

Diversity, Biosynthesis and Bioactivity of Aeruginosins, a Family of Cyanobacteria-Derived Nonribosomal Linear Tetrapeptides
Abstract
:1. Introduction
2. Biogenesis
2.1. Genus Microcystis
2.2. Genus Planktothrix
2.3. Genus Nostoc
2.4. Genus Nodularia
2.5. Sponges
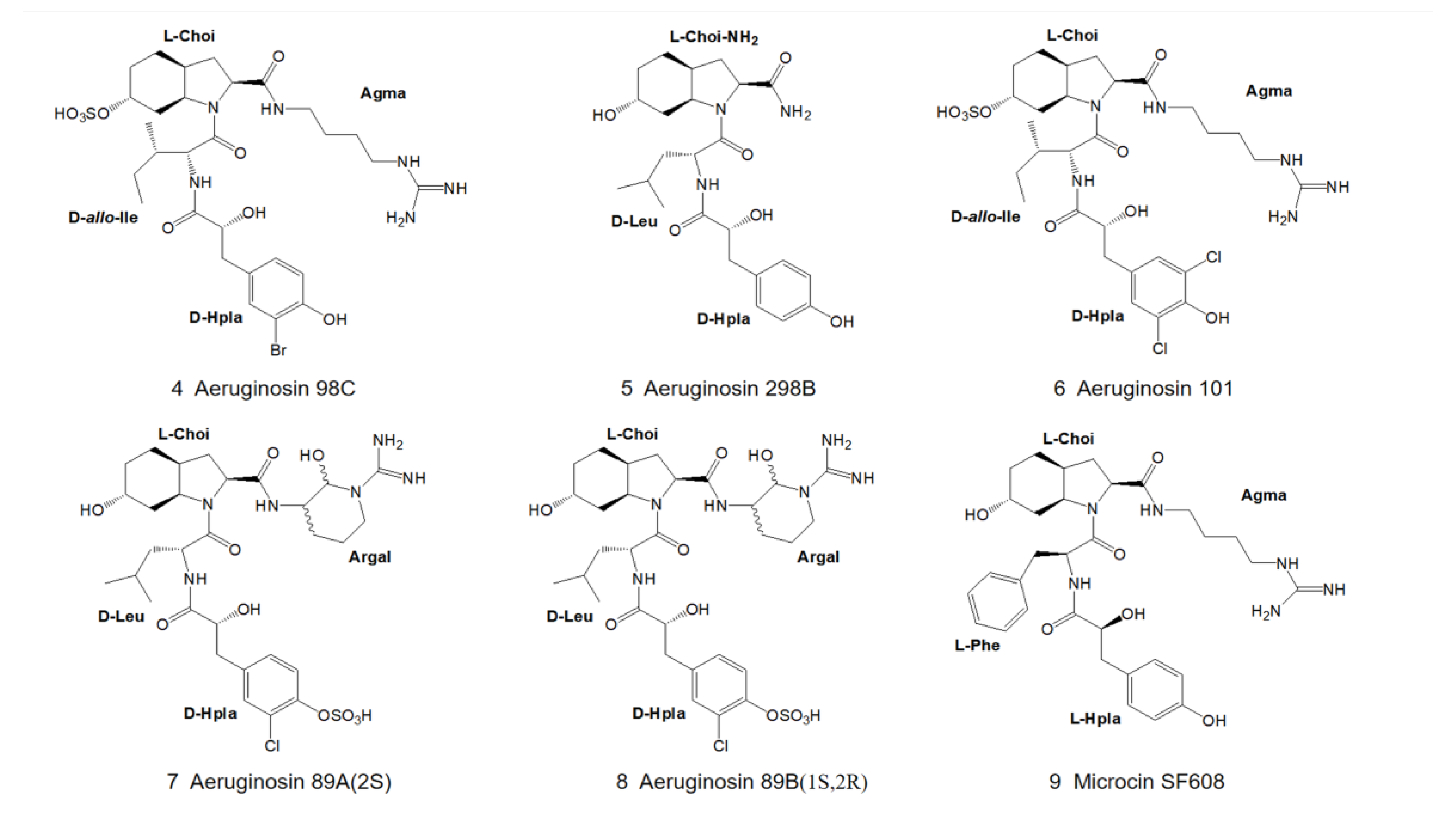
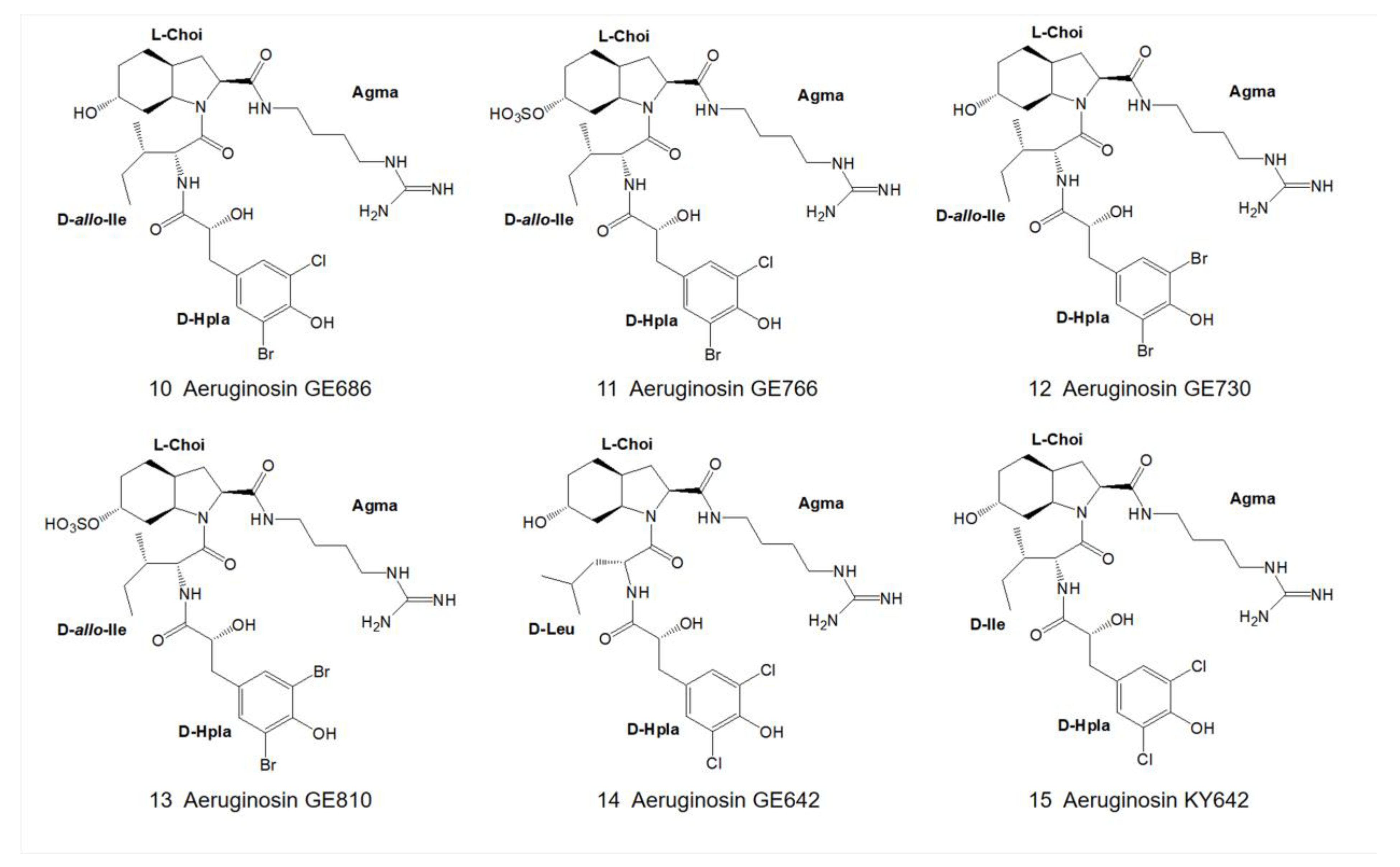
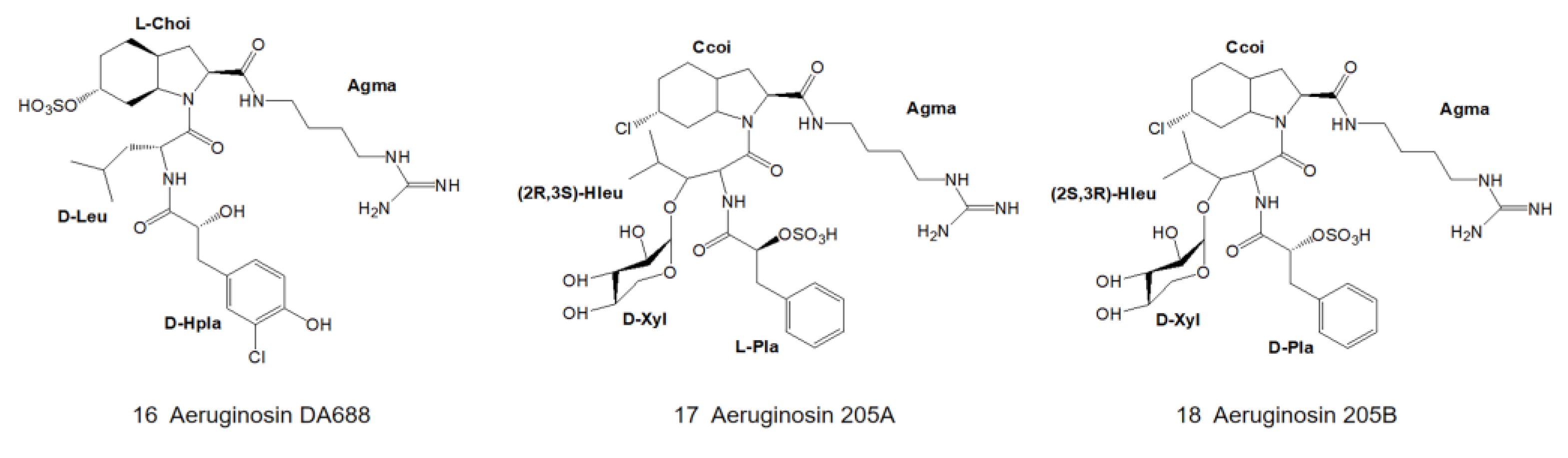
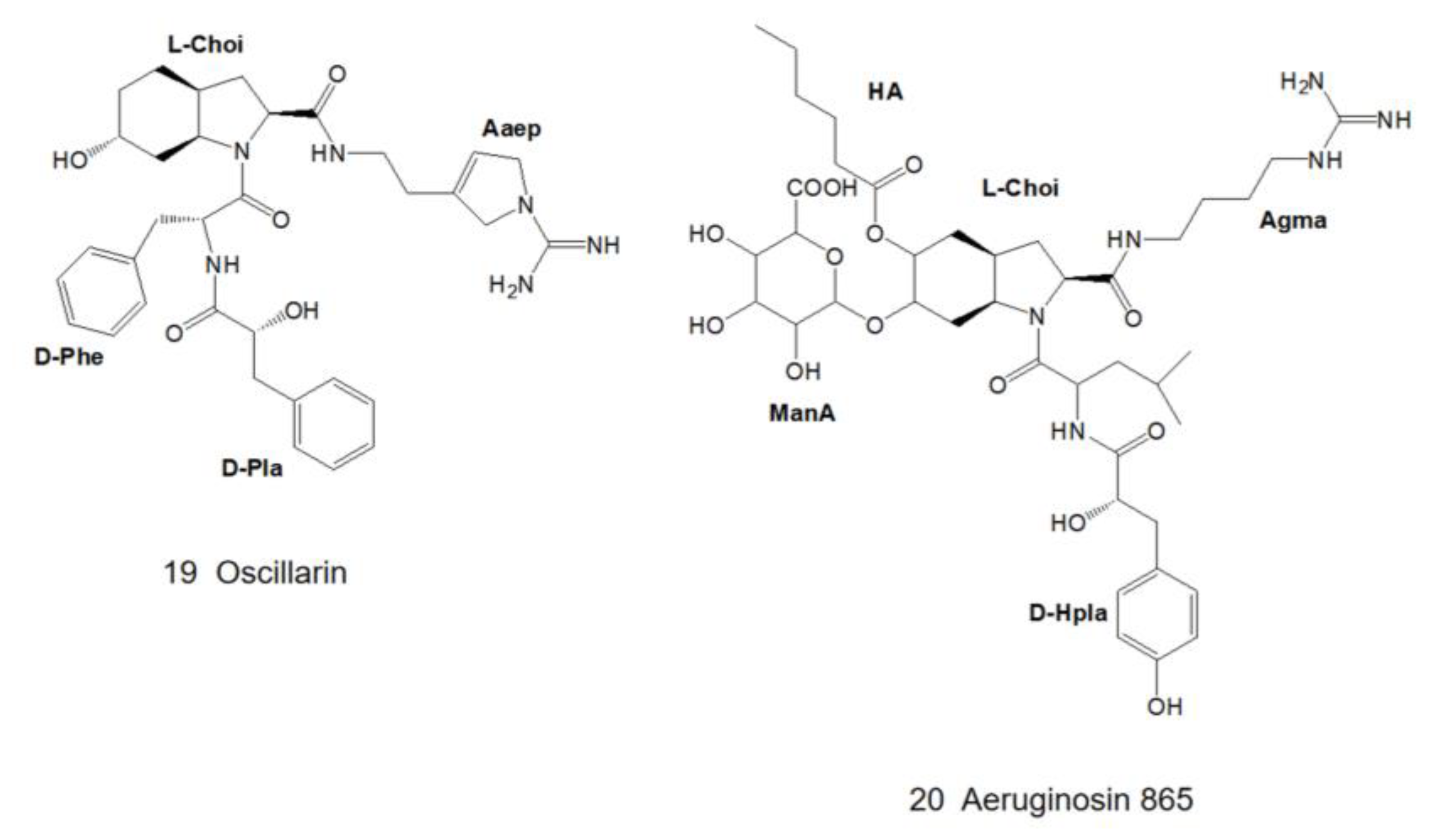
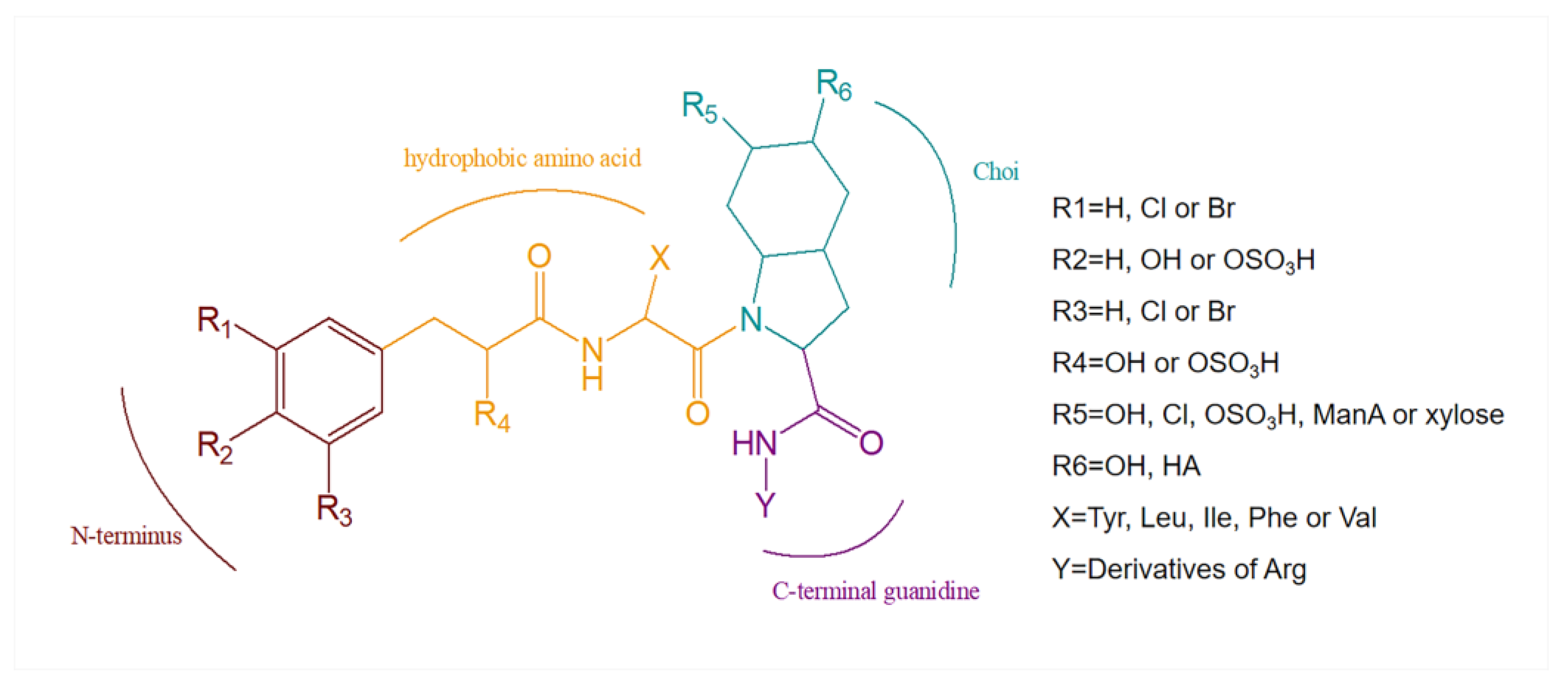
3. Structural Diversity
3.1. Diversity of N-Terminal Residue
3.2. Diversity of the Side Chain of the Second Residue
3.3. Modifications of Choi Moiety
3.4. Diversity of the Side Chain of C-Terminal Residue
4. Biosynthetic Pathways
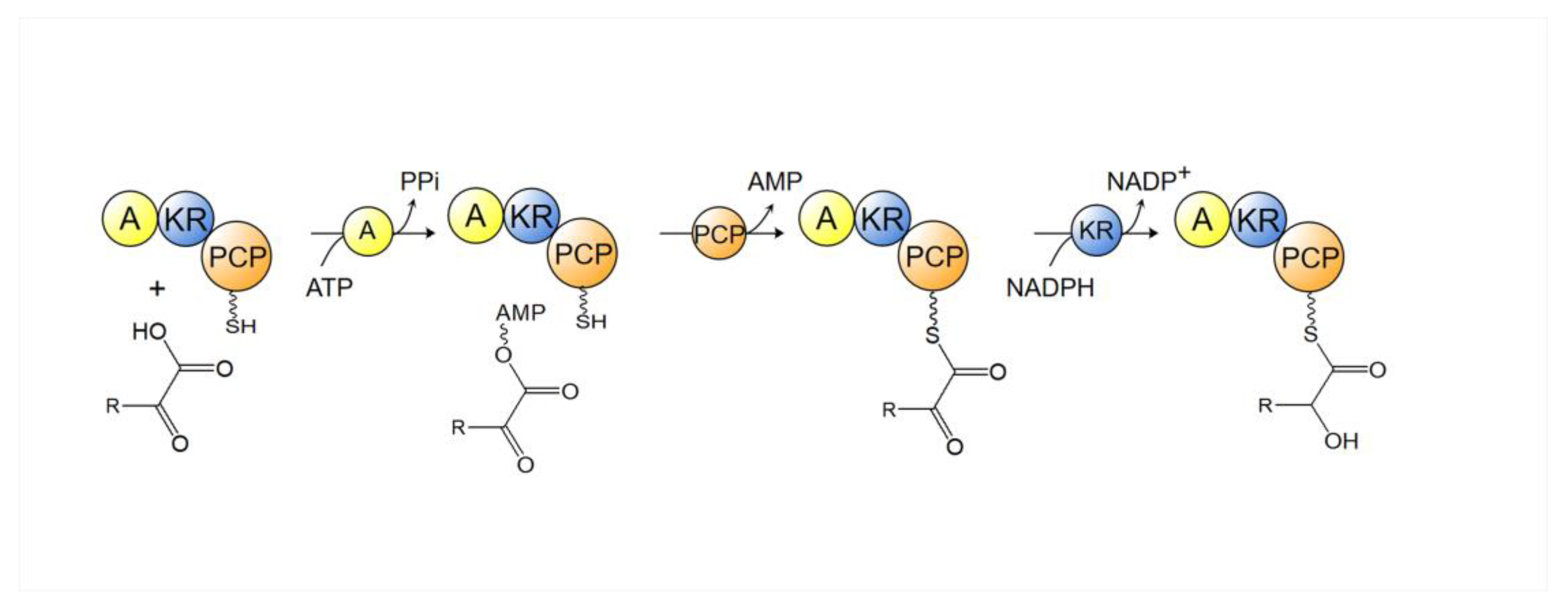
4.1. Polyketide Synthase (PKS)
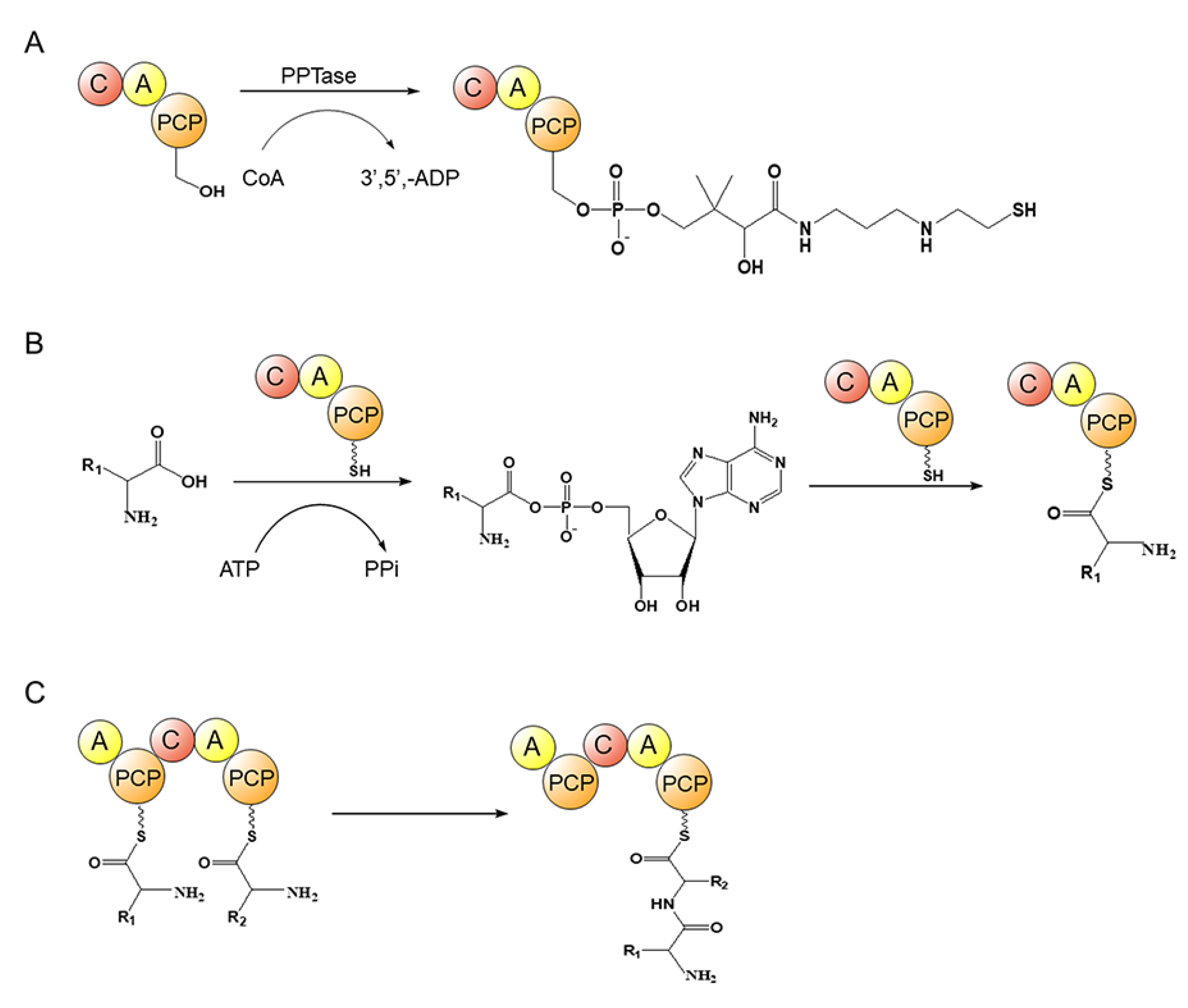
4.2. Nonribosomal Polypeptide Synthase (NRPS)
4.3. NRPS-PKS Hybrid
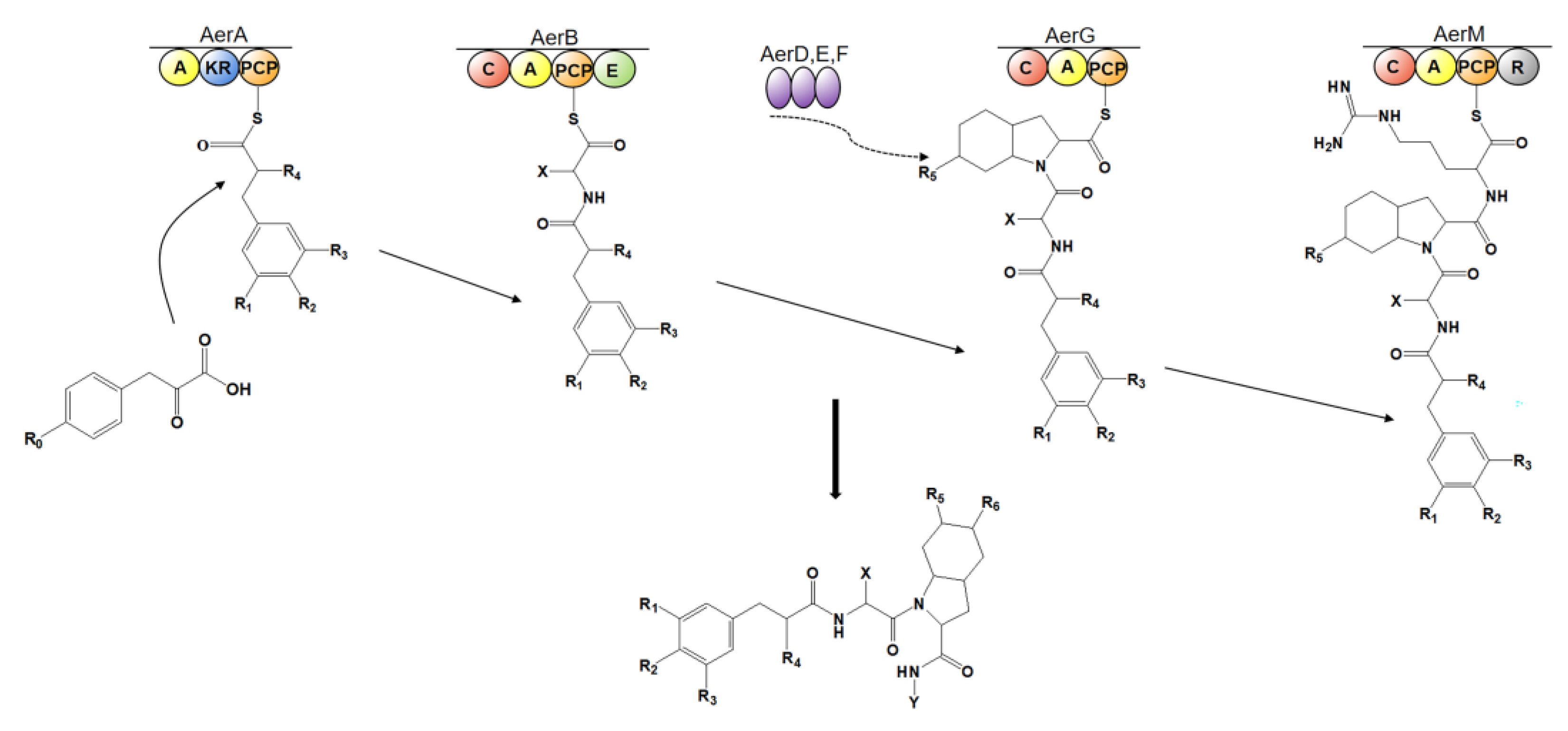
4.4. Biosynthesis of Aeruginosin
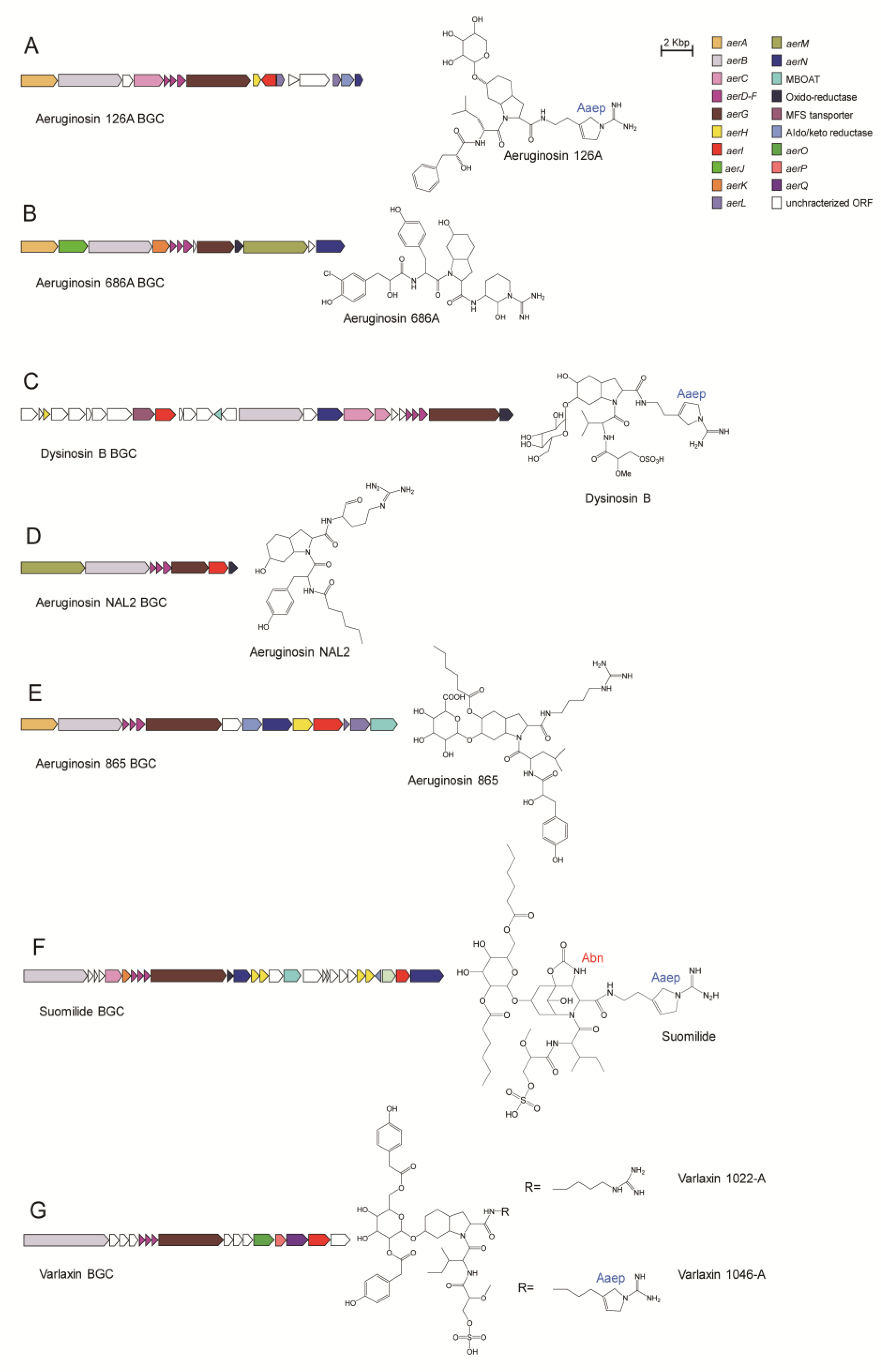
4.4.1. Aeruginosin 126A
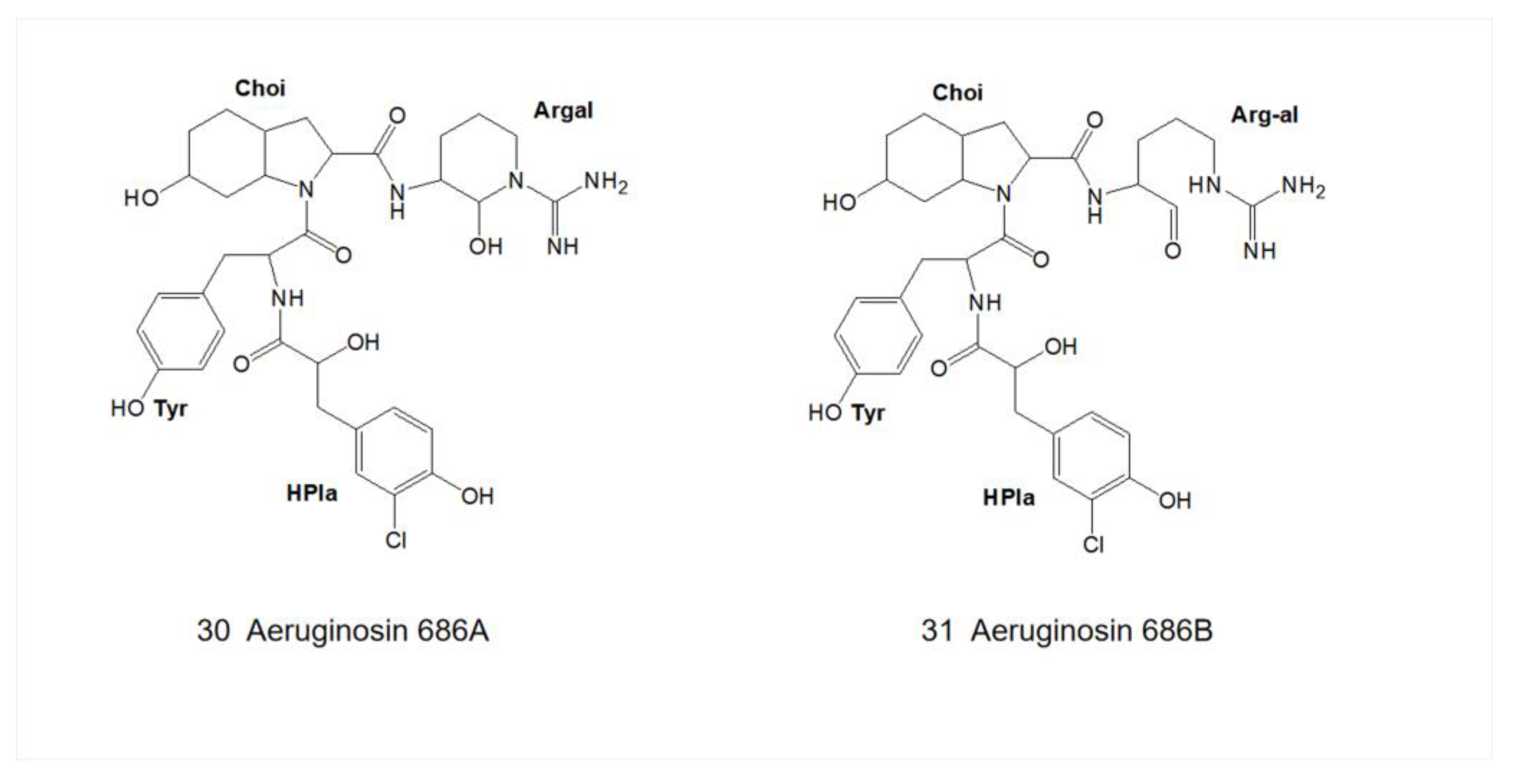
4.4.2. Aeruginosin 686A
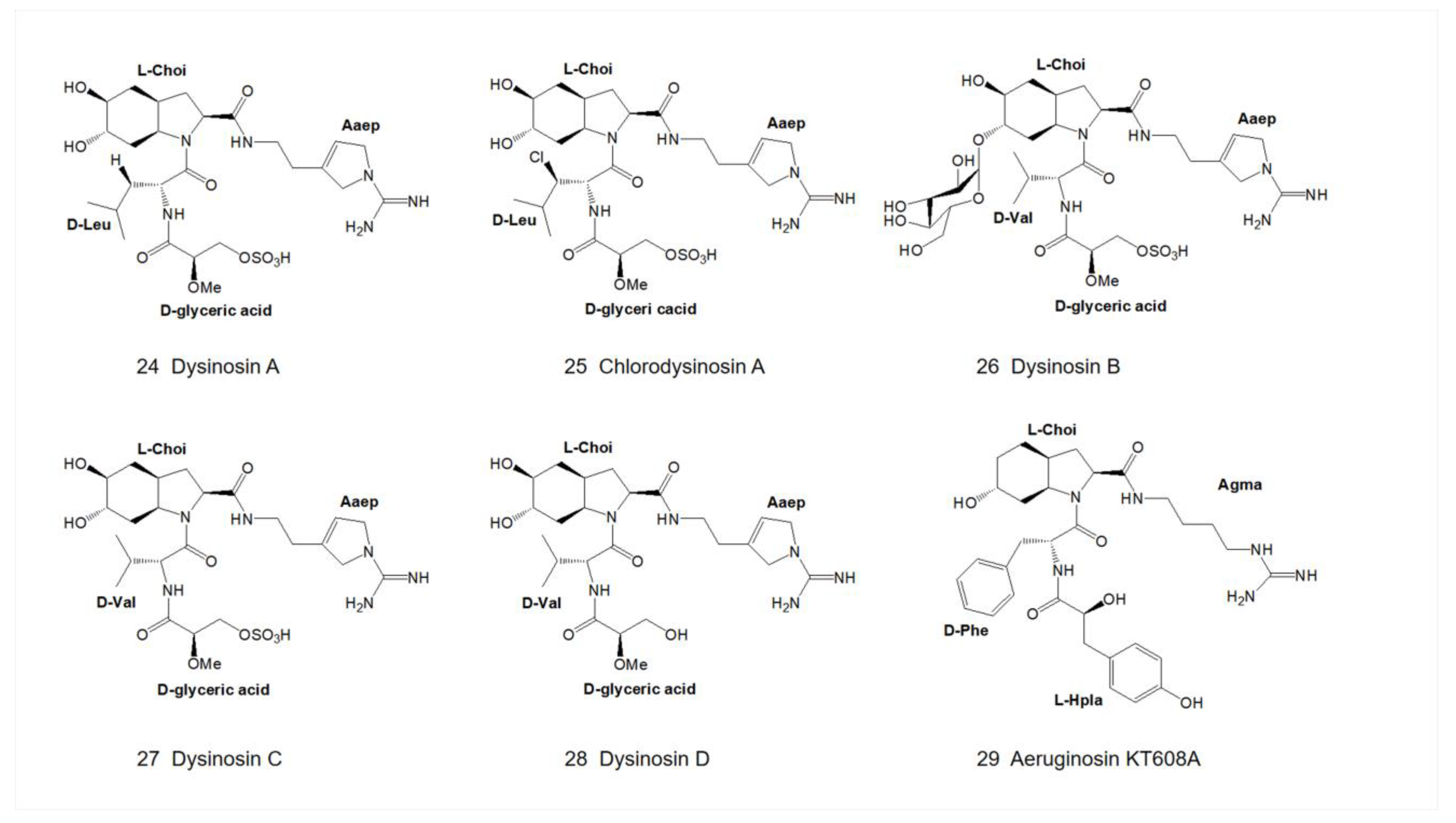
4.4.3. Dysinosin B
4.4.4. Aeruginosin NAL2
4.4.5. Aeruginosin 865
4.4.6. Suomilide
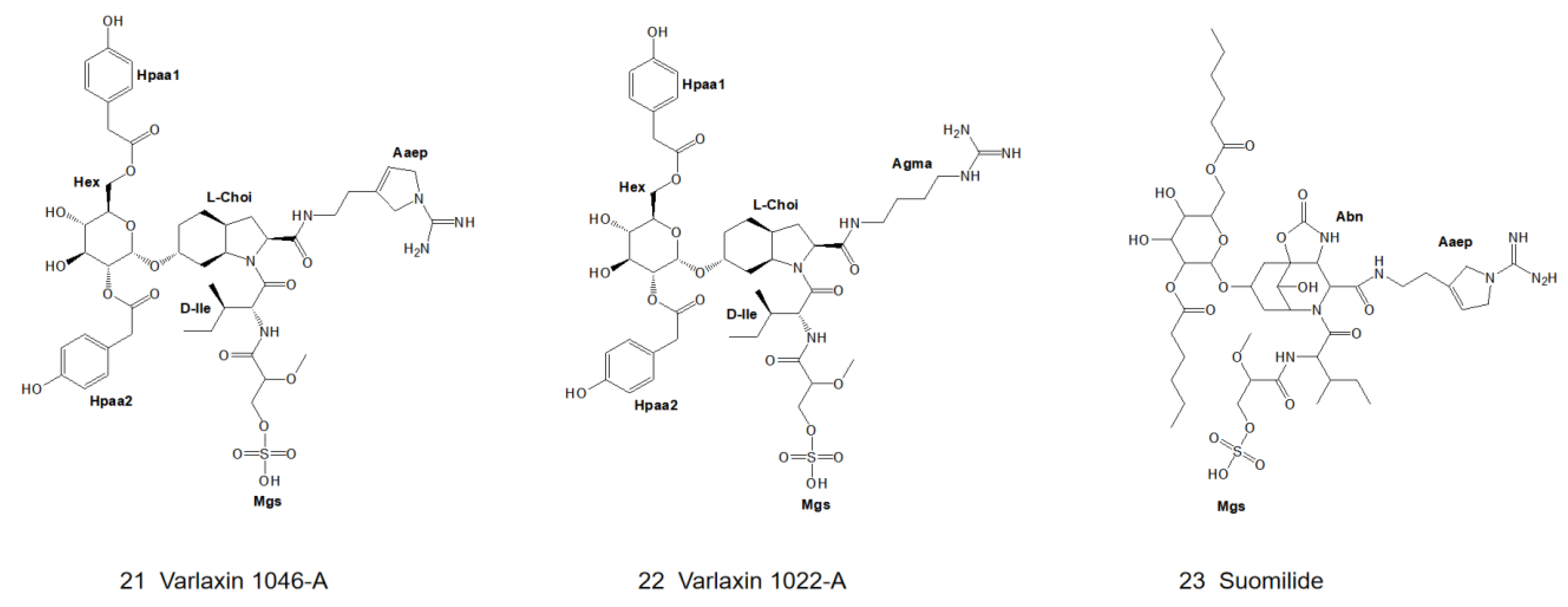
4.4.7. Varlaxin
5. Bioactivity
5.1. Thrombin Inhibitory Activity
5.2. Trypsin Inhibitory Activity
5.3. Other Bioactivities
6. Conclusions and Prospects
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Paerl, H.W.; Millie, D.F. Physiological ecology of toxic aquatic cyanobacteria. Phycologiacal 1996, 35, 160–167. [Google Scholar] [CrossRef]
- Qin, B.; Li, W.; Zhu, G.; Wu, T.; Gao, G. Cyanobacterial bloom management through integrated monitoring and forecasting in large shallow eutrophic Lake Taihu. J. Hazard. Mater. 2015, 287, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Luesch, H.; Harrigan, G.G.; Goetz, G.; Horgen, F.D. The Cyanobacterial origin of potent anticancer agents originally isolated from Sea Hares. Curr. Med. Chem. 2002, 9, 1791–1806. [Google Scholar] [CrossRef] [PubMed]
- Ishida, K.; Okita, Y.; Mastsuda, H.; Okino, T.; Murakami, M. Aeruginosins, protease inhibitors from the cyanobacterium Microcystis aeruginosa. Tetrahedron 1999, 55, 10971–10988. [Google Scholar] [CrossRef]
- Elkobi-Peer, S.; Faigenbaum, R.; Carmeli, S. Bromine- and Chlorine-containing aeruginosins from Microcystis aeruginosa bloom material collected in Kibbutz Geva, Israel. J. Nat. Prod. 2012, 75, 2144–2151. [Google Scholar] [CrossRef]
- Valls, N.; Vallribera, M.; López-Canet, M.; Bonjoch, J. Synthesis of Microcin SF608. J. Org. Chem. 2002, 67, 4945–4950. [Google Scholar] [CrossRef]
- Raveh, A.; Carmeli, S. Two novel biological active modified peptides from the cyanobacterium Microcystis sp. Phytochem. Lett. 2009, 2, 10–14. [Google Scholar] [CrossRef]
- Adiv, S.; Carmeli, S. Protease inhibitors from Microcystis aeruginosa bloom Material Collected from the Dalton Reservoir. J. Nat. Prod. 2013, 76, 2307–2315. [Google Scholar] [CrossRef]
- Shin, H.J.; Matsuda, H.; Murakami, M.; Yamaguchi, K. Aeruginosins 205A and -B, serine protease inhibitory glycopeptides from the cyanobacterium Oscillatoria agardhii (NIES-205). J. Org. Chem. 1997, 62, 1810–1813. [Google Scholar] [CrossRef]
- Engh, R.; Konetschny-Rapp, S.; Krell, H.-W.; Martin, U.; Tsaklakidis, C. PCT WO97/21725. Chem. Abst. 1997, 127, 122002. [Google Scholar]
- Kapuścik, A.; Hrouzek, P.; Kuzma, M.; Bártová, S.; Novák, P.; Jokela, J.; Pflüger, M.; Eger, A.; Hundsberger, H.; Kopecký, J. Novel aeruginosin-865 from Nostoc sp. as a potent anti-inflammatory agent. ChemBioChem. 2013, 14, 2329–2337. [Google Scholar] [CrossRef] [PubMed]
- Heinilä, L.M.P.; Jokela, J.; Ahmed, M.N.; Wahlsten, M.; Kumar, S.; Hrouzek, P.; Permi, P.; Koistinen, H.; Fewer, D.P.; Sivonen, K. Discovery of varlaxins, new aeruginosin-type inhibitors of human trypsins. Org. Biomol. Chem. 2022, 20, 2681–2692. [Google Scholar] [CrossRef]
- Fujii, K.; Sivonen, K.; Adachi, K.; Noguchi, K.; Shimizu, Y.; Sano, H.; Hirayama, K.; Suzuki, M.; Harada, K. Comparative study of toxic and non-toxic cyanobacterial products: A novel glycoside, suomilide, from non-toxic Nodularia spumigena HKVV. Tetrahedron Lett. 1997, 38, 5529–5532. [Google Scholar] [CrossRef]
- Ahmed, M.N.; Wahlsten, M.; Jokela, J.; Nees, M.; Stenman, U.H.; Alvarenga, D.O.; Strandin, T.; Sivonen, K.; Poso, A.; Permi, P.; et al. Potent inhibitor of human trypsins from the aeruginosin family of natural products. ACS Chem. Biol. 2021, 16, 2537–2546. [Google Scholar] [CrossRef] [PubMed]
- Carroll, A.R.; Pierens, G.K.; Fechner, G.; De Almeida, L.P.; Ngo, A.; Simpson, M.; Hyde, E.; Hooper, J.N.; Boström, S.L.; Musil, D.; et al. Dysinosin A: A novel inhibitor of factor VIIa and thrombin from a new genus and species of Australian sponge of the family dysideidae. J. Am. Chem. Soc. 2002, 124, 13340–13341. [Google Scholar] [CrossRef]
- Goetz, G.H.; Harrigan, G.G.; Likos, J.J.; Kasten, T.P. Isolation of chlorodysinosin A. PCT WO03/051831. Chem. Abst. 2003, 139, 47155. [Google Scholar]
- Carroll, A.R.; Buchanan, M.S.; Edser, A.; Hyde, E.; Simpson, M.; Quinn, R.J. Dysinosins B−D, inhibitors of Factor VIIa and thrombin from the Australian sponge Lamellodysidea chlorea. J. Nat. Prod. 2004, 67, 1291–1294. [Google Scholar] [CrossRef]
- Murakami, M.; Okita, Y.; Matsuda, H.; Okino, T.; Yamaguchi, K. Aeruginosin 298-A, a thrombin and trypsin inhibitor from the blue-green alga Microcystis aeruginosa (NIES-298). Tetrahedron Lett. 1994, 19, 3129–3132. [Google Scholar] [CrossRef]
- Ersmark, K.; Del, V.J.R.; Hanessian, S. Chemistry and biology of the aeruginosin family of serine protease inhibitors. Angew. Chem. Int. Ed. 2008, 47, 1202–1223. [Google Scholar] [CrossRef]
- Namikoshi, M.; Rinehart, K. Bioactive compounds produced by cyanobacteria. J. Ind. Microbiol. Biotechnol. 1996, 17, 373–384. [Google Scholar] [CrossRef]
- Fewer, D.P.; Jokela, J.; Paukku, E.; Österholm, J.; Wahlsten, P.; Permi, P.; Aitio, O.; Rouhiainen, L.; Gomez-Saez, G.V.; Sivonen, K. New Structural variants of aeruginosin produced by the toxic bloom forming cyanobacterium Nodularia spumigena. PLoS ONE 2013, 8, e73618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandler, B.; Murakami, M.; Clardy, J. Atomic structure of the trypsin−aeruginosin 98-B complex. J. Am. Chem. Soc. 1998, 120, 595–596. [Google Scholar] [CrossRef]
- Hanessian, S.; Ersmark, K.; Wang, X.T.; Del Valle, J.R.; Blomberg, N.; Xue, Y.F.; Fjellstrom, O. Structure-based organic synthesis of unnatural aeruginosin hybrids as potent inhibitors of thrombin. Bioorg. Med. Chem. Lett. 2007, 17, 3480–3485. [Google Scholar] [CrossRef]
- Valls, N.; López-Canet, M.; Vallribera, M.; Bonjoch, J. First total syntheses of aeruginosin 298-A and aeruginosin 298-B, based on a stereocontrolled route to the new amino acid 6-hydroxyoctahydroindole-2-carboxylic acid. Chem. Eur. J. 2001, 7, 3446. [Google Scholar] [CrossRef] [PubMed]
- Hanessian, S.; Del, V.J.R.; Xue, Y.; Blomberg, N. Total synthesis and structural confirmation of chlorodysinosin A. J. Am. Chem. Soc. 2006, 128, 10491–10495. [Google Scholar] [CrossRef] [PubMed]
- Hanessian, S.; Tremblay, M.; Petersen, J.F. The N-Acyloxyiminium Ion Aza-Prins Route to Octahydroindoles: Total Synthesis and Structural Confirmation of the Antithrombotic Marine Natural Product Oscillarin. J. Am. Chem. Soc. 2004, 126, 6064–6071. [Google Scholar] [CrossRef] [PubMed]
- Scherer, M.; Gademann, K. Total Synthesis and Structural Revision of Aeruginosin KT608A. Org. Lett. 2017, 19, 3915–3918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jorge, L.R.; Masahiro, M.; Alexander, T. Structure of Thrombin Inhibited by Aeruginosin 298-A from a Blue-Green Alga. J. Am. Chem. Soc. 1998, 120, 597–598. [Google Scholar]
- Murakami, M.; Ishida, K.; Okino, T.; Okita, Y.; Matsuda, H.; Yamaguchi, K. Aeruginosins 98-A and B, trypsin inhibitors from the blue-green alga Microcystis aeruginosa (NIES-98). Tetrahedron Lett. 1995, 36, 2785–2788. [Google Scholar] [CrossRef]
- Ishida, K.; Welker, M.; Christiansen, G.; Cadel-Six, S.; Bouchier, C.; Dittmann, E.; Hertweck, C.; de Tandeau, M.N. Plasticity and Evolution of Aeruginosin Biosynthesis in Cyanobacteria. Appl. Environ. Microbiol. 2009, 75, 2017–2026. [Google Scholar] [CrossRef] [Green Version]
- Welker, M.; Marsalek, B.; Sejnohova, L.; von Dohren, H. Detection and identification of oligopeptides in Microcystis (cyanobacteria) colonies: Toward an understanding of metabolic diversity. Peptides. 2006, 27, 2090–2103. [Google Scholar] [CrossRef]
- Skrzypczak-Jankun, E.; Carperos, V.E.; Ravichandran, K.G.; Tulinsky, A.; Westbrook, M.; Maraganore, J.M. Structure of the hirugen and hirulog 1 complexes of alpha-thrombin. J. Mol. Biol. 1991, 221, 1379–1393. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Jokela, J.; Wahlsten, M.; Nowruzi, B.; Permi, P.; Zhang, Y.Z.; Xhaard, H.; Fewer, D.P.; Sivonen, K. Nostosins, Trypsin Inhibitors Isolated from the Terrestrial Cyanobacterium Nostoc sp. Strain FSN. J. Nat. Prod. 2014, 77, 1784–1790. [Google Scholar] [CrossRef]
- Welker, M.; von Döhren, H. Cyanobacterial peptides-nature’s own combinatorial biosynthesis. FEMS Microbiol. Rev. 2006, 30, 530–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazur-Marzec, H.; Kaczkowska, M.J.; Blaszczyk, A.; Akcaalan, R.; Spoof, L.; Meriluoto, J. Diversity of Peptides Produced by Nodularia spumigena from Various Geographical Regions. Mar. Drugs 2013, 11, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahlstedt, S.; Fielding, E.N.; Moore, B.S.; Walsh, C.T. Prephenate decarboxylases: A new prephenate-utilizing enzyme family that performs nonaromatizing decarboxylation en route to diverse secondary metabolites. Biochemistry 2010, 49, 9021–9023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Omura, S.; Fujimoto, T.; Otoguro, K.; Matsuzaki, K.; Moriguchi, R.; Tanaka, H.; Sasaki, Y. Lactacystin, a novel microbial metabolite, induces neuritogenesis of neuroblastoma cells. J. Antibiot. 1991, 44, 113–116. [Google Scholar] [CrossRef] [Green Version]
- Cane, D.E.; Walsh, C.T. The parallel and convergent universes of polyketide synthases and nonribosomal peptide synthetases. Chem. Biol. 1999, 6, R319–R325. [Google Scholar] [CrossRef]
- Marahiel, M.A.; Stachelhaus, T.; Mootz, H.D. Modular Peptide Synthetases Involved in Nonribosomal Peptide Synthesis. Chem. Rev. 1997, 97, 2651–2674. [Google Scholar] [CrossRef]
- Cande, D.E.; Walsh, C.T.; Khosla, C. Harnessing the Biosynthetic Code: Combinations, Permutations, and Mutations. Science 1998, 282, 63–68. [Google Scholar] [CrossRef] [Green Version]
- Ishida, K.; Christiansen, G.; Yoshida, W.Y.; Kurmayer, R.; Welker, M.; Valls, N.; Bonjoch, J.; Hertweck, C.; Börner, T.; Hemscheidt, T.; et al. Biosynthesis and Structure of Aeruginosides 126A and 126B, Cyanobacterial Peptide Glycosides Bearing a 2-carboxy-6hydroxyoctahydroindole moiety. Chem. Biol. 2007, 14, 565–576. [Google Scholar] [CrossRef] [Green Version]
- Cox, R.J. Polyketides, Proteins and Genes in Fungi: Programmed Nano-Machines Begin to Reveal Their Genes. Org. Biomol. Chem. 2007, 38, 268. [Google Scholar] [CrossRef]
- Chiang, Y.M.; Oakley, B.R.; Keller, N.P.; Wang, C.C. Unraveling polyketide synthesis in members of the genus Aspergillus. Appl. Microbiol. Biotechnol. 2010, 86, 1719–1736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Döhren, H.; Keller, U.; Vater, J.; Zocher, R. Multifunctional Peptide Synthetases. Chem. Rev. 1997, 97, 2675–2706. [Google Scholar] [CrossRef] [PubMed]
- Mootz, H.D.; Marahiel, M.A. Biosynthetic systems for nonribosomal peptide antibiotic assembly. Curr. Opin. Chem. Biol. 1997, 1, 543–551. [Google Scholar] [CrossRef]
- Sung, C.T.; Chang, S.L.; Entwistle, R.; Ahn, G.; Lin, T.S.; Petrova, V.; Yeh, H.H.; Praseuth, M.B.; Chiang, Y.M.; Oakley, B.R.; et al. Overexpression of a three-gene conidial pigment biosynthetic pathway in Aspergillus nidulans reveals the first NRPS known to acetylate tryptophan. Fungal Genet. Biol. 2017, 101, 1–6. [Google Scholar] [CrossRef]
- Süssmuth, R.D.; Mainz, A. Nonribosomal Peptide Synthesis-Principles and Prospects. Angew. Chem. Int. Ed. 2017, 56, 3770–3821. [Google Scholar] [CrossRef]
- Becker, J.E.; Moore, R.E.; Moore, B.S. Cloning, sequencing, and biochemical characterization of the nostocyclopeptide biosynthetic gene cluster: Molecular basis for imine macrocyclization. Gene 2004, 325, 35–42. [Google Scholar] [CrossRef]
- Ross, A.C.; Xu, Y.; Lu, L.; Kersten, R.D.; Shao, Z.; Al-Suwailem, A.M.; Dorrestein, P.C.; Qian, P.Y.; Moore, B.S. Biosynthetic Multitasking Facilitates Thalassospiramide Structural Diversity in Marine Bacteria. J. Am. Chem. Soc. 2013, 135, 1155–1162. [Google Scholar] [CrossRef] [Green Version]
- Mootz, H.D.; Marahiel, M.A. The tyrocidine biosynthesis operon of Bacillus brevis: Complete nucleotide sequence and biochemical characterization of functional internal adenylation domains. J. Bacteriol. 1997, 179, 6843–6850. [Google Scholar] [CrossRef] [Green Version]
- Drake, E.J.; Miller, B.R.; Shi, C.; Tarrasch, J.T.; Sundlov, J.A.; Allen, C.L.; Skiniotis, G.; Aldrich, C.C.; Gulick, A.M. Structures of two distinct conformations of holo-non-ribosomal peptide synthetases. Nature 2016, 529, 235–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, C.T.; Gehring, A.M.; Weinreb, P.H.; Quadri, L.E.; Weinreb, P.H. Post-translational modification of polyketide and nonribosomal peptide synthases. Curr. Opin. Chem. Biol. 1997, 1, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, F.; Tanabe, G. Chemical strategies for visualizing and analyzing endogenous NRPS megasynthetases. ChemBioChem 2019, 20, 2032–2040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trauger, J.W.; Kohli, R.M.; Mootz, H.D.; Marahiel, M.A.; Walsh, C.T. Peptide cyclization catalysed by the thioesterase domain of tyrocidine synthetase. Nature 2000, 407, 215–218. [Google Scholar] [CrossRef]
- Grünewald, J.; Kopp, F.; Mahlert, C.; Linne, U.; Sieber, S.A.; Marahiel, M.A. Fluorescence resonance energy transfer as a probe of peptide cyclization catalyzed by nonribosomal thioesterase domains. Chem. Biol. 2005, 12, 873–881. [Google Scholar] [CrossRef] [Green Version]
- Alonzo, D.A.; Chiche-Lapierre, C.; Tarry, M.J.; Wang, J.; Schmeing, T.M. Structural basis of keto acid utilization in nonribosomal depsipeptide synthesis. Nat. Chem. Biol. 2020, 16, 493–496. [Google Scholar] [CrossRef]
- Magarvey, N.A.; Ehling-Schulz, M.; Walsh, C.T. Characterization of the Cereulide NRPS α-Hydroxy Acid Specifying Modules: Activation of α-Keto Acids and Chiral Reduction on the Assembly Line. J. Am. Chem. Soc. 2006, 128, 10698–10699. [Google Scholar] [CrossRef]
- Qiu, X.; Zhu, W.; Wang, W.; Jin, H.; Zhu, P.; Zhuang, R.; Yan, X. Structural and functional insights into the role of a cupin superfamily isomerase in the biosynthesis of Choi moiety of aeruginosin. J. Struct. Biol. 2019, 205, 44–52. [Google Scholar] [CrossRef]
- Qiu, X.; Wei, Y.; Zhu, W.; Fu, J.; Duan, X.; Jin, H.; Zhu, P.; Zhou, C.; Yan, X. Structural and functional investigation of AerF, a NADPH-dependent alkenal double bond reductase participating in the biosynthesis of Choi moiety of aeruginosin. J. Struct. Biol. 2020, 209, 107415. [Google Scholar] [CrossRef]
- Tan, K.; Zhou, M.; Jedrzejczak, R.P.; Wu, R.; Higuera, R.A.; Borek, D.; Babnigg, G.; Joachimiak, A. Structures of teixobactin-producing nonribosomal peptide synthetase condensation and adenylation domains. Curr. Res. Struct. Biol. 2020, 2, 14–24. [Google Scholar] [CrossRef]
- Schorn, M.A.; Jordan, P.A.; Podell, S.; Blanton, J.M.; Agarwal, V.; Biggs, J.S.; Allen, E.E.; Moore, B.S. Comparative Genomics of Cyanobacterial Symbionts Reveals Distinct, Specialized Metabolism in Tropical Dysideidae Sponges. mBio 2019, 10, e00821-e19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- May, D.S.; Crnkovic, C.M.; Krunic, A.; Wilson, T.A.; Fuchs, J.R.; Orjala, J.E. 15N Stable Isotope Labeling and Comparative Metabolomics Facilitates Genome Mining in Cultured Cyanobacteria. ACS Chem. Biol. 2020, 15, 758–765. [Google Scholar] [CrossRef]
- Auerbach, D.; Yan, F.; Zhang, Y.M.; Muller, R. Characterization of an Unusual Glycerate Esterification Process in Vioprolide Biosynthesis. ACS Chem. Biol. 2018, 13, 3123–3130. [Google Scholar] [CrossRef]
- Eckel, R.H.; Kahn, R.; Robertson, R.M.; Rizza, R.A. Preventing cardiovascular disease and diabetes: A call to action from the American Diabetes Association and the American Heart Association. Circulation 2006, 29, 1697–1699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Townsend, N.; Nichols, M.; Scarborough, P.; Rayner, M. Cardiovascular disease in Europe--epidemiological update 2015. Eur. Heart J. 2015, 36, 2696–2705. [Google Scholar] [CrossRef] [Green Version]
- Leung, D.; Abbenante, G.; Fairlie, D.P. Protease inhibitors: Current status and future prospects. J. Med. Chem. 2000, 43, 305–341. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Goswami, L.N.; Dikshit, D.K. Progress in the design of low molecular weight thrombin inhibitors. Med. Res. Rev. 2005, 25, 66–92. [Google Scholar] [CrossRef]
- Turk, B. Targeting proteases: Successes, failures and future prospects. Nat. Rev. Drug Discovery 2006, 5, 785–799. [Google Scholar] [CrossRef]
- Wang, G.; Goyal, N. Aeruginosin analogs and other compounds with rigid bicyclic structure as potential antithrombotic agents. Cardiovasc. Hematol. Agents. Med. Chem. 2009, 7, 147–165. [Google Scholar] [CrossRef]
- Nyberg, P.; Ylipalosaari, M.; Sora, T.; Salo, T. Trypsins and their role in carcinoma growth. Exp. Cell Res. 2006, 312, 1219–1228. [Google Scholar] [CrossRef]
- Hockla, A.; Radisky, D.C.; Radisky, E.S. Mesotrypsin promotes malignant growth of breast cancer cells through shedding of CD109. Breast Cancer Res. Treat. 2010, 124, 27–38. [Google Scholar] [CrossRef] [Green Version]
- Jiang, G.; Cao, F.; Ren, G.; Gao, D.; Bhakta, V.; Zhang, Y.; Cao, H.; Dong, Z.; Zang, W.; Zhang, S.; et al. PRSS3 promotes tumour growth and metastasis of human pancreatic cancer. Gut 2010, 59, 1535–1544. [Google Scholar] [CrossRef] [PubMed]
- Nativitat, V.; Mercè, V.; Shmuel, C.; Josep, B. Syntheses of both the putative and revised structures of aeruginosin EI461 bearing a new bicyclic alpha-amino acid. Org. Lett. 2003, 5, 447–450. [Google Scholar]
- Sidelmann, J.J.; Gram, J.; Jespersen, J.; Kluft, C. Fibrin Clot Formation and Lysis: Basic Mechanisms. Semin. Thromb. Hemost. 2000, 26, 605–618. [Google Scholar] [CrossRef]
- Zabczyk, M.; Ariens, R.A.S.; Undas, A. Fibrin clot properties in cardiovascular disease: From basic mechanisms to clinical practice. Cardiovasc. Res. 2023, 119, 94–111. [Google Scholar] [CrossRef] [PubMed]
- Danø, K.; Rømer, j.; Nielsen, B.S.; Bjørn, S.; Pyke, C.; Rygaard, J.; Lund, L.R. Cancer invasion and tissue remodeling-cooperation of protease systems and cell types. APMIS. 1999, 107, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Lieb, W.; Gona, P.; Lason, M.G.; Massaro, J.M.; Lipinska, I.; Keaney, J.F., Jr.; Rong, J.; Corey, D.; Hoffmann, U.; Fox, C.S.; et al. Biomarkers of the osteoprotegerin pathway: Clinical Correlates, subclinical disease, incident cardiovascular disease, and mortality. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1849–1854. [Google Scholar] [CrossRef]
- Hsu, P.C.; Chen, Y.H.; Cheng, C.F.; Kuo, C.Y.; Sytwu, H.K. Interleukin-6 and Interleukin-8 regulate STAT3 activation migration/invasion and EMT in chrysophanol-treated oral cancer cell lines. Life 2021, 11, 423. [Google Scholar] [CrossRef]
- Schirbel, A.; Kessler, S.; Rieder, F.; West, G.; Rebert, N.; Asosingh, K.; McDonald, C.; Fiocchi, C. Pro-Angiogenic Activity of TLRs and NLRs: A Novel link between gut microbiota and intestinal angiogenesis. Gastroenterology 2013, 144, 613–623. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Froio, R.M.; Sciuto, T.E.; Dvorak, A.M.; Alon, R.; Luscinskas, F.W. ICAM-1 regulates neutrophil adhesion and transcellular migration of TNF-α-activated vascular endothelium under flow. Blood. 2005, 106, 584–592. [Google Scholar] [CrossRef] [Green Version]
- Singh, N.; Baby, D.; Rajguru, J.P.; Patil, P.B.; Thakkannavar, S.S.; Pujari, V.B. Inflammation and cancer. Ann. Afr. Med. 2019, 18, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Bella, J.; Kolatkar, P.R.; Marlor, C.W.; Greve, J.M.; Rossmann, M.G. The structure of the two amino-terminal domains of human ICAM-1 suggests how it functions as a rhinovirus receptor and as an LFA-1 integrin ligand. Proc. Natl. Acad. Sci. USA 1998, 95, 4140–4145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanz, M.; Andreote, A.P.; Fiore, M.F.; Dörr, F.A.; Pinto, E. Structural characterization of new peptide variants produced by cyanobacteria from the Brazilian Atlantic coastal forest using liquid chromatography coupled to quadrupole time-of-flight tandem mass spectrometry. Mar. Drugs 2015, 13, 3892–3919. [Google Scholar] [CrossRef] [PubMed]
- Stachelhaus, T.; Schneider, A.; Marahiel, M.A. Rational design of peptide antibiotics by targeted replacement of bacterial and fungal domains. Science 1995, 269, 69–72. [Google Scholar] [CrossRef] [PubMed]
- Crüsemann, M.; Kohlhaas, C.; Piel, J. Evolution-guided engineering of nonribosomal peptide synthetase adenylation domains. Chem. Sci. 2013, 4, 1041–1045. [Google Scholar] [CrossRef]
- Stanisic, A.; Husken, A.; Stephan, P.; Niquille, D.L.; Reinstein, J.; Kries, H. Engineered nonribosomal peptide synthetase shows opposite amino acid loading and condensation specificity. ACS Catal. 2021, 11, 8692–8700. [Google Scholar] [CrossRef]
- Bozhuyuk, K.A.J.; Fleischhacker, F.; Linck, A.; Wesche, F.; Tietze, A.; Niesert, C.P.; Bode, H.B. De novo design and engineering of non-ribosomal peptide synthetases. Nat. Chem. 2018, 10, 275–281. [Google Scholar] [CrossRef]
- AKegler, C.; Bode, H.B. Artificial Splitting of a Non-Ribosomal Peptide Synthetase by Inserting Natural Docking Domains. Angew. Chem. Int. Ed. 2020, 59, 13463–13467. [Google Scholar] [CrossRef]
- Hahn, M.; Stachelhaus, T. Harnessing the potential of communication-mediating domains for the biocombinatorial synthesis of nonribosomal peptides. Proc. Natl. Acad. Sci. USA 2006, 103, 275–280. [Google Scholar] [CrossRef] [Green Version]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Name | Source | Structure Diversity | IC50 | Refs. | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| The N Terminal Residue | The Second Residue | The Third Residue | The C Terminal Residue | Trypsin | Thrombin | Plasmin | ||||
| 1 | Aeruginosin 298A | Microcystis aeruginosa NIES-298 | R2=OH, R4=OH D-Hpla | X = side chain of D-Leu | R5=OH L-Choi | L-Argol | 1.38 μM | 0.42 μM | >14 μM | [4,28] |
| 2 | Aeruginosin 98A | Microcystis aeruginosa NIES-98 | R1=Cl, R2=OH, R4=OH D-Hpla | X = side chain of D-allo-Ile | R5=OSO3H L-Choi | Agma | 0.87 μM | 10.17 μM | 8.72 μM | [20,29] |
| 3 | Aeruginosin 98B | Microcystis aeruginosa NIES-98 | R1=H, R2=OH, R4=OH D-Hpla | X = side chain of D-allo-Ile | R5=OSO3H L-Choi | Agma | 0.92 μM | 15.28 μM | 10.7 μM | [20,29] |
| 4 | Aeruginosin 98-C | Microcystis aeruginosa NIES-98 | R1=Br, R2=OH, R4=OH D-Hpla | X = side chain of D-allo-Ile | R5=OSO3H L-Choi | Agma | 5.33 μM | 4.5 μM | 6.83 μM | [29] |
| 5 | Aeruginosin 298B | Microcystis aeruginosa NIES-298 | R2=OH, R4=OH D-Hpla | X = side chain of D-Leu | R5=OH L-Choi-NH2 | - | >100 μM | >100 μM | >100 μM | [4] |
| 6 | Aeruginosin 101 | Microcystis aeruginosa NIES-101 | R1=Cl, R2=OH, R3=Cl, R4=OH D-Hpla | X = side chain of D-allo-Ile | R5=OSO3H L-Choi | Agma | 4.15 μM | 4.43 μM | 4.57 μM | [4] |
| 7 | Aeruginosin 89A | Microcystis aeruginosa NIES-89 | R1=Cl, R2=OSO3H, R4=OH D-Hpla | X = side chain of D-Leu | R5=OH L-Choi | L-Argal | 0.48 μM | 0.04 μM | 0.02 μM | [4] |
| 8 | Aeruginosin 89B | Microcystis aeruginosa NIES-89 | R1=Cl, R2=OSO3H, R4=OH D-Hpla | X = side chain of D-Leu | R5=OH L-Choi | D-Argal | 7.9 μM | 0.06 μM | 0.55 μM | [4] |
| 9 | Microcin SF608 | Microcystis aeruginosa | R2=OH, R4=OH L-Hpla | X = side chain of L-Phe | R5=OH L-Choi | Agma | 0.82 μM | - | - | [29] |
| 10 | Aeruginosin GE686 | Microcystis aeruginosa from bloom material | R1=Br, R2=OH, R3=Cl, R4=OH D-Hpla | X = side chain of D-allo-Ile | R5=OH L-Choi | Agma | 3.2 μM | 12.8 μM | - | [5] |
| 11 | Aeruginosin GE766 | Microcystis aeruginosa from bloom material | R1=Br, R2=OH, R3=Cl, R4=OH D-Hpla | X = side chain of D-allo-Ile | R5=OSO3H L-Choi | Agma | 12.2 μM | >45.5 μM | - | [5] |
| 12 | Aeruginosin GE730 | Microcystis aeruginosa from bloom material | R1=Br, R2=OH, R3=Br, R4=OH D-Hpla | X = side chain of D-allo-Ile | R5=OH L-Choi | Agma | 2.3 μM | 12.9 μM | - | [5] |
| 13 | Aeruginosin GE810 | Microcystis aeruginosa from bloom material | R1=Br, R2=OH, R3=Br, R4=OH D-Hpla | X = side chain of D-allo-Ile | R5=OSO3H L-Choi | Agma | 18.2 μM | >45.5 μM | - | [5] |
| 14 | Aeruginosin GE642 | Microcystis aeruginosa from bloom material | R1=Cl, R2=OH, R3=Cl, R4=OH D-Hpla | X = side chain of D-Leu | R5=OH L-Choi | Agma | 8.5 μM | >45.5 μM | - | [5] |
| 15 | Aeruginosin KY642 | Microcystis aeruginosa from bloom material | R1=Cl, R2=OH, R3=Cl, R4=OH D-Hpla | X = side chain of D-Ile | R5=OH L-Choi | Agma | 1.85 μM | - | - | [5,7] |
| 16 | Aeruginosin DA688 | Microcystis aeruginosa from bloom material | R1=Cl, R2=OH, R3=H, R4=OH D-Hpla | X = side chain of D-Leu | R5=OSO3H L-Choi | Agma | 9.5 μM | >45.5 μM | - | [8] |
| 17 | Aeruginosin 205A | Planktothrix agardhii NIES-205 | R4=OSO3H L-Pla | X = side chain of (2R,3S)-Hleu | R5=Cl Ccoi | Agma | 0.08 μM | 1.65 μM | - | [4,9] |
| 18 | Aeruginosin 205B | Planktothrix agardhii NIES-205 | R4=OSO3H D-Pla | X = side chain of (2S,3R)-Hleu | R5=Cl Ccoi | Agma | 0.08 μM | 0.19 μM | - | [4,9] |
| 19 | Oscillarin | Planktothrix agardhii B2 83 | R4=OH, D-Pla | X = side chain of D-Phe | R5=OH L-Choi | Aaep | 0.03 μM | 0.02 μM | >300 μM | [10,26] |
| 20 | Aeruginosin 865 | Nostoc sp. Lukešová 30/93 | R2=OH, R4=OH D-Hpla | X = side chain of D-Leu | R5=ManA,R6=HA Choi | Agma | - | - | - | [11] |
| 21 | Varlaxin 1046A | Nostoc sp. UHCC 0870 | Mgs | X = side chain of D-Ile | Hex | Aaep | 0.62–3.6 nM | - | - | [12] |
| 22 | Varlaxin 1022A | Nostoc sp. UHCC 0870 | Mgs | X = side chain of D-Ile | Hex | Agma | 97–230 nM | - | - | [12] |
| 23 | Suomilide | Nodularia sphaerocarpa UHCC 0038 | Mgs | X = side chain of allo-Ile | Abn | Aaep | 1.8 μM | - | - | [12,13] |
| 24 | Dysinosin A | Species in the family Dysideidae | R4=H D-glyceric acid | X = side chain of D-Leu | R5=OH R6=OH L-Choi | Aaep | - | 0.38 μM | - | [15,19] |
| 25 | Chlorodysinosin A | - | D-glyceric acid | X = side chain of D-Leu | R5=OH L-Choi | Aaep | 0.03 μM | 0.004 μM | - | [19,25] |
| 26 | Dysinosin B | Lamellodysidea chlorea | D-glyceric acid | X = side chain of D-Val | R5=xylose,R6=OH L-Choi | Aaep | - | 0.17 μM | - | [17] |
| 27 | Dysinosin C | Lamellodysidea chlorea | D-glyceric acid | X = side chain of D-Val | R5=OH,R6=OH L-Choi | Aaep | - | 0.55 μM | - | [17] |
| 28 | Dysinosin D | Lamellodysidea chlorea | D-glyceric acid | X = side chain of D-Val | R5=OH,R6=OH L-Choi | Aaep | - | >5.1 μM | - | [17] |
| 29 | Aeruginosin KT608A | Microcystis aeruginosa from bloom material | R2=OH, R4=OH L-Hpla | X = side chain of D-Phe | R5=OH L-Choi | Agma | 1.9 μM | - | - | [12,27] |
| 30 | Aeruginosin 686A | Microcystis aeruginosa PCC 7806 | R1=Cl, R2=OH, R4=OH | X = side chain of D-Tyr | R5=OH L-Choi | Argal | - | - | - | [30,31] |
| 31 | Aeruginosin 686B | Microcystis aeruginosa PCC 7806 | R1=Cl, R2=OH, R4=OH | X = side chain of D-Tyr | R5=OH L-Choi | Arg | - | - | - | [30,31] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, J.; Zhang, M.; Huang, Z.; Fang, J.; Wang, Z.; Zhou, C.; Qiu, X. Diversity, Biosynthesis and Bioactivity of Aeruginosins, a Family of Cyanobacteria-Derived Nonribosomal Linear Tetrapeptides. Mar. Drugs 2023, 21, 217. https://doi.org/10.3390/md21040217
Liu J, Zhang M, Huang Z, Fang J, Wang Z, Zhou C, Qiu X. Diversity, Biosynthesis and Bioactivity of Aeruginosins, a Family of Cyanobacteria-Derived Nonribosomal Linear Tetrapeptides. Marine Drugs. 2023; 21(4):217. https://doi.org/10.3390/md21040217
Chicago/Turabian StyleLiu, Jiameng, Mengli Zhang, Zhenkuai Huang, Jiaqi Fang, Zhongyuan Wang, Chengxu Zhou, and Xiaoting Qiu. 2023. "Diversity, Biosynthesis and Bioactivity of Aeruginosins, a Family of Cyanobacteria-Derived Nonribosomal Linear Tetrapeptides" Marine Drugs 21, no. 4: 217. https://doi.org/10.3390/md21040217
APA StyleLiu, J., Zhang, M., Huang, Z., Fang, J., Wang, Z., Zhou, C., & Qiu, X. (2023). Diversity, Biosynthesis and Bioactivity of Aeruginosins, a Family of Cyanobacteria-Derived Nonribosomal Linear Tetrapeptides. Marine Drugs, 21(4), 217. https://doi.org/10.3390/md21040217