
Marine-Fungi-Derived Gliotoxin Promotes Autophagy to Suppress Mycobacteria tuberculosis Infection in Macrophage

 ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
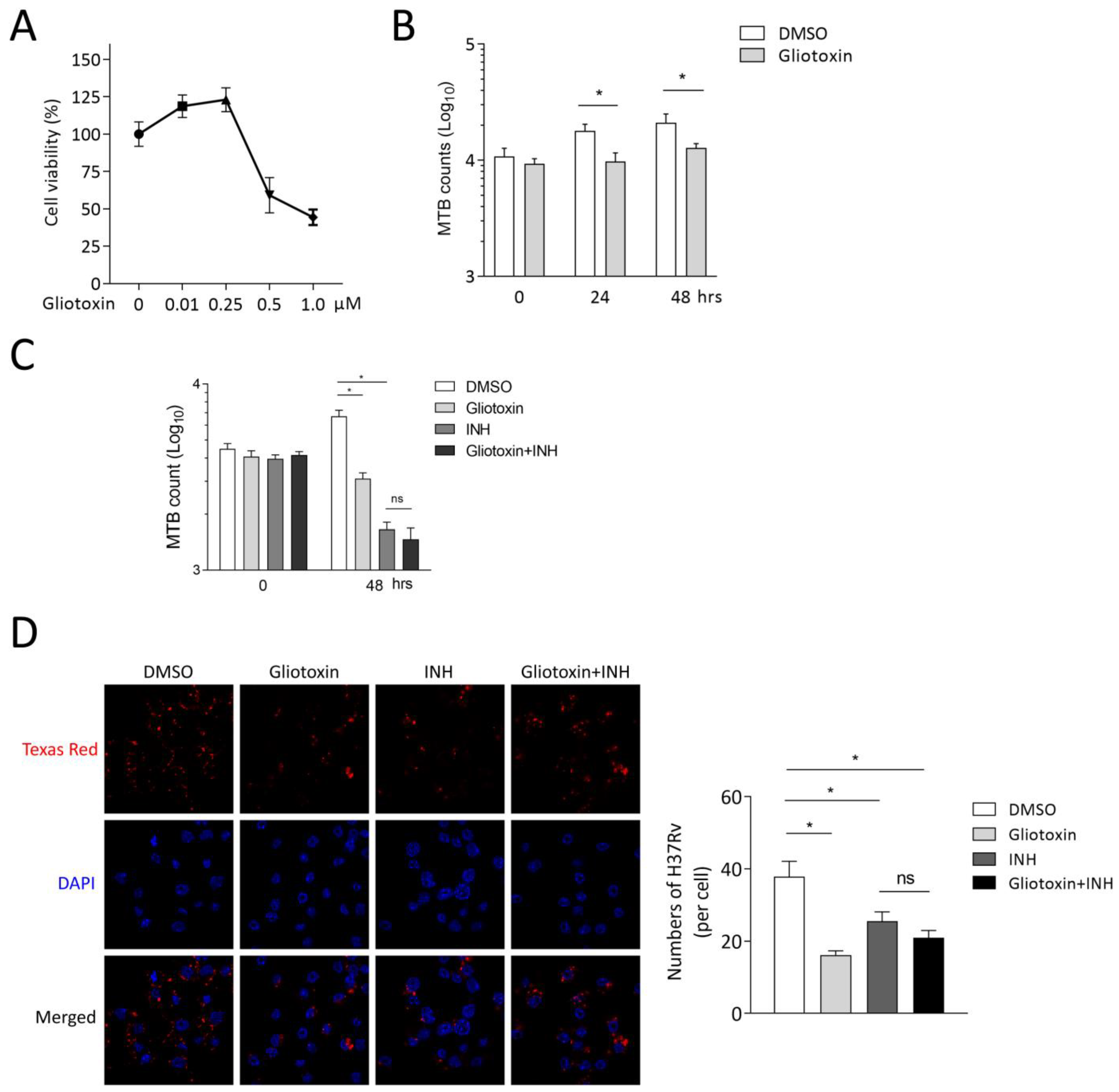
2.1. Gliotoxin Inhibits MTB Infection in THP-1-Derived Macrophages
2.2. Gliotoxin Suppresses MTB Infection in RAW 264.7
2.3. Gliotoxin Promotes Autophagy upon MTB Infection
2.4. Gliotoxin Inhibits MTB Infection by Promoting Autophagy Machinery
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Reagents
4.3. CCK-8 Assay
4.4. Colony-Forming Units (CFU) Assay
4.5. RNA Extraction and Real-Time Quantitative Reverse-Transcription Polymerase Chain Reaction (qRT-PCR)
4.6. Western Blot Analysis
4.7. Reactive-Oxygen-Species (ROS) Detection
4.8. Confocal Laser Scanning Microscopy Analysis
4.9. Flow Cytometry (FCM) Analysis
4.10. Enzyme-Linked Immunosorbent Assay (ELISA) Analysis
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Hameed, H.M.A.; Islam, M.M.; Chhotaray, C.; Wang, C.; Liu, Y.; Tan, Y.J.; Li, X.J.; Tan, S.Y.; Delorme, V.; Yew, W.W.; et al. Molecular targets related drug resistance mechanisms in MDR-, XDR-, and TDR-Mycobacterium tuberculosis strains. Front. Cell Infect. Microbiol. 2018, 8, 114. [Google Scholar] [CrossRef]
- Bussi, C.; Gutierrez, M.G. Mycobacterium tuberculosis infection of host cells in space and time. FEMS Microbiol. Rev. 2019, 43, 341–361. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef]
- Daletos, G.; Ancheeva, E.; Chaidir, C.; Kalscheuer, R.; Proksch, P. Antimycobacterial metabolites from marine invertebrates. Arch. Pharm. 2016, 349, 763–773. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.W.; Zhou, X.F.; Lin, X.P.; Qin, X.C.; Zhang, T.Y.; Wang, J.F.; Tu, Z.C.; Yang, B.; Liao, S.G.; Tian, Y.Q.; et al. Antituberculosis compounds from a deep-sea-derived fungus Aspergillus sp. SCSIO Ind09F01. Nat. Prod. Res. 2017, 31, 1958–1962. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Yi, M.; Ding, L.; He, S. A review of anti-inflammatory compounds from marine fungi, 2000–2018. Mar Drugs. 2019, 17, 636. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Singh, P.; Kolloli, A.; Shi, L.B.; Bushkin, Y.R.; Tyagi, S.; Subbian, S. Immunometabolism of phagocytes during Mycobacterium tuberculosis infection. Front. Mol. Biosci. 2019, 6, 105. [Google Scholar] [CrossRef] [PubMed]
- Mayer-Barber, K.D.; Barber, D.L. Innate and adaptive cellular immune responses to Mycobacterium tuberculosis infection. Cold Spring Harb. Perspect. Med. 2015, 5, a018424. [Google Scholar] [CrossRef]
- Khan, A.; Singh, V.K.; Hunter, R.L.; Jagannath, C. Macrophage heterogeneity and plasticity in tuberculosis. J. Leukoc. Biol. 2019, 106, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Guirado, E.; Schlesinger, L.S.; Kaplan, G. Macrophages in tuberculosis: Friend or foe. Semin. Immunopathol. 2013, 35, 563–583. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Zhang, K.; Singh, V.K.; Mishra, A.; Kachroo, P.; Bing, T.; Won, J.H.; Mani, A.; Papanna, R.; Mann, L.K.; et al. Human M1 macrophages express unique innate immune response genes after mycobacterial infection to defend against tuberculosis. Commun. Biol. 2022, 5, 480. [Google Scholar] [CrossRef]
- Cooper, A.M. Cell-mediated immune responses in tuberculosis. Annu. Rev. Immunol. 2009, 27, 393–422. [Google Scholar] [CrossRef]
- Jo, E.K. Autophagy as an innate defense against mycobacteria. Pathog. Dis. 2013, 67, 108–118. [Google Scholar] [CrossRef]
- Paik, S.; Kim, J.K.; Chung, C.; Jo, E.K. Autophagy: A new strategy for host-directed therapy of tuberculosis. Virulence 2019, 10, 448–459. [Google Scholar] [CrossRef]
- Liu, C.H.; Liu, H.Y.; Ge, B.X. Innate immunity in tuberculosis: Host defense vs pathogen evasion. Cell. Mol. Immunol. 2017, 14, 963–975. [Google Scholar] [CrossRef] [PubMed]
- Lange, C.; Kalsdorf, B.; Maurer, F.P.; Heyckendorf, J. Tuberculosis. Internist 2019, 60, 1155–1175. [Google Scholar] [CrossRef] [PubMed]
- Nguta, J.M.; Appiah-Opong, R.; Nyarko, A.K.; Yeboah-Manu, D.; Addo, P.G. Current perspectives in drug discovery against tuberculosis from natural products. Int. J. Mycobacteriol. 2015, 4, 165–183. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, T.; Reker, D.; Schneider, P.; Schneider, G. Counting on natural products for drug design. Nat. Chem. 2016, 8, 531–541. [Google Scholar] [CrossRef]
- Rehman, M.U.; Wali, A.F.; Ahmad, A.; Shakeel, S.; Rasool, S.; Ali, R.; Rashid, S.M.; Madkhali, H.; Ganaie, M.A.; Khan, R. Neuroprotective strategies for neurological disorders by natural products: An update. Curr. Neuropharmacol. 2019, 17, 247–267. [Google Scholar] [CrossRef]
- Xu, L.; Li, Y.; Dai, Y.; Peng, J.Y. Natural products for the treatment of type 2 diabetes mellitus: Pharmacology and mechanisms. Pharmacol. Res. 2018, 130, 451–465. [Google Scholar] [CrossRef]
- Deng, L.J.; Qi, M.; Li, N.; Lei, Y.H.; Zhang, D.M.; Chen, J.X. Natural products and their derivatives: Promising modulators of tumor immunotherapy. J. Leukoc. Biol. 2020, 108, 493–508. [Google Scholar] [CrossRef] [PubMed]
- Cary, D.C.; Peterlin, B.M. Natural products and HIV/AIDS. AIDS Res. Hum. Retroviruses 2018, 34, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Nguta, J.M.; Appiah-Opong, R.; Nyarko, A.K.; Yeboah-Manu, D.; Addo, P.G. Medicinal plants used to treat TB in Ghana. Int. J. Mycobacteriol. 2015, 4, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.M.; Wang, C.Y.; Gerwick, W.H.; Shao, C.L. Marine natural products as potential anti-tubercular agents. Eur. J. Med. Chem. 2019, 165, 273–292. [Google Scholar] [CrossRef]
- Khan, M.T.; Kaushik, A.C.; Bhatti, A.I.; Zhang, Y.J.; Zhang, S.; Wei, A.J.; Malik, S.I.; Wei, D.Q. Marine natural products and drug resistance in latent tuberculosis. Mar Drugs. 2019, 17, 549. [Google Scholar] [CrossRef] [PubMed]
- Rao, T.; Tan, Z.R.; Peng, J.B.; Guo, Y.; Chen, Y.; Zhou, H.H.; Ouyang, D.S. The pharmacogenetics of natural products: A pharmacokinetic and pharmacodynamic perspective. Pharmacol. Res. 2019, 146, 104283. [Google Scholar] [CrossRef]
- Bok, J.W.; Chung, D.; Balajee, S.A.; Marr, K.A.; Andes, D.; Nielsen, K.F.; Frisvad, J.C.; Kirby, K.A.; Keller, N.P. GliZ, a transcriptional regulator of gliotoxin biosynthesis, contributes to Aspergillus fumigatus virulence. Infect. Immun. 2006, 74, 6761–6768. [Google Scholar] [CrossRef]
- Stanley, S.A.; Grant, S.S.; Kawate, T.; Iwase, N.; Shimizu, M.; Wivagg, C.; Silvis, M.; Kazyanskaya, E.; Aquadro, J.; Golas, A.; et al. Identification of novel inhibitors of M. tuberculosis growth using whole cell based high-throughput screening. ACS Chem. Biol. 2012, 7, 1377–1384. [Google Scholar] [CrossRef]
- Kolloli, A.; Subbian, S. Host-directed therapeutic strategies for tuberculosis. Front. Med. 2017, 4, 171. [Google Scholar] [CrossRef]
- Zumla, A.; Maeurer, M.; Chakaya, J.; Hoelscher, M.; Ntoumi, F.; Rustomjee, R.; Vilaplana, C.; Yeboah-Manu, D.; Rasolofo, V.; Munderi, P.; et al. Host-directed therapies for tackling multi-drug resistant Tuberculosis: Learning from the Pasteur-Bechamp debates. Clin. Infect. Dis. 2015, 61, 1432–1438. [Google Scholar] [CrossRef]
- Khader, S.A.; Divangahi, M.; Hanekom, W.; Hill, P.C.; Maeurer, M.; Makar, K.W.; Mayer-Barber, K.D.; Mhlanga, M.M.; Nemes, E.; Schlesinger, L.S.; et al. Targeting innate immunity for tuberculosis vaccination. J. Clin. Investig. 2019, 129, 3482–3491. [Google Scholar] [CrossRef]
- Weiss, G.; Schaible, U.E. Macrophage defense mechanisms against intracellular bacteria. Immunol. Rev. 2015, 264, 182–203. [Google Scholar] [CrossRef]
- Zhou, X.Y.; Zhang, L.J.; Lie, L.M.; Zhang, Z.L.; Zhu, B.; Yang, J.H.; Gao, Y.C.; Li, P.F.; Huang, Y.Q.; Xu, H.; et al. MxA suppresses TAK1-IKKalpha/beta-NF-kappaB mediated inflammatory cytokine production to facilitate Mycobacterium tuberculosis infection. J. Infect. 2020, 81, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.B.; Davis, A.S.; Taylor, G.A.; Deretic, V. Human IRGM induces autophagy to eliminate intracellular mycobacteria. Science 2006, 313, 1438–1441. [Google Scholar] [CrossRef]
- Gao, Y.C.; Wen, Q.; Hu, S.F.; Zhou, X.Y.; Xiong, W.J.; Du, X.L.; Zhang, L.J.; Fu, Y.L.; Yang, J.H.; Zhou, C.Y.; et al. IL-36gamma Promotes Killing of Mycobacterium tuberculosis by macrophages via WNT5A-induced noncanonical WNT signaling. J. Immunol. 2019, 203, 922–935. [Google Scholar] [CrossRef] [PubMed]
- Xiong, W.J.; Wen, Q.; Du, X.L.; Wang, J.L.; He, W.T.; Wang, R.N.; Hu, S.F.; Zhou, X.Y.; Yang, J.H.; Gao, Y.C.; et al. Novel function of cyclooxygenase-2: Suppressing Mycobacteria by promoting autophagy via the protein Kinase B/Mammalian target of rapamycin pathway. J. Infect. Dis. 2018, 217, 1267–1279. [Google Scholar] [CrossRef]
- Huang, J.; Canadien, V.; Lam, G.Y.; Steinberg, B.E.; Dinauer, M.C.; Magalhaes, M.A.; Glogauer, M.; Grinstein, S.; Brumell, J.H. Activation of antibacterial autophagy by NADPH oxidases. Proc. Natl. Acad. Sci. USA 2009, 106, 6226–6231. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.T.; Stenger, S.; Li, H.; Wenzel, L.; Tan, B.H.; Krutzik, S.R.; Ochoa, M.T.; Schauber, J.; Wu, K.; Meinken, C.; et al. Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science 2006, 311, 1770–1773. [Google Scholar] [CrossRef]
- Chandra, V.; Bhagyaraj, E.; Nanduri, R.; Ahuja, N.; Gupta, P. NR1D1 ameliorates Mycobacterium tuberculosis clearance through regulation of autophagy. Autophagy 2015, 11, 1987–1997. [Google Scholar] [CrossRef]
- Kim, Y.S.; Lee, H.M.; Kim, J.K.; Yang, C.S.; Kim, T.S.; Jung, M.; Jin, H.S.; Kim, S.; Jang, J.; Oh, G.T.; et al. PPAR-alpha activation mediates innate host defense through induction of TFEB and lipid catabolism. J. Immunol. 2017, 198, 3283–3295. [Google Scholar] [CrossRef]
- Kim, S.Y.; Yang, C.S.; Lee, H.M.; Kim, J.K.; Kim, Y.S.; Kim, Y.R.; Kim, J.S.; Kim, T.S.; Yuk, J.M.; Dufour, C.R.; et al. ESRRA (estrogen-related receptor alpha) is a key coordinator of transcriptional and post-translational activation of autophagy to promote innate host defense. Autophagy 2018, 14, 152–168. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fu, J.; Luo, X.; Lin, M.; Xiao, Z.; Huang, L.; Wang, J.; Zhu, Y.; Liu, Y.; Tao, H. Marine-Fungi-Derived Gliotoxin Promotes Autophagy to Suppress Mycobacteria tuberculosis Infection in Macrophage. Mar. Drugs 2023, 21, 616. https://doi.org/10.3390/md21120616
Fu J, Luo X, Lin M, Xiao Z, Huang L, Wang J, Zhu Y, Liu Y, Tao H. Marine-Fungi-Derived Gliotoxin Promotes Autophagy to Suppress Mycobacteria tuberculosis Infection in Macrophage. Marine Drugs. 2023; 21(12):616. https://doi.org/10.3390/md21120616
Chicago/Turabian StyleFu, Jun, Xiaowei Luo, Miaoping Lin, Zimin Xiao, Lishan Huang, Jiaxi Wang, Yongyan Zhu, Yonghong Liu, and Huaming Tao. 2023. "Marine-Fungi-Derived Gliotoxin Promotes Autophagy to Suppress Mycobacteria tuberculosis Infection in Macrophage" Marine Drugs 21, no. 12: 616. https://doi.org/10.3390/md21120616
APA StyleFu, J., Luo, X., Lin, M., Xiao, Z., Huang, L., Wang, J., Zhu, Y., Liu, Y., & Tao, H. (2023). Marine-Fungi-Derived Gliotoxin Promotes Autophagy to Suppress Mycobacteria tuberculosis Infection in Macrophage. Marine Drugs, 21(12), 616. https://doi.org/10.3390/md21120616