat. Prod. 2003, 66, 968–975. [Google Scholar] [CrossRef]
- Valls, R.; Piovetti, L.; Banaigs, B.; Praud, A. Secondary Metabolites from Morocco Brown Algae of the Genus Cystoseira. Phytochemistry 1993, 32, 961–966. [Google Scholar] [CrossRef]
- de Sousa, C.B.; Gangadhar, K.N.; Morais, T.R.; Conserva, G.A.A.; Vizetto-Duarte, C.; Pereira, H.; Laurenti, M.D.; Campino, L.; Levy, D.; Uemi, M.; et al. Antileishmanial Activity of Meroditerpenoids from the Macroalgae Cystoseira Baccata. Exp. Parasitol. 2017, 174, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Amico, V.; Cunsolo, F.; Oriente, G.; Piattelli, M.; Ruberto, G. Cystoketal, a New Metabolite From the Brown Alga Cystoseira Balearica. J. Nat. Prod. 1984, 47, 947–952. [Google Scholar] [CrossRef]
- Nath, N.; Schmidt, M.; Gil, R.R.; Williamson, R.T.; Martin, G.E.; Navarro-Vázquez, A.; Griesinger, C.; Liu, Y. Determination of Relative Configuration from Residual Chemical Shift Anisotropy. J. Am. Chem. Soc. 2016, 138, 9548–9556. [Google Scholar] [CrossRef]
- Gayathri, C.; Tsarevsky, N.V.; Gil, R.R. Residual Dipolar Couplings (RDCs) Analysis of Small Molecules Made Easy: Fast and Tuneable Alignment by Reversible Compression/Relaxation of Reusable PMMA Gels. Chem. –A Eur. J. 2010, 16, 3622–3626. [Google Scholar] [CrossRef]
- Matsumori, N.; Kaneno, D.; Murata, M.; Nakamura, H.; Tachibana, K. Stereochemical Determination of Acyclic Structures Based on Carbon-Proton Spin-Coupling Constants. A Method of Configuration Analysis for Natural Products. J. Org. Chem. 1999, 64, 866–876. [Google Scholar] [CrossRef]
- Matsumori, N.; Murata, M.; Tachibana, K. Conformational Analysis of Natural Products Using Long-Range Carbon-Proton Coupling Constants: Three-Dimensional Structure of Okadaic Acid in Solution. Tetrahedron 1995, 51, 12229–12238. [Google Scholar] [CrossRef]
- Pachler, K.G.R. Nuclear Magnetic Resonance Study of Some α-Amino Acids—II. Rotational Isomerism. Spectrochim. Acta 1964, 20, 581–587. [Google Scholar] [CrossRef]
- Pachler, K.G.R. Nuclear Magnetic Resonance Study of Some α-Amino Acids—I. Spectrochim. Acta 1963, 19, 2085–2092. [Google Scholar] [CrossRef]
- Nath, N.; Fuentes-Monteverde, J.C.; Pech-Puch, D.; Rodríguez, J.; Jiménez, C.; Noll, M.; Kreiter, A.; Reggelin, M.; Navarro-Vázquez, A.; Griesinger, C. Relative Configuration of Micrograms of Natural Compounds Using Proton Residual Chemical Shift Anisotropy. Nat. Commun. 2020, 11, 1–9. [Google Scholar] [CrossRef]
- Teles, R.R.; França, J.A.; Navarro-Vázquez, A.; Hallwass, F. Atribuição da Estereoquímica da α-santonina através das medidas do Acoplamento Dipolar Residual. Química Nova 2015, 38, 1345–1350. [Google Scholar]
- Butts, C.P.; Jones, C.R.; Harvey, J.N. High Precision NOEs as a Probe for Low Level Conformers—A Second Conformation of Strychnine. Chem. Commun. 2011, 47, 1193–1195. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Jordan, K.D. Comparison of Density Functional and MP2 Calculations on the Water Monomer and Dimer. J. Phys. Chem. 1994, 98, 10089–10094. [Google Scholar] [CrossRef]
- Ernzerhof, M.; Scuseria, G.E. Assessment of the Perdew–Burke–Ernzerhof Exchange-Correlation Functional. J. Chem. Phys. 1999, 110, 5029–5036. [Google Scholar] [CrossRef] [Green Version]
- Adamo, C.; Barone, V. Exchange Functionals with Improved Long-Range Behavior and Adiabatic Connection Methods without Adjustable Parameters: The MPW and MPW1PW Models. J. Chem. Phys. 1998, 108, 664–675. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward Reliable Density Functional Methods without Adjustable Parameters: The PBE0 Model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Heyd, J.; Scuseria, G.E.; Ernzerhof, M. Hybrid Functionals Based on a Screened Coulomb Potential. J. Chem. Phys. 2003, 118, 8207–8215. [Google Scholar] [CrossRef] [Green Version]
- Handy, N.C.; Cohen, A.J. Left-Right Correlation Energy. Mol. Phys. 2001, 99, 403–412. [Google Scholar] [CrossRef]
- Feller, D. The Role of Databases in Support of Computational Chemistry Calculations. J. Comput. Chem. 1996, 17, 1571–1586. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Weigend, F. Accurate Coulomb-Fitting Basis Sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Klamt, A.; Schüürmann, G. COSMO: A New Approach to Dielectric Screening in Solvents with Explicit Expressions for the Screening Energy and Its Gradient. J. Chem. Soc. Perkin Trans. 2 1993, 2, 799–805. [Google Scholar] [CrossRef]
- Miertuš, S.; Scrocco, E.; Tomasi, J. Electrostatic Interaction of a Solute with a Continuum. A Direct Utilization of AB Initio Molecular Potentials for the Prevision of Solvent Effects. Chem. Phys. 1981, 55, 117–129. [Google Scholar] [CrossRef]
- Grimblat, N.; Gavín, J.A.; Hernández Daranas, A.; Sarotti, A.M. Combining the Power of J Coupling and DP4 Analysis on Stereochemical Assignments: The J-DP4 Methods. Org. Lett. 2019, 21, 4003–4007. [Google Scholar] [CrossRef] [PubMed]
- Tarazona, G.; Fernández, R.; Pérez, M.; Millán, R.E.; Jiménez, C.; Rodríguez, J.; Cuevas, C. Enigmazole C: A Cytotoxic Macrocyclic Lactone and Its Ring-Opened Derivatives from a New Species of Homophymia Sponge. J. Nat. Prod. 2022, 85, 1059–1066. [Google Scholar] [CrossRef]
- Smith, S.G.; Goodman, J.M. Assigning Stereochemistry to Single Diastereoisomers by GIAO NMR Calculation: The DP4 Probability. J. Am. Chem. Soc. 2010, 132, 12946–12959. [Google Scholar] [CrossRef]
- Troche-Pesqueira, E.; Anklin, C.; Gil, R.R.; Navarro-Vázquez, A. Computer-Assisted 3D Structure Elucidation of Natural Products Using Residual Dipolar Couplings. Angew. Chem. Int. Ed. 2017, 56, 3660–3664. [Google Scholar] [CrossRef]
- Navarro-Vázquez, A.; Gil, R.R.; Blinov, K. Computer-Assisted 3D Structure Elucidation (CASE-3D) of Natural Products Combining Isotropic and Anisotropic NMR Parameters. J. Nat. Prod. 2018, 81, 203–210. [Google Scholar] [CrossRef]
- Liu, Y.; Cohen, R.D.; Gustafson, K.R.; Martin, G.E.; Williamson, R.T. Enhanced Measurement of Residual Chemical Shift Anisotropy for Small Molecule Structure Elucidation. Chem. Commun. 2018, 54, 4254–4257. [Google Scholar] [CrossRef]
- Liu, Y.; Saurí, J.; Mevers, E.; Peczuh, M.W.; Hiemstra, H.; Clardy, J.; Martin, G.E.; Williamson, R.T. Unequivocal Determination of Complex Molecular Structures Using Anisotropic NMR Measurements. Science (1979) 2017, 356, eaam5349. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, M.; Sun, H.; Rogne, P.; Scriba, G.K.E.; Griesinger, C.; Kuhn, L.T.; Reinscheid, U.M. Determining the Absolute Configuration of (+)−Mefloquine HCl, the Side-Effect-Reducing Enantiomer of the Antimalaria Drug Lariam. J. Am. Chem. Soc. 2012, 134, 3080–3083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tukey, J.W. Abstracts of Papers. Ann. Math. Stat. 1958, 29, 614–623. [Google Scholar] [CrossRef]
- Thiele, C.M.; Bermel, W. Speeding up the Measurement of One-Bond Scalar (1J) and Residual Dipolar Couplings (1D) by Using Non-Uniform Sampling (NUS). J. Magn. Reson. 2012, 216, 134–143. [Google Scholar] [CrossRef]
- Furrer, J.; John, M.; Kessler, H.; Luy, B. J-Spectroscopy in the Presence of Residual Dipolar Couplings: Determination of One-Bond Coupling Constants and Scalable Resolution. J. Biomol. NMR 2007, 37, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Antus, S.; Snatzke, G.; Steinke, I. Circulardichroismus, LXXXI. Synthese Und Circulardichroismus von Steroiden Mit Isochromanon-Chromophor. Liebigs Ann. Der Chem. 1983, 1983, 2247–2261. [Google Scholar] [CrossRef]
- Nozoe, S.; Hirai, K.; Snatzke, F.; Snaizke, G. Circular Dichroism-LXIV, On The Chiroptical Properties Of Siccamn Derivatives And The Absolute Configuration Of Siccanochromene-A. Tetrahedron 1974, 30, 2773–2776. [Google Scholar] [CrossRef]
- Polavarapu, P.L.; Chakraborty, D.K. Absolute Stereochemistry of Chiral Molecules from Ab Initio Theoretical and Experimental Molecular Optical Rotations. J. Am. Chem. Soc. 1998, 120, 6160–6164. [Google Scholar] [CrossRef]
- Stephens, P.J.; McCann, D.M.; Cheeseman, J.R.; Frisch, M.J. Determination of Absolute Configurations of Chiral Molecules Using Ab Initio Time-Dependent Density Functional Theory Calculations of Optical Rotation: How Reliable Are Absolute Configurations Obtained for Molecules with Small Rotations? Chirality 2005, 17, S52–S64. [Google Scholar] [CrossRef]
- Polavarapu, P.L. Optical Rotation: Recent Advances in Determining the Absolute Configuration. Chirality 2002, 14, 768–781. [Google Scholar] [CrossRef]
- Górecki, M.; Suszczyńska, A.; Woźnica, M.; Baj, A.; Wolniak, M.; Cyrański, M.K.; Witkowski, S.; Frelek, J. Chromane Helicity Rule-Scope and Challenges Based on an ECD Study of Various Trolox Derivatives. Org. Biomol. Chem. 2014, 12, 2235–2254. [Google Scholar] [CrossRef]
- Batista, J.M.; Batista, A.N.L.; Rinaldo, D.; Vilegas, W.; Cass, Q.B.; Bolzani, V.S.; Kato, M.J.; López, S.N.; Furlan, M.; Nafie, L.A. Absolute Configuration Reassignment of Two Chromanes from Peperomia Obtusifolia (Piperaceae) Using VCD and DFT Calculations. Tetrahedron Asymmetry 2010, 21, 2402–2407. [Google Scholar] [CrossRef]
- Pescitelli, G.; Bruhn, T. Good Computational Practice in the Assignment of Absolute Configurations by TDDFT Calculations of ECD Spectra. Chirality 2016, 28, 466–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melo Sousa, C.M.; Giordani, R.B.; Almeida, W.A.M.; Griesinger, C.; Gil, R.R.; Navarro-Vázquez, A.; Hallwass, F. Effect of the Solvent on the Conformation of Monocrotaline as Determined by Isotropic and Anisotropic NMR Parameters. Magn. Reson. Chem. 2021, 59, 561–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolmer, A.; Edwards, L.J.; Kuprov, I.; Thiele, C.M. Conformational Analysis of Small Organic Molecules Using NOE and RDC Data: A Discussion of Strychnine and α-Methylene-γ-Butyrolactone. J. Magn. Reson. 2015, 261, 101–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stephens, P.J.; Devlin, F.J.; Cheeseman, J.R.; Frisch, M.J. Calculation of Optical Rotation Using Density Functional Theory. J. Phys. Chem. A 2001, 105, 5356–5371. [Google Scholar] [CrossRef]
- Bruhn, T.; Schaumlöffel, A.; Hemberger, Y.; Bringmann, G. SpecDis: Quantifying the Comparison of Calculated and Experimental Electronic Circular Dichroism Spectra. Chirality 2013, 25, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An Advanced Semantic Chemical Editor, Visualization, and Analysis Platform. J. Cheminformatics 2012, 4, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Anta, C.; González, N.; Rodríguez, J.; Jiménez, C. A New Secosterol from the Indonesian Octocoral Pachyclavularia Violacea. J. Nat. Prod. 2002, 65, 1357–1359. [Google Scholar] [CrossRef]
- Palermo, G.; Riccio, R.; Bifulco, G. Effect of Electronegative Substituents and Angular Dependence on the Heteronuclear Spin−Spin Coupling Constant 3 J C−H: An Empirical Prediction Equation Derived by Density Functional Theory Calculations. J. Org. Chem. 2010, 75, 1982–1991. [Google Scholar] [CrossRef]
- Haasnoot, C.A.G.; de Leeuw, F.A.A.M.; Altona, C. The Relationship between Proton-Proton NMR Coupling Constants and Substituent Electronegativities—I. Tetrahedron 1980, 36, 2783–2792. [Google Scholar] [CrossRef]
- Hallwass, F.; Schmidt, M.; Sun, H.; Mazur, A.; Kummerlöwe, G.; Luy, B.; Navarro-Vázquez, A.; Griesinger, C.; Reinscheid, U.M. Residual Chemical Shift Anisotropy (RCSA): A Tool for the Analysis of the Configuration of Small Molecules. Angew. Chem. Int. Ed. 2011, 50, 9487–9490. [Google Scholar] [CrossRef] [PubMed]
- Hallwass, F.; Teles, R.R.; Hellemann, E.; Griesinger, C.; Gil, R.R.; Navarro-Vázquez, A. Measurement of Residual Chemical Shift Anisotropies in Compressed Polymethylmethacrylate Gels. Automatic Compensation of Gel Isotropic Shift Contribution. Magn. Reson. Chem. 2018, 56, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Cornilescu, G.; Marquardt, J.L.; Ottiger, M.; Bax, A. Validation of Protein Structure from Anisotropic Carbonyl Chemical Shifts in a Dilute Liquid Crystalline Phase. J. Am. Chem. Soc. 1998, 120, 6836–6837. [Google Scholar] [CrossRef]
- Losonczi, J.A.; Andrec, M.; Fischer, M.W.F.; Prestegard, J.H. Order Matrix Analysis of Residual Dipolar Couplings Using Singular Value Decomposition. J. Magn. Reson. 1999, 138, 334–342. [Google Scholar] [CrossRef] [Green Version]
- Navarro-Vázquez, A. MSpin-RDC. A Program for the Use of Residual Dipolar Couplings for Structure Elucidation of Small Molecules. Magn. Reson. Chem. 2012, 50, S73–S79. [Google Scholar] [CrossRef]
- Hellemann, E.; Teles, R.R.; Hallwass, F.; Barros, W.; Navarro-Vázquez, A.; Gil, R.R. Mechanical Behavior of Polymer Gels for RDCs and RCSAs Collection: NMR Imaging Study of Buckling Phenomena. Chem. –A Eur. J. 2016, 22, 16632–16635. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1b | ||
|---|---|---|
| Position | δC, mult. a,b | δH, mult., J (in Hz) c |
| 1 | 23.06 CH2 | 2.774 (t, 6.9) |
| 2 | 34.04 CH2 | H2a: 1.859 (dt, 13.5, 6.9) |
| H2b: 1.805 (dt, 13.5, 6.9) | ||
| 3 | 76.80 qC | - |
| 4 | 45.15 CH2 | H4b: 2.705 (d, 13.7) |
| H4a: 2.514 (d, 13.7) | ||
| 5 | 155.10 qC | - |
| 6 | 44.77 CH2 | H6b: 3.028 (d, 18.7) |
| H6a: 2.237 (d, 18.7) | ||
| 7 | 45.25 qC | - |
| 8 | 35.38 CH2 | H8b: 1.754 (m) |
| H8a: 1.520 (m) | ||
| 9 | 19.32 CH2 | 1.744 (m) |
| 10 | 30.03 CH2 | H10a: 1.944 (td, 12.0, 11.9, 6.8) |
| H10b: 1.441 (ddd, 13.1, 8.4, 3.1) | ||
| 11 | 55.42 qC | - |
| 12 | 209.32 qC | - |
| 13 | 133.45 qC | - |
| 14 | 40.37 CH2 | H14b: 2.570 (d, 14.3) |
| H14a: 2.506 (d, 14.3) | ||
| 15 | 71.17 qC | - |
| Me16- | 31.88 CH3 | 1.235 (s) |
| Me17- | 29.08 CH3 | 1.041 (s) |
| Me18- | 21.51 CH3 | 1.105 (s) |
| Me19- | 22.71 CH3 | 0.804 (s) |
| Me20- | 24.28 CH3 | 1.225 (s) |
| 1′ | 145.77 qC | - |
| 2′ | 121.16 qC | - |
| 3′ | 111.58 CH | 6.450 (d, 3.0) |
| 4′ | 153.13 qC | - |
| 5′ | 115.64 CH | 6.563 (d, 3.0) |
| 6′ | 127.43 qC | - |
| MeO-4′ | 55.97 CH3 | 3.701 (s) |
| Me-6′ | 17.08 CH3 | 2.165 (s) |
| OH | - | 3.983 (br s) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fuentes-Monteverde, J.C.C.; Nath, N.; Forero, A.M.; Balboa, E.M.; Navarro-Vázquez, A.; Griesinger, C.; Jiménez, C.; Rodríguez, J. Connection of Isolated Stereoclusters by Combining 13C-RCSA, RDC, and J-Based Configurational Analyses and Structural Revision of a Tetraprenyltoluquinol Chromane Meroterpenoid from Sargassum muticum. Mar. Drugs 2022, 20, 462. https://doi.org/10.3390/md20070462
Fuentes-Monteverde JCC, Nath N, Forero AM, Balboa EM, Navarro-Vázquez A, Griesinger C, Jiménez C, Rodríguez J. Connection of Isolated Stereoclusters by Combining 13C-RCSA, RDC, and J-Based Configurational Analyses and Structural Revision of a Tetraprenyltoluquinol Chromane Meroterpenoid from Sargassum muticum. Marine Drugs. 2022; 20(7):462. https://doi.org/10.3390/md20070462
Chicago/Turabian StyleFuentes-Monteverde, Juan Carlos C., Nilamoni Nath, Abel M. Forero, Elena M. Balboa, Armando Navarro-Vázquez, Christian Griesinger, Carlos Jiménez, and Jaime Rodríguez. 2022. "Connection of Isolated Stereoclusters by Combining 13C-RCSA, RDC, and J-Based Configurational Analyses and Structural Revision of a Tetraprenyltoluquinol Chromane Meroterpenoid from Sargassum muticum" Marine Drugs 20, no. 7: 462. https://doi.org/10.3390/md20070462
APA StyleFuentes-Monteverde, J. C. C., Nath, N., Forero, A. M., Balboa, E. M., Navarro-Vázquez, A., Griesinger, C., Jiménez, C., & Rodríguez, J. (2022). Connection of Isolated Stereoclusters by Combining 13C-RCSA, RDC, and J-Based Configurational Analyses and Structural Revision of a Tetraprenyltoluquinol Chromane Meroterpenoid from Sargassum muticum. Marine Drugs, 20(7), 462. https://doi.org/10.3390/md20070462