
New Bioactive β-Resorcylic Acid Derivatives from the Alga-Derived Fungus Penicillium antarcticum KMM 4685
 ,
,  , , and
, , and 
Abstract
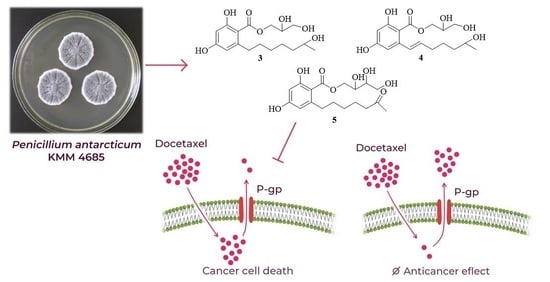
:
1. Introduction
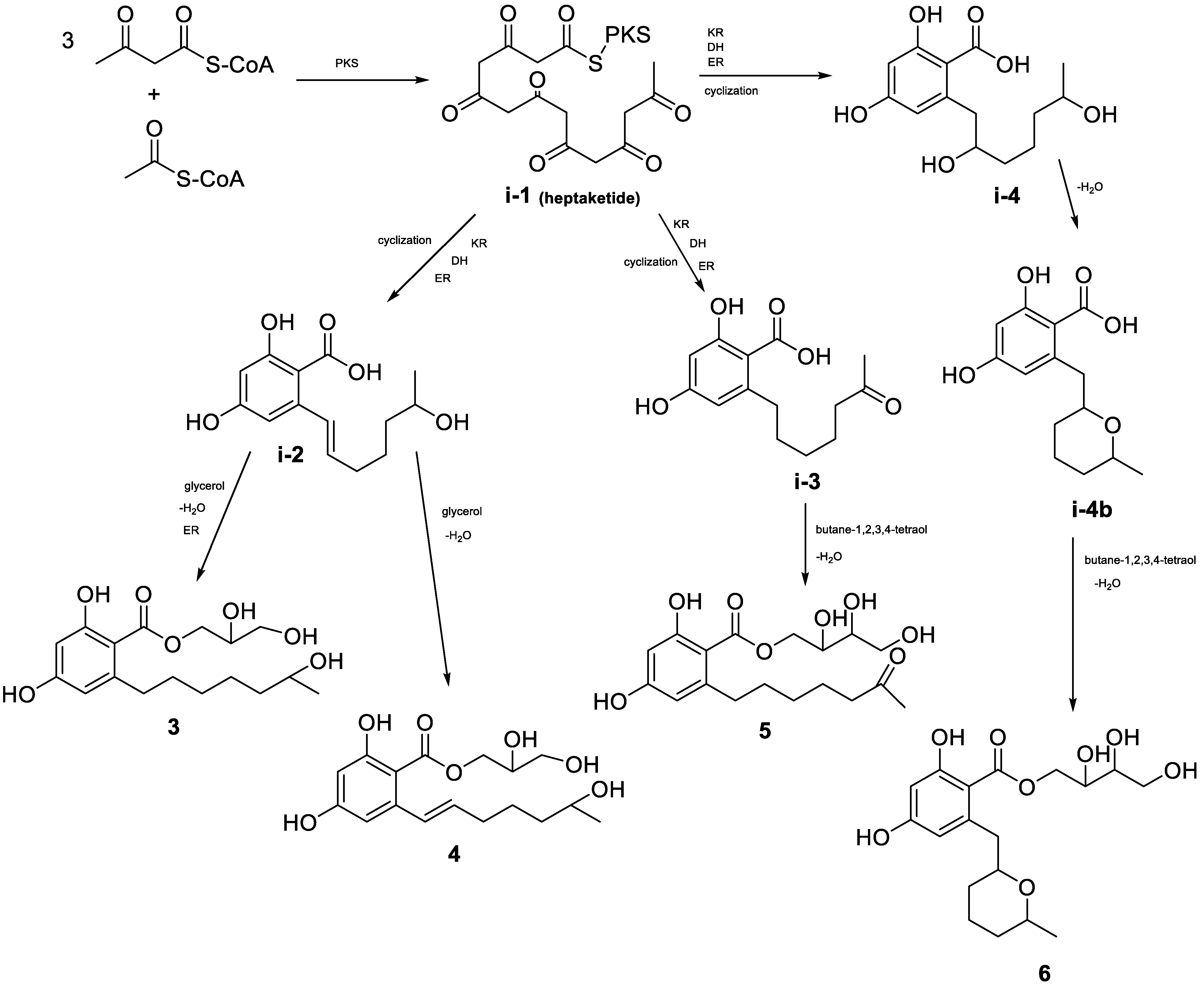
2. Results and Discussion
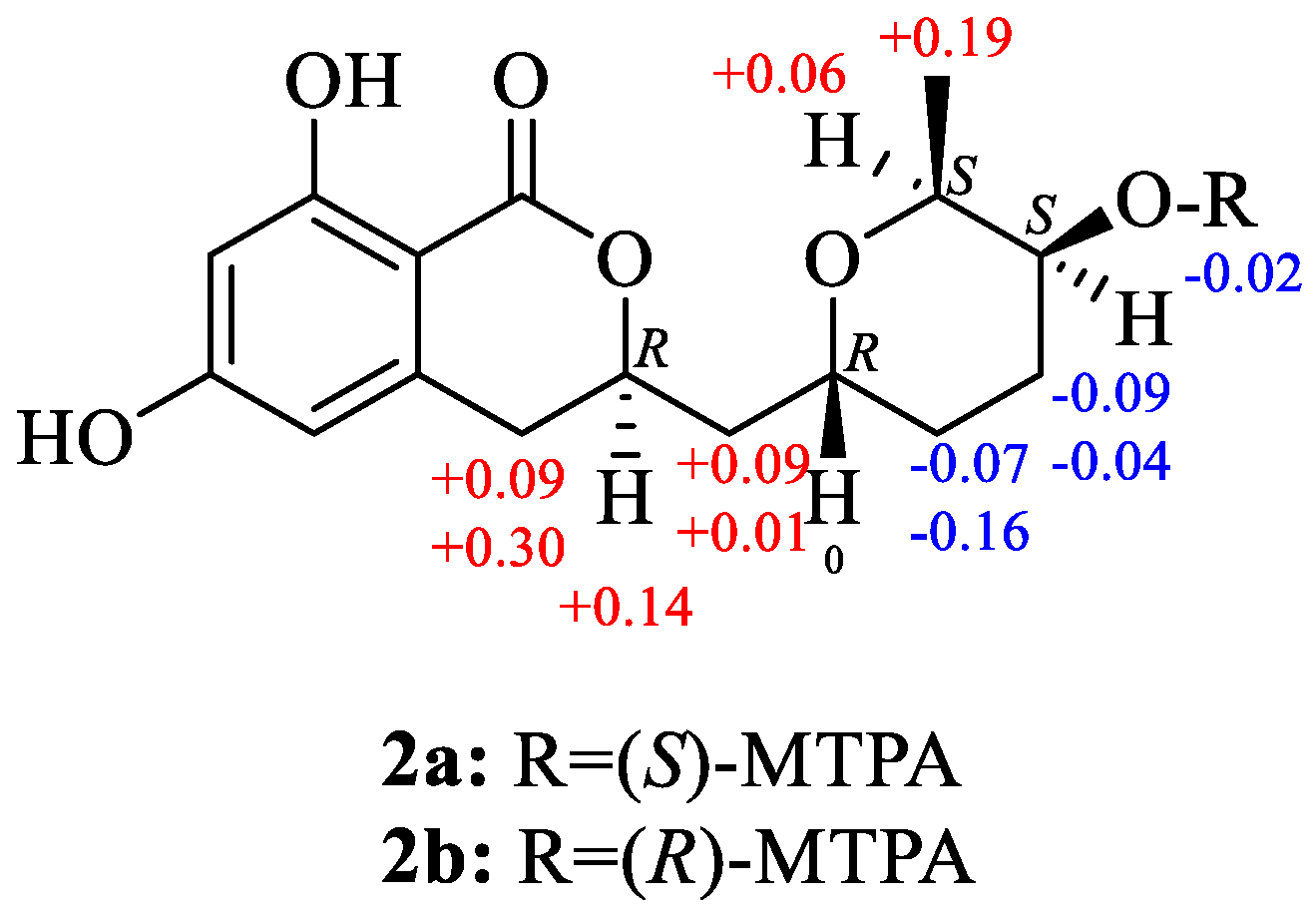
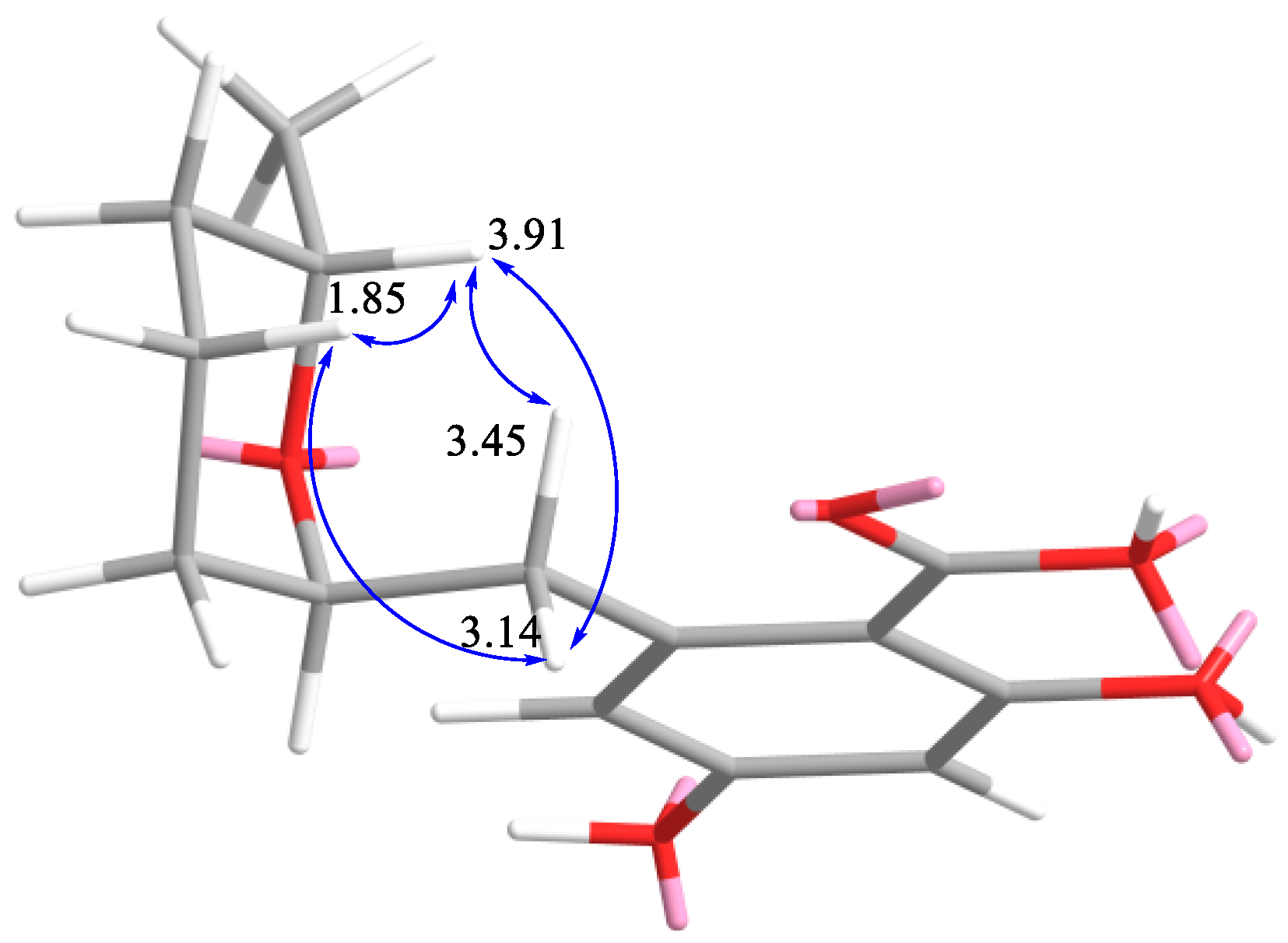
2.1. Structure Determination
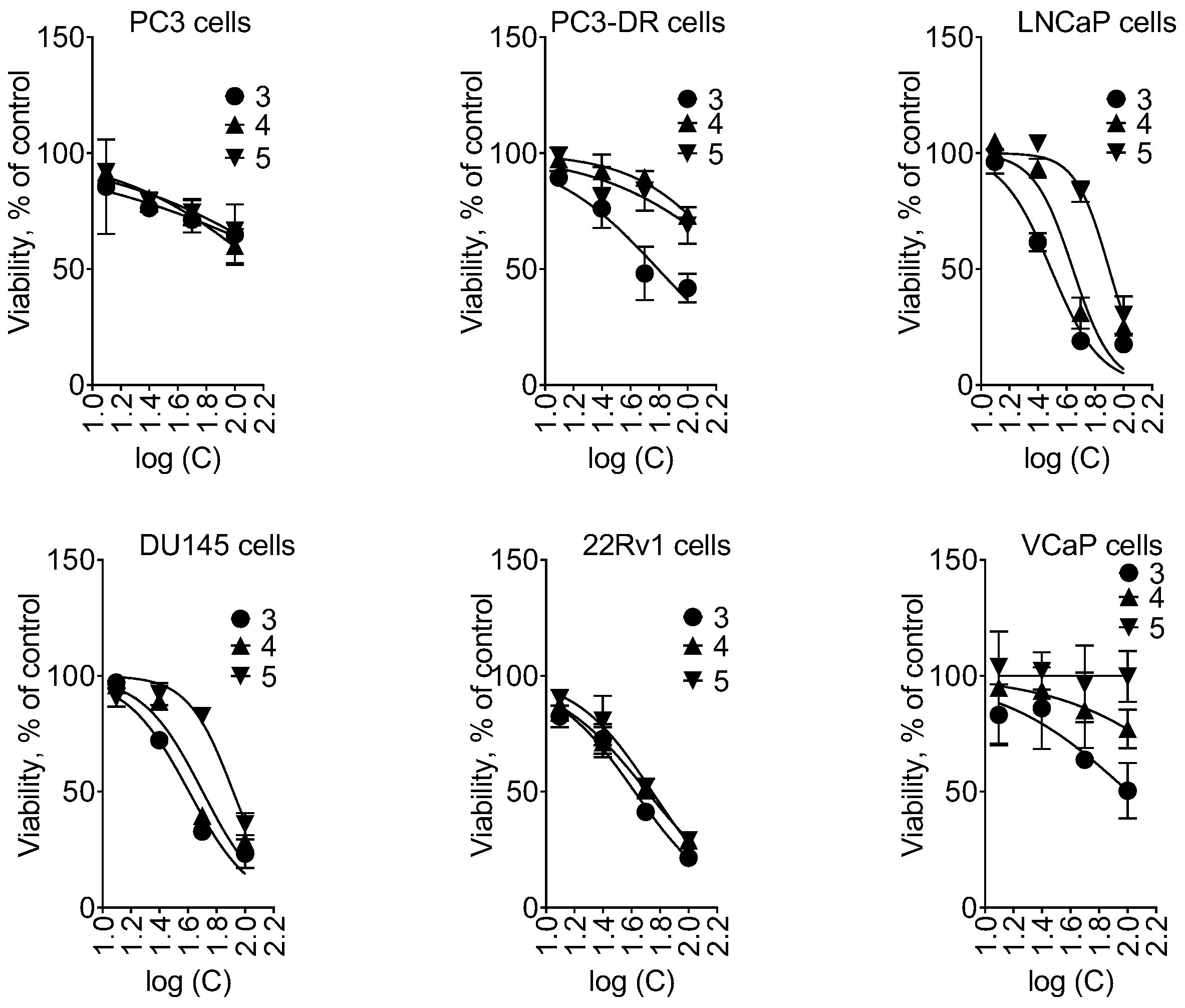
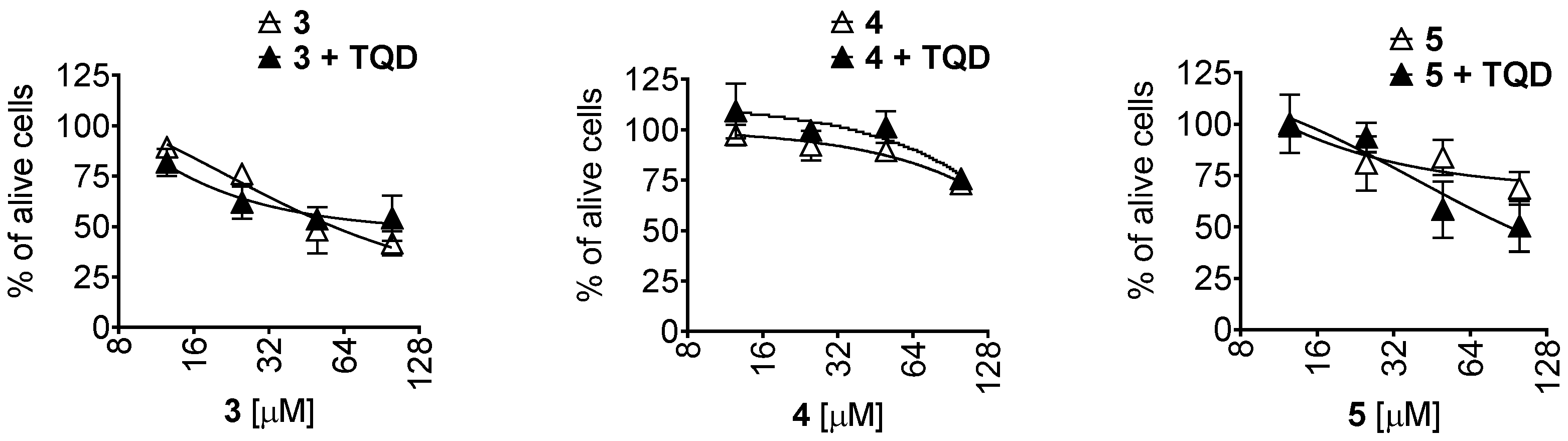
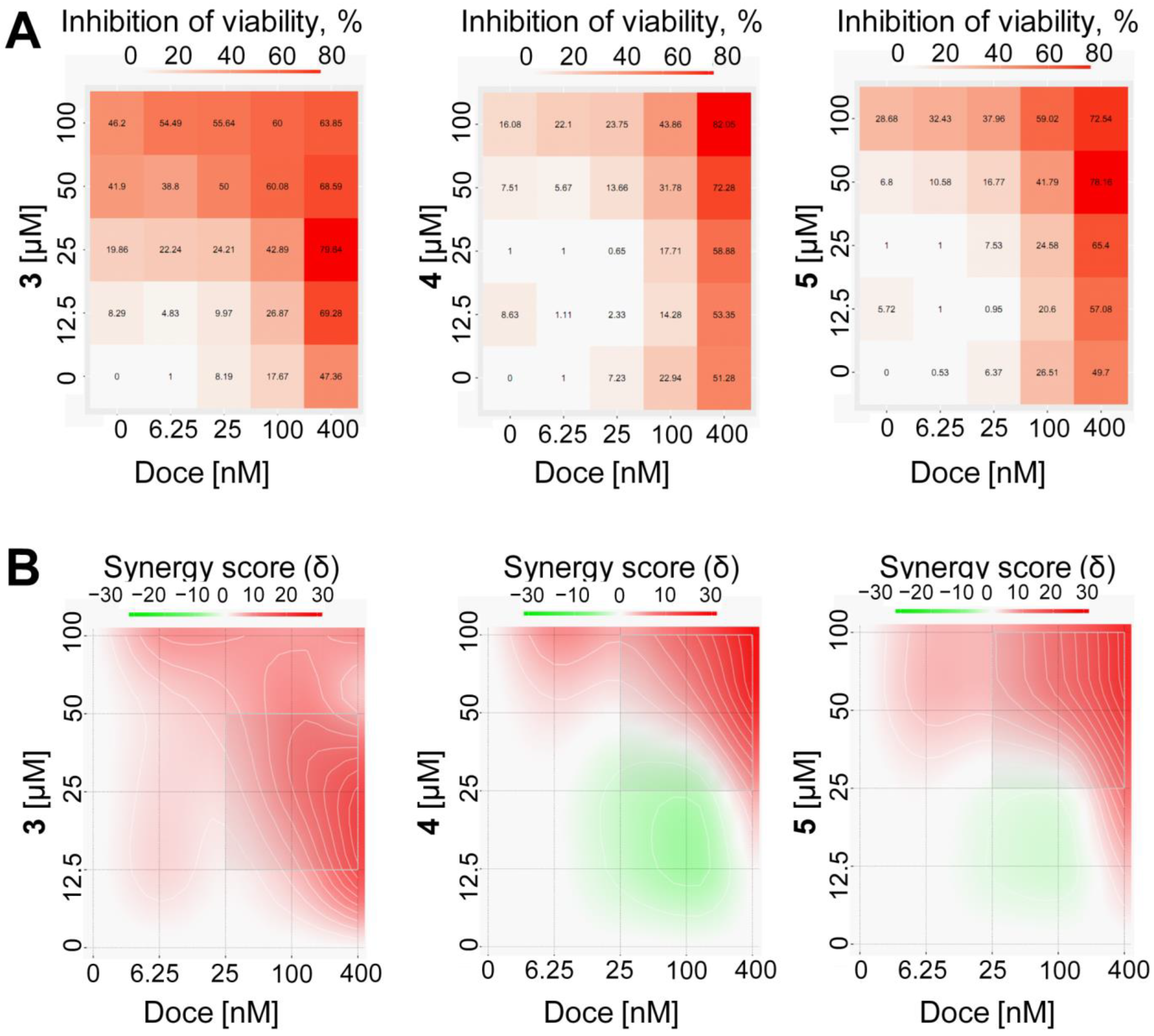
2.2. Activity and Selectivity in Prostate Cancer Cells
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Fungal Strain
3.3. Cultivation of P. thomii
3.4. Extraction and Isolation
3.5. Spectral Data
3.6. Preparation of Pentaacetate β-Resoantarctine C (6a)
3.7. Preparation of (S)-MTPA and (R)-MTPA Esters of 2
3.8. Reagents and Antibodies for Biological Experiments
3.9. Cell Lines and Culture Conditions
3.10. MTT Assay
3.11. Drug Combination Studies
3.12. P-Glycoprotein Activity Assay
3.13. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2021, 38, 362–413. [Google Scholar] [CrossRef]
- Yang, X.; Liu, J.; Mei, J.; Jiang, R.; Tu, S.; Deng, H.; Liu, J.; Yang, S.; Li, J. Origins, structures, and bioactivities of secondary metabolites from marine-derived Penicillium fungi. Mini-Rev. Med. Chem. 2021, 21, 2000–2019. [Google Scholar] [CrossRef]
- McRae, C.F.; Seppelt, R.D.; Hocking, A.D. Penicillium species from terrestrial habitats in the Windmill Islands, East Antarctica, including a new species, Penicillium antarcticum. Polar Biol. 1999, 21, 97–111. [Google Scholar] [CrossRef]
- Park, M.S.; Eom, J.E.; Fong, J.J.; Lim, Y.W. New record and enzyme activity of four species in Penicillium section Citrina from marine environments in Korea. J. Microbiol. 2015, 53, 219–225. [Google Scholar] [CrossRef]
- Visagie, C.M.; Frisvad, J.C.; Houbraken, J.; Visagie, A.; Samson, R.A.; Jacobs, K. A re-evaluation of Penicillium section Canescentia, including the description of five new species. Pers. Mol. Phylogeny Evol. Fungi 2021, 46, 163–187. [Google Scholar] [CrossRef]
- Grijseels, S.; Nielsen, J.; Nielsen, J.C.; Randelovic, M.; Nielsen, K.F.; Workman, M.; Frisvad, J.C. Penicillium arizonense, a new, genome sequenced fungal species, reveals a high chemical diversity in secreted metabolites. Sci. Rep. 2016, 6, 35112. [Google Scholar] [CrossRef] [Green Version]
- Zang, Y.; Gong, Y.; Shi, Z.; Qi, C.; Chen, C.; Tong, Q.; Liu, J.; Wang, J.; Zhu, H.; Zhang, Y. Multioxidized aromatic polyketides produced by a soil-derived fungus Penicillium canescens. Phytochemistry 2022, 193, 113012. [Google Scholar] [CrossRef] [PubMed]
- Zang, Y.; Gong, Y.; Gong, J.; Liu, J.; Chen, C.; Gu, L.; Zhou, Y.; Wang, J.; Zhu, H.; Zhang, Y. Fungal polyketides with three distinctive ring skeletons from the fungus Penicillium canescens uncovered by OSMAC and Molecular Networking Strategies. J. Org. Chem. 2020, 85, 4980. [Google Scholar] [CrossRef]
- Zang, Y.; Gong, Y.H.; Li, X.W.; Li, X.N.; Liu, J.J.; Chen, C.M.; Zhou, Y.; Gu, L.H.; Luo, Z.W.; Wang, J.P.; et al. Canescones A-E: Aromatic polyketide dimers with PTP1B inhibitory activity from Penicillium canescens. Org. Chem. Front. 2019, 6, 3274–3281. [Google Scholar] [CrossRef]
- Frank, M.; Hartmann, R.; Plenker, M.; Mándi, A.; Kurtán, T.; Özkaya, F.C.; Müller, W.E.G.; Kassack, M.U.; Hamacher, A.; Lin, W.; et al. Brominated azaphilones from the sponge-associated fungus Penicillium canescens strain 4.14.6a. J. Nat. Prod. 2019, 82, 2159–2166. [Google Scholar] [CrossRef] [PubMed]
- Bertinetti, B.V.; Peña, N.I.; Cabrera, G.M. An antifungal tetrapeptide from the culture of Penicillium canescens. Chem. Biodivers. 2009, 6, 1178–1184. [Google Scholar] [CrossRef]
- Shiono, Y.; Seino, Y.; Koseki, T.; Murayama, T.; Kimura, K.I. Antarones A and B, Two polyketides from an endophytic Penicillium antarcticum. Z. Naturforsch. Sect. B 2008, 63, 909–914. [Google Scholar] [CrossRef]
- Wiese, J.; Aldemir, H.; Schmaljohann, R.; Gulder, T.A.M.; Imhoff, J.F.; Kerr, R. Asperentin B, a new inhibitor of the protein tyrosine phosphatase 1B. Mar. Drugs 2017, 15, 191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Sakai, K.; Nagano, Y.; Sakaguchi, S.O.; Lima, A.O.; Pellizari, V.H.; Iwatsuki, M.; Takishita, K.; Nonaka, K.; Fujikura, K.; et al. Cladomarine, a new anti-saprolegniasis compound isolated from the deep-sea fungus, Penicillium coralligerum YK-247. J. Antibiot. 2017, 70, 911–914. [Google Scholar] [CrossRef]
- Bang, S.; Shim, S.H. Beta resorcylic acid lactones (RALs) from fungi: Chemistry, biology, and biosynthesis. Arch. Pharm. Res. 2020, 43, 1093–1113. [Google Scholar] [CrossRef] [PubMed]
- Bang, S.; Kim, J.; Oh, J.; Kim, J.S.; Yu, S.R.; Deyrup, S.T.; Bahn, Y.S.; Shim, S.H. Rare β-Resorcylic acid derivatives from a halophyte-associated fungus Colletotrichum gloeosporioides JS0419 and their antifungal activities. Mar. Drugs 2022, 20, 195. [Google Scholar] [CrossRef] [PubMed]
- Kuttikrishnan, S.; Prabhu, K.S.; Al Sharie, A.H.; Al Zu’bi, Y.O.; Alali, F.Q.; Oberlies, N.H.; Ahmad, A.; El-Elimat, T.; Uddin, S. Natural resorcylic acid lactones: A chemical biology approach for anticancer activity. Drug Discov. Today 2022, 27, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Winssinger, N.; Barluenga, S. Chemistry and biology of resorcylic acid lactones. Chem. Commun. 2007, 8, 22–36. [Google Scholar] [CrossRef]
- Kinkade, C.W.; Rivera-Núñez, Z.; Gorcyzca, L.; Aleksunes, L.M.; Barrett, E.S. Impact of fusarium-derived mycoestrogens on female reproduction: A systematic review. Toxins 2021, 13, 373. [Google Scholar] [CrossRef]
- Moulin, E.; Zoete, V.; Barluenga, S.; Karplus, M.; Winssinger, N. Design, synthesis, and biological evaluation of HSP90 inhibitors based on conformational analysis of radicicol and its analogues. J. Am. Chem. Soc. 2005, 127, 6999–7004. [Google Scholar] [CrossRef] [PubMed]
- Leshchenko, E.V.; Antonov, A.S.; Dyshlovoy, S.A.; Berdyshev, D.V.; Hauschild, J.; Zhuravleva, O.; Borkunov, G.V.; Menshov, A.S.; Kirichuk, N.N.; Popov, R.S.; et al. Meroantarctines A–C, meroterpenoids with rearranged skeletons from the alga-derived fungus Penicillium antarcticum KMM 4685 with potent p-glycoprotein inhibitory activity. J. Nat. Prod. 2022, 85, 2746–2752. [Google Scholar] [CrossRef]
- Afiyatullov, S.S.; Zhuravleva, O.I.; Antonov, A.S.; Leshchenko, E.V.; Pivkin, M.V.; Khudyakova, Y.V.; Denisenko, V.A.; Pislyagin, E.A.; Kim, N.Y.; Berdyshev, D.V.; et al. Piltunines A–F from the marine-derived fungus Penicillium piltunense KMM 4668. Mar. Drugs 2019, 17, 647. [Google Scholar] [CrossRef] [Green Version]
- Tang, Q.; Guo, K.; Li, X.Y.; Zheng, X.Y.; Kong, X.J.; Zheng, Z.H.; Xu, Q.Y.; Deng, X. Three new asperentin derivatives from the algicolous fungus Aspergillus sp. Mar. Drugs 2014, 12, 5993–6002. [Google Scholar] [CrossRef] [Green Version]
- Grove, J.F. New metabolic products of Aspergillus flavus. Part I. Asperentin, its methyl ethers, and 5′-hydroxyasperentin. J. Chem. Soc. Perkin Trans. 1972, 1, 2400–2406. [Google Scholar] [CrossRef]
- Grove, J.F. New metabolic products of Aspergillus flavus. Part IV. 4′-Hydroxyasperentin and 5′-hydroxyasperentin 8-methyl ether. J. Chem. Soc. Perkin Trans. 1973, 1, 2704–2706. [Google Scholar] [CrossRef] [PubMed]
- Kimura, Y.; Shimomura, N.; Tanigawa, F.; Fujioka, S.; Shimada, A. Plant growth activities of aspyran, asperentin, and its analogues produced by the fungus Aspergillus sp. Z. Naturforsch C 2012, 67, 587–593. [Google Scholar] [CrossRef] [PubMed]
- Yehia, R.S.; Osman, G.H.; Assaggaf, H.; Salem, R.; Mohamed, M.S.M. Isolation of potential antimicrobial metabolites from endophytic fungus Cladosporium cladosporioides from endemic plant Zygophyllum mandavillei. S. Afr. J. Bot. 2020, 134, 296–302. [Google Scholar] [CrossRef]
- Dyshlovoy, S.A.; Shubina, L.K.; Makarieva, T.N.; Hauschild, J.; Strewinsky, N.; Guzii, A.G.; Menshov, A.S.; Popov, R.S.; Grebnev, B.B.; Busenbender, T.; et al. New diterpenes from the marine sponge Spongionella sp. overcome drug resistance in prostate cancer by inhibition of P-glycoprotein. Sci. Rep. 2022, 12, 13570. [Google Scholar] [CrossRef]
- O’Neill, A.J.; Prencipe, M.; Dowling, C.; Fan, Y.; Mulrane, L.; Gallagher, W.M.; O’Connor, D.; O’Connor, R.; Devery, A.; Corcoran, C.; et al. Characterisation and manipulation of docetaxel resistant prostate cancer cell lines. Mol. Cancer 2011, 10, 126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puhr, M.; Hoefer, J.; Schäfer, G.; Erb, H.H.H.; Oh, S.J.; Klocker, H.; Heidegger, I.; Neuwirt, H.; Culig, Z. Epithelial-to-mesenchymal transition leads to docetaxel resistance in prostate cancer and is mediated by reduced expression of miR-200c and miR-205. Am. J. Pathol. 2012, 181, 2188–2201. [Google Scholar] [CrossRef] [Green Version]
- Dyshlovoy, S.A.; Kaune, M.; Hauschild, J.; Kriegs, M.; Hoffer, K.; Busenbender, T.; Smirnova, P.A.; Zhidkov, M.E.; Poverennaya, E.V.; Oh-Hohenhorst, S.J.; et al. Efficacy and mechanism of action of marine alkaloid 3,10-dibromofascaplysin in drug-resistant prostate cancer cells. Mar. Drugs 2020, 18, 609. [Google Scholar] [CrossRef]
- Dyshlovoy, S.A.; Pelageev, D.N.; Hauschild, J.; Borisova, K.L.; Kaune, M.; Krisp, C.; Venz, S.; Sabutskii, Y.E.; Khmelevskaya, E.A.; Busenbender, T.; et al. Successful targeting of thewarburg effect in prostate cancer by glucose-conjugated 1,4-naphthoquinones. Cancers 2019, 11, 1690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ianevski, A.; Giri, A.K.; Aittokallio, T. SynergyFinder 2.0: Visual analytics of multi-drug combination synergies. Nucleic Acids Res. 2021, 48, W488–W493. [Google Scholar] [CrossRef] [PubMed]
- Yadav, B.; Wennerberg, K.; Aittokallio, T.; Tang, J. Searching for drug synergy in complex dose-response landscapes using an interaction potency model. Comput. Struct. Biotechnol. J. 2015, 13, 504–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No | 1 | 2 | ||
|---|---|---|---|---|
| δC, Type | δH, Mult. (J in Hz) | δc, Type | δH, Mult. (J in Hz) | |
| 1 | 171.8, C | 171.4, C | ||
| 2 | 100.2, C | 101.2, C | ||
| 3 | 159.1, C | 166.3, C | ||
| 4 | 101.8, CH | 6.26, s | 102.2, CH | 6.19, d (2.2) |
| 5 | 155.6, C | 165.6, C | ||
| 6 | 135.7, C | 107.8, C | 6.20, brs | |
| 7 | 126.1, C | 143.5, C | ||
| 8 | 28.6, CH2 | 3.15, dd (16.7, 3.3) 2.63, dd (16.7, 11.0) | 34.4, CH2 | 2.88, dd (16.5, 4.0) 2.85, dd (16.5, 10.7) |
| 9 | 77.7, CH | 4.61, m | 77.8, CH | 4.65, m |
| 10 | 40.5, CH2 | 1.92, m 1.85, m | 40.3, CH2 | 1.89, m 1.83, m |
| 11 | 66.4, CH | 4.00, m | 66.5, CH | 3.99, dt (9.0, 3.3) |
| 12 | 30.0, CH2 | 1.87, m 1.33. m | 30.0, CH2 | 1.87, m 1.33. m |
| 13 | 27.2, CH2 | 1.80, m 1.72, m | 27.2, CH2 | 1.79, m 1.73, m |
| 14 | 69.0, CH | 3.70, dt (9.0, 4.3) | 69.0, CH | 3.70, dt (9.0, 4.3) |
| 15 | 72.3, CH | 3.96, qd (6.6, 4.2) | 72.4, CH | 3.96, qd (6.6, 4.2) |
| 16 | 13.1, CH3 | 1.20, d (6.6) | 13.1, CH3 | 1.20, d (6.6) |
| Position | δC, Type | |||
|---|---|---|---|---|
| 3 * | 4 * | 5 ** | 6 *** | |
| 1 | 172.4, C | 172.3, C | 172.5, C | 172.6, C |
| 2 | 106.0, C | 105.1, C | 106.2, C | 105.1, C |
| 3 | 165.0, C | 165.0, C | 165.3, C | 166.8, C |
| 4 | 102.6, CH | 102.6, CH | 101.9, CH | 102.4, CH |
| 5 | 163.0, C | 163.6, C | 163.5, C | 163.2, C |
| 6 | 111.7, CH | 108.9, CH | 111.7, CH | 113.2, CH |
| 7 | 149.2, C | 144.9, C | 149.1, C | 145.4, C |
| 8 | 37.3, CH2 | 132.3, CH | 37.2, CH2 | 39.9, CH2 |
| 9 | 33.1, CH2 | 133.6, CH | 32.8, CH2 | 74.2, CH |
| 10 | 30.8, CH2 | 34.0, CH2 | 30.2, CH2 | 27.2, CH2 |
| 11 | 26.8, CH2 | 26.6, CH2 | 24.8, CH2 | 18.7, CH2 |
| 12 | 40.1, CH2 | 39.7, CH2 | 44.3, CH2 | 33.3, CH2 |
| 13 | 68.6, CH | 68.4, CH | 209.0, C | 67.8, CH |
| 14 | 23.5, CH3 | 23.5, CH3 | 29.8, CH3 | 21.3, CH3 |
| 1′ | 67.2, CH2 | 67.1, CH2 | 68.1, CH2 | 70.0, CH2 |
| 2′ | 71.1, CH | 71.0, CH | 71.3, CH | 70.8, CH |
| 3′ | 64.5, CH2 | 64.3, CH2 | 73.8, CH | 73.1, CH |
| 4′ | 64.6, CH2 | 64.8, CH2 | ||
| Position | δH, Mult. (J in Hz) | |||
|---|---|---|---|---|
| 3 * | 4 * | 5 ** | 6 *** | |
| 4 | 6.15, d (2.5) | 6.20, d (2.4) | 6.16, d (2.5) | 6.28, brs |
| 6 | 6.20, d (2.5) | 6.38, d (2.4) | 6.20, d (2.5) | 6.28, brs |
| 8 | 2.88, m 2.84, m | 6.98, dt (15.5, 1.5) | 2.90, m 2.84, m | 3.45, dd (12.5, 3.9) 3.14, dd (12.4, 9.1) |
| 9 | 1.58, m 1.55, m | 5.91, dt (15.5, 7.0) | 1.57, m (2H) | 4.03, m |
| 10 | 1.38, m (2H) | 2.24, m (2H) | 1.36, m (2H) | 1.54, m 1.51, m |
| 11 | 1.39, m (2H) | 1.60, m 1.52, m | 1.57, m (2H) | 1.85, m 1.65, m |
| 12 | 1.41, m (2H) | 1.51, m 1.45, m | 2.46, m (2H) | 1.66, m 1.24, m |
| 13 | 3.70, ddd (12.0, 9.2, 5.6) | 3.75, dd (11.9, 6.0) | 3.91, m | |
| 14 | 1.13, d (6.0) | 1.16, d (6.2) | 2.11, s | 1.13, d (6.3) |
| 1′ | 4.41, ddd (11.5, 4.3, 1.3) 4.30, ddd (11.5, 6.6, 1.3) | 4.39, dd (11.4, 4.5) 4.31, dd (11.4, 5.9) | 4.57, dd (11.5, 4.3, 1.3) 4.38, dd (11.5, 6.6, 1.3) | 4.62, dd (11.2, 2.1) 4.33, dd (11.2, 7.8) |
| 2′ | 3.96, m | 3.95, m | 3.88, td (7.0, 2,6) | 3.99, m |
| 3′ | 3.62, ddd (11.4, 5.5, 0.6) 3.60, ddd (11.4, 5.5, 1.0) | 3.64, dd (11.2, 5.5) 3.60, dd (11.2, 5.5) | 3.61, m | 3.62, ddd (8.0, 5.6, 4.0) |
| 4′ | 3.77, dd (10.6, 3.1) 3.64, m | 3.80, dd (10.8, 4.0) 3.66, dd (10.8, 5.7) | ||
| C-3-OH | 11.7, s | |||
| C-5-OH | 9.17, s | |||
| C-2′-OH | 5.00 d (5.3) | |||
| C-3′-OH | 3.99 d (5.8) | |||
| C-4′-OH | 3.62 d (5.4) | |||
| Cell Line | Compound, IC50 [µM] | Control, IC50 [nM] | ||
|---|---|---|---|---|
| 3 | 4 | 5 | Doce | |
| PC3 | >100 | >100 | >100 | 7.5 ± 7.1 |
| PC3-DR | 21.4 ± 18.8 | >100 | >100 | 325 ± 22 |
| LNCaP | 31 ± 2 | 44.1 ± 3.8 | 79.2 ± 2.8 | 5.1 ± 0.6 |
| DU145 | 40.4 ± 2.7 | 50.4 ± 3.8 | 82.5 ± 4.1 | 2.2 ± 0.6 |
| 22Rv1 | 41.9 ± 2.1 | 51.3 ± 2.8 | 55.4 ± 3.1 | 0.6 ± 0.1 |
| VCaP | >100 | >100 | >100 | 0.23 ± 0.22 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leshchenko, E.V.; Antonov, A.S.; Borkunov, G.V.; Hauschild, J.; Zhuravleva, O.I.; Khudyakova, Y.V.; Menshov, A.S.; Popov, R.S.; Kim, N.Y.; Graefen, M.; et al. New Bioactive β-Resorcylic Acid Derivatives from the Alga-Derived Fungus Penicillium antarcticum KMM 4685. Mar. Drugs 2023, 21, 178. https://doi.org/10.3390/md21030178
Leshchenko EV, Antonov AS, Borkunov GV, Hauschild J, Zhuravleva OI, Khudyakova YV, Menshov AS, Popov RS, Kim NY, Graefen M, et al. New Bioactive β-Resorcylic Acid Derivatives from the Alga-Derived Fungus Penicillium antarcticum KMM 4685. Marine Drugs. 2023; 21(3):178. https://doi.org/10.3390/md21030178
Chicago/Turabian StyleLeshchenko, Elena V., Alexandr S. Antonov, Gleb V. Borkunov, Jessica Hauschild, Olesya I. Zhuravleva, Yuliya V. Khudyakova, Alexander S. Menshov, Roman S. Popov, Natalya Yu Kim, Markus Graefen, and et al. 2023. "New Bioactive β-Resorcylic Acid Derivatives from the Alga-Derived Fungus Penicillium antarcticum KMM 4685" Marine Drugs 21, no. 3: 178. https://doi.org/10.3390/md21030178
APA StyleLeshchenko, E. V., Antonov, A. S., Borkunov, G. V., Hauschild, J., Zhuravleva, O. I., Khudyakova, Y. V., Menshov, A. S., Popov, R. S., Kim, N. Y., Graefen, M., Bokemeyer, C., von Amsberg, G., Yurchenko, A. N., & Dyshlovoy, S. A. (2023). New Bioactive β-Resorcylic Acid Derivatives from the Alga-Derived Fungus Penicillium antarcticum KMM 4685. Marine Drugs, 21(3), 178. https://doi.org/10.3390/md21030178