

Synthesis and Antimalarial Evaluation of Halogenated Analogues of Thiaplakortone A
 and
and
Abstract
:1. Introduction
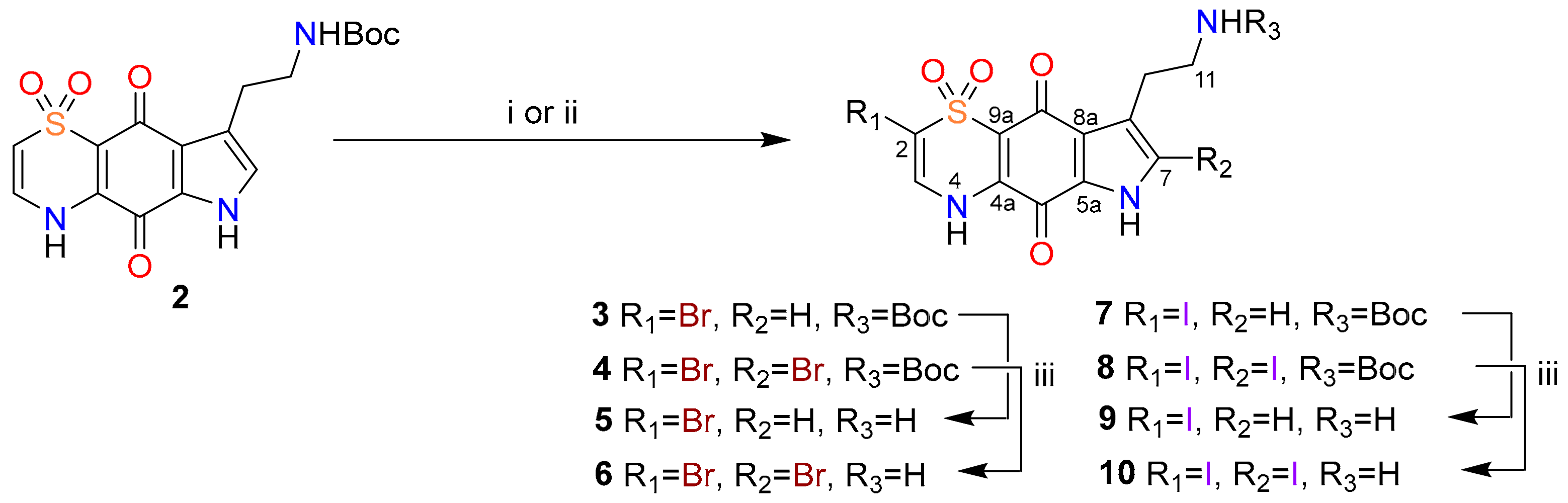
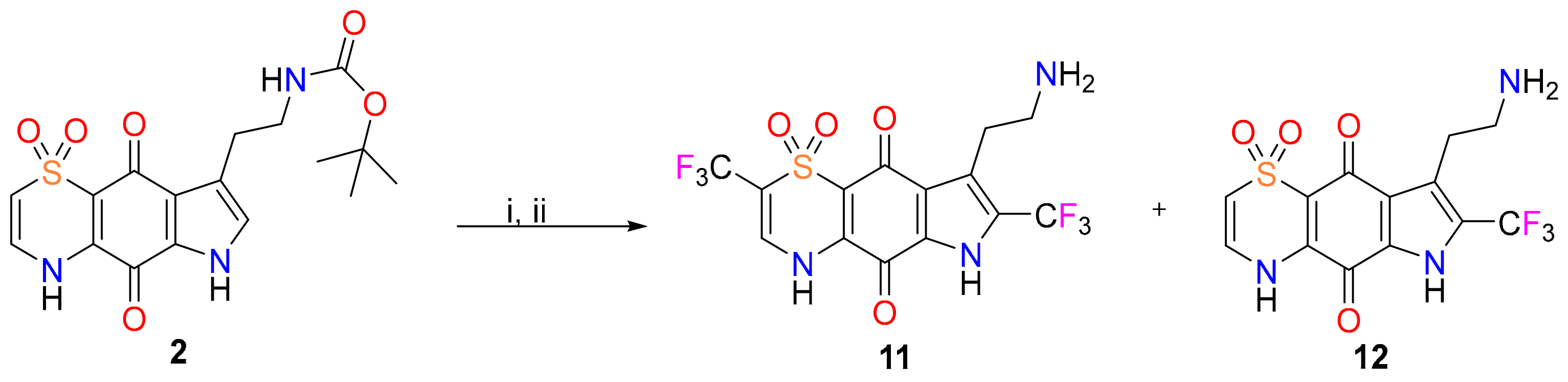
2. Results and Discussion
3. Materials and Methods
3.1. Chemistry Procedures
3.2. Halogenation Reactions
3.3. Boc Deprotection Procedure
3.4. Diversinate™ Synthetic Procedure
3.5. HPLC Purification of Reaction Products
3.6. Experimental Data for Compounds 3–11
3.7. In Vitro Antimalarial Image-Based Asexual Assay
3.8. In Vitro Cytotoxicity Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chinnappanna, N.K.R.; Yennam, G.; Chaitanya, C.B.H.N.V.; Pottathil, S.; Borah, P.; Venugopala, K.N.; Deb, P.K.; Mailavaram, R.P. Recent approaches in the drug research and development of novel antimalarial drugs with new targets. Acta Pharm. 2023, 73, 1–27. [Google Scholar] [CrossRef]
- Available online: https://www.who.int/teams/global-malaria-programme/reports/world-malaria-report-2022 (accessed on 1 March 2023).
- Mishra, M.; Mishra, V.K.; Kashaw, V.; Iyer, A.K.; Kashaw, S.K. Comprehensive review on various strategies for antimalarial drug discovery. Eur. J. Med. Chem. 2016, 125, 1300–1320. [Google Scholar] [CrossRef]
- Klein, E. Antimalarial drug resistance: A review of the biology and strategies to delay emergence and spread. Int. J. Antimicrob. Agents 2013, 41, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Dondorp, A.M.; Nosten, F.; Yi, P.; Das, D.; Phyo, A.P.; Tarning, J.; Lwin, K.M.; Ariey, F.; Hanpithakpong, W.; Lee, S.J.; et al. Artemisinin resistance in Plasmodium falciparum malaria. N. Engl. J. Med. 2009, 361, 455–467. [Google Scholar] [CrossRef] [PubMed]
- Balikagala, B.; Fukuda, N.; Ikeda, M.; Katuro, O.T.; Tachibana, S.-I.; Yamauchi, M.; Opio, W.; Emoto, S.; Anywar, D.A.; Kimura, E.; et al. Evidence of artemisinin-resistant malaria in Africa. N. Engl. J. Med. 2021, 385, 1163–1171. [Google Scholar] [CrossRef]
- Available online: https://www.griffith.edu.au/institute-drug-discovery/unique-resources/naturebank (accessed on 3 March 2023).
- Duffy, S.; Avery, V.M. Development and optimization of a novel 384-well anti-malarial imaging assay validated for high-throughput screening. Am. J. Trop. Med. Hyg. 2012, 86, 84–92. [Google Scholar] [CrossRef] [PubMed]
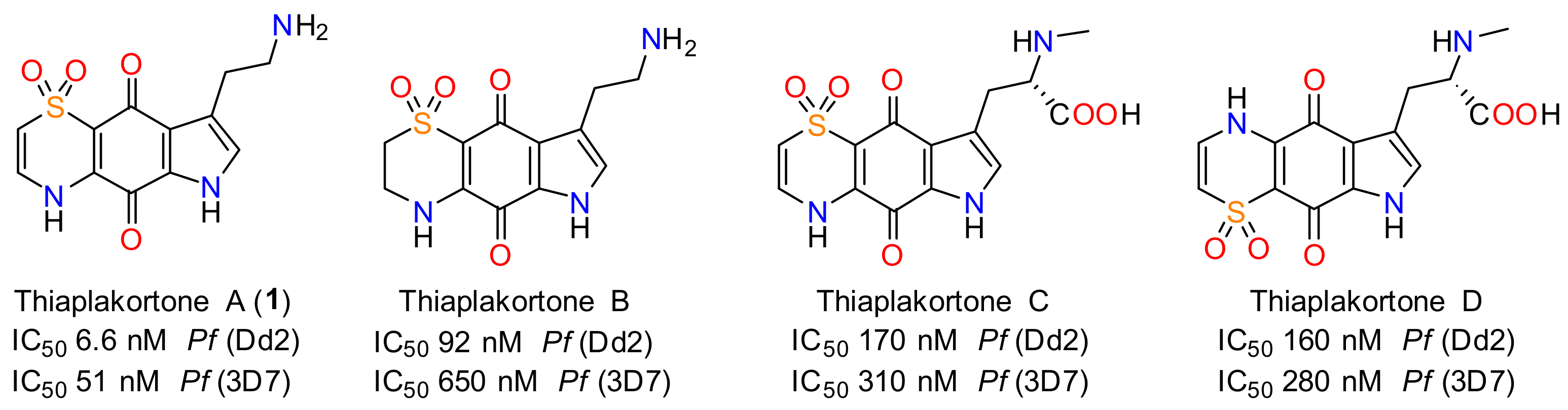
- Davis, R.A.; Duffy, S.; Fletcher, S.; Avery, V.M.; Quinn, R.J. Thiaplakortones A–D: Antimalarial thiazine alkaloids from the Australian marine sponge Plakortis lita. J. Org. Chem. 2013, 78, 9608–9613. [Google Scholar] [CrossRef]
- Pouwer, R.H.; Deydier, S.M.; Le, P.V.; Schwartz, B.D.; Franken, N.C.; Davis, R.A.; Coster, M.J.; Charman, S.A.; Edstein, M.D.; Skinner-Adams, T.S.; et al. Total synthesis of thiaplakortone A: Derivatives as metabolically stable leads for the treatment of malaria. ACS Med. Chem. Lett. 2014, 5, 178–182. [Google Scholar] [CrossRef]
- Schwartz, B.D.; Skinner-Adams, T.S.; Andrews, K.T.; Coster, M.J.; Edstein, M.D.; MacKenzie, D.; Charman, S.A.; Koltun, M.; Blundell, S.; Campbell, A.; et al. Synthesis and antimalarial evaluation of amide and urea derivatives based on the thiaplakortone A natural product scaffold. Org. Biomol. Chem. 2015, 13, 1558–1570. [Google Scholar] [CrossRef]
- Hernandes, M.Z.; Cavalcanti, S.M.T.; Moreira, D.R.M.; de Azevedo, J.; Filgueira, W.; Leite, A.C.L. Halogen atoms in the modern medicinal chemistry: Hints for the drug design. Curr. Drug Targets 2010, 11, 303–314. [Google Scholar] [CrossRef]
- Buchini, S.; Buschiazzo, A.; Withers, S.G. A new generation of specific Trypanosoma cruzi trans-sialidase inhibitors. Angew. Chem. Int. Ed. 2008, 47, 2700–2703. [Google Scholar] [CrossRef] [PubMed]
- Bonnefous, C.L.; Payne, J.E.; Roppe, J.; Zhuang, H.; Chen, X.; Symons, K.T.; Nguyen, P.M.; Sablad, M.; Rozenkrants, N.; Zhang, Y. Discovery of inducible nitric oxide synthase (iNOS) inhibitor development candidate KD7332, part 1: Identification of a novel, potent, and selective series of quinolinone iNOS dimerization inhibitors that are orally active in rodent pain models. J. Med. Chem. 2009, 52, 3047–3062. [Google Scholar] [CrossRef] [PubMed]
- Leite, A.C.L.; Moreira, D.R.d.M.; Cardoso, M.V.d.O.; Hernandes, M.Z.; Pereira, V.R.A.; Silva, R.O.; Kiperstok, A.C.; Lima, M.d.S.; Soares, M.B.P. Synthesis, Cruzain Docking, and in vitro Studies of aryl-4-oxothiazolylhydrazones against Trypanosoma cruzi. ChemMedChem 2007, 2, 1339–1345. [Google Scholar] [CrossRef]
- Gerebtzoff, G.; Li-Blatter, X.; Fischer, H.; Frentzel, A.; Seelig, A. Halogenation of drugs enhances membrane binding and permeation. ChemBioChem 2004, 5, 676–684. [Google Scholar] [CrossRef] [PubMed]
- Gentry, C.; Egleton, R.; Gillespie, T.; Abbruscato, T.; Bechowski, H.; Hruby, V.; Davis, T. The effect of halogenation on blood–brain barrier permeability of a novel peptide drug. Peptides 1999, 20, 1229–1238. [Google Scholar] [CrossRef]
- Geronikaki, A.A.; Lagunin, A.A.; Hadjipavlou-Litina, D.I.; Eleftheriou, P.T.; Filimonov, D.A.; Poroikov, V.V.; Alam, I.; Saxena, A.K. Computer-aided discovery of anti-inflammatory thiazolidinones with dual cyclooxygenase/lipoxygenase inhibition. J. Med. Chem. 2008, 51, 1601–1609. [Google Scholar] [CrossRef]
- La Motta, C.; Sartini, S.; Mugnaini, L.; Salerno, S.; Simorini, F.; Taliani, S.; Marini, A.M.; Da Settimo, F.; Lavecchia, A.; Novellino, E. Exploiting the pyrazolo [3, 4-d] pyrimidin-4-one ring system as a useful template to obtain potent adenosine deaminase inhibitors. J. Med. Chem. 2009, 52, 1681–1692. [Google Scholar] [CrossRef]
- Monforte, A.-M.; Logoteta, P.; Ferro, S.; De Luca, L.; Iraci, N.; Maga, G.; De Clercq, E.; Pannecouque, C.; Chimirri, A. Design, synthesis, and structure–activity relationships of 1, 3-dihydrobenzimidazol-2-one analogues as anti-HIV agents. Bioorg. Med. Chem. 2009, 17, 5962–5967. [Google Scholar] [CrossRef]
- Metrangolo, P.; Neukirch, H.; Pilati, T.; Resnati, G. Halogen bonding based recognition processes: A world parallel to hydrogen bonding. Acc. Chem. Res. 2005, 38, 386–395. [Google Scholar] [CrossRef]
- Metrangolo, P.; Resnati, G. Halogen versus hydrogen. Science 2008, 321, 918–919. [Google Scholar] [CrossRef]
- Gunaydin, H.; Altman, M.D.; Ellis, J.M.; Fuller, P.; Johnson, S.A.; Lahue, B.; Lapointe, B. Strategy for extending half-life in drug design and its significance. ACS Med. Chem. Lett. 2018, 9, 528–533. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.H.; Chen, J.S. Synthesis of breitfussin B by late-stage bromination. Org. Lett. 2015, 17, 3718–3721. [Google Scholar] [CrossRef]
- Liu, R.; Zhang, P.; Gan, T.; Cook, J.M. Regiospecific bromination of 3-methylindoles with NBS and its application to the concise synthesis of optically active unusual tryptophans present in marine cyclic peptides. J. Org. Chem. 1997, 62, 7447–7456. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, Y.; Dixon, J.A.; O’Hara, F.; Funder, E.D.; Dixon, D.D.; Rodriguez, R.A.; Baxter, R.D.; Herle, B.; Sach, N.; Collins, M.R.; et al. Practical and innate carbon–hydrogen functionalization of heterocycles. Nature 2012, 492, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Egbewande, F.A.; Coster, M.J.; Jenkins, I.A.; Davis, R.A. Reaction of papaverine with Baran Diversinates™. Molecules 2019, 24, 3938. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://chemicalize.com (accessed on 20 March 2023).
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Hughes, J.P.; Rees, S.; Kalindjian, S.B.; Philpott, K.L. Principles of early drug discovery. Br. J. Pharmacol. 2011, 162, 1239–1249. [Google Scholar] [CrossRef]
- Tietjen, I.; Cassel, J.; Register, E.T.; Zhou, X.Y.; Messick, T.E.; Keeney, F.; Lu, L.D.; Beattie, K.D.; Rali, T.; Tebas, P.; et al. The natural stilbenoid (–)-hopeaphenol inhibits cellular entry of SARS-CoV-2 USA-WA1/2020, B.1.1.7, and B.1.351 variants. Antimicrob. Agents Chemother. 2021, 65, e00772-21. [Google Scholar] [CrossRef]
- Varricchio, A.; Khan, S.; Price, Z.K.; Davis, R.A.; Ramesh, S.A.; Yool, A.J. Pharmacological inhibition of membrane signaling mechanisms reduces the invasiveness of U87-MG and U251 MG glioblastoma cells in vitro. Cancers 2023, 15, 1027. [Google Scholar] [CrossRef]


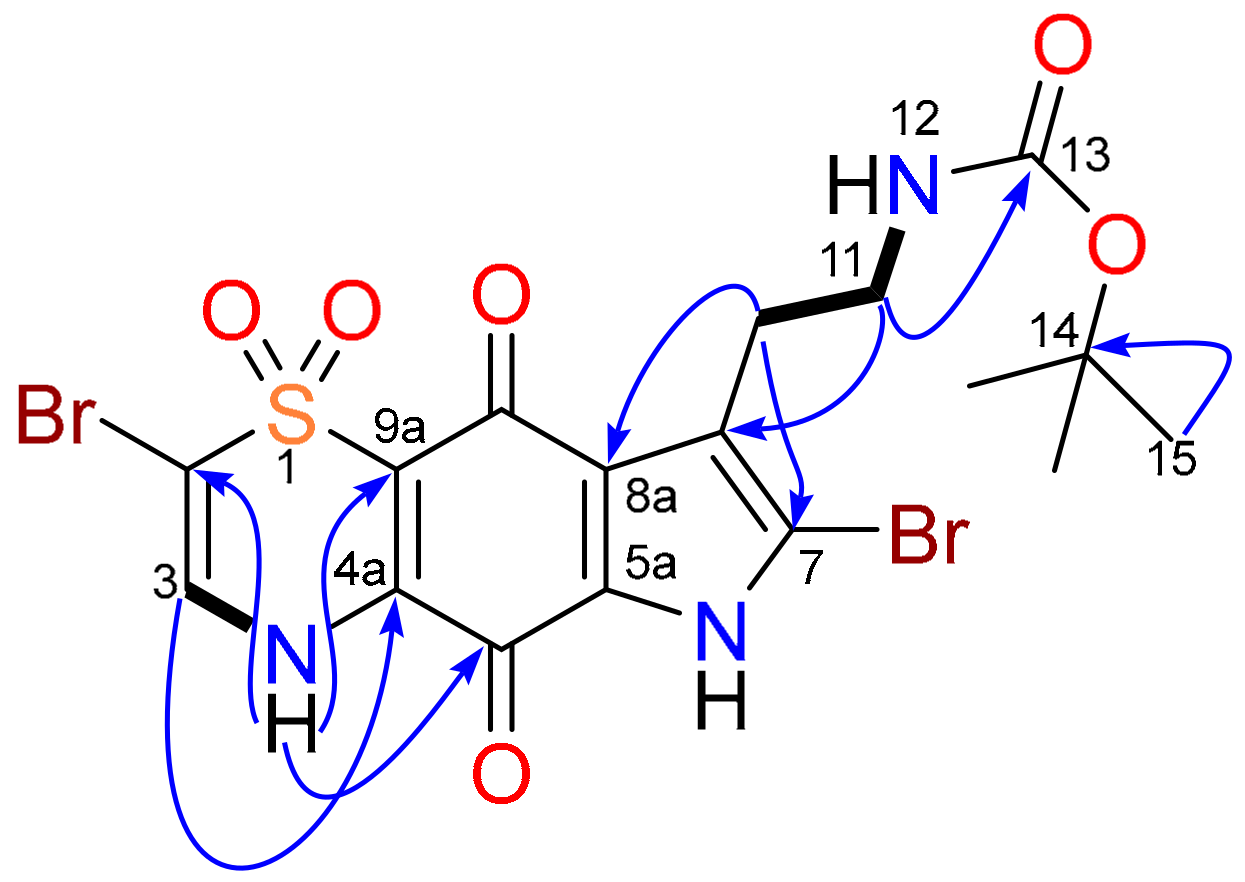
 ) and key HMBC (
) and key HMBC ( ) correlations for compound 4.
) correlations for compound 4.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | MW | cLog P | cLog D7.4 | HBD | HBA | Ro5 Violations |
|---|---|---|---|---|---|---|
| 1 | 293 | −1.52 | −3.19 | 3 | 6 | 0 |
| 2 | 393 | −0.02 | −0.02 | 3 | 6 | 0 |
| 3 | 471 | 1.56 | 1.56 | 3 | 6 | 0 |
| 4 | 549 | 2.02 | 1.95 | 3 | 6 | 0 |
| 5 | 371 | 0.06 | −1.61 | 3 | 6 | 0 |
| 6 | 449 | 0.15 | −0.94 | 3 | 6 | 0 |
| 7 | 519 | 1.72 | 1.72 | 3 | 6 | 1 |
| 8 | 645 | 2.27 | 2.21 | 3 | 6 | 1 |
| 9 | 419 | 0.22 | −1.45 | 3 | 6 | 0 |
| 10 | 545 | 0.50 | −0.61 | 3 | 6 | 1 |
| 11 | 429 | −0.42 | −1.50 | 3 | 6 | 0 |
| Compound | Pf3D7 a IC50 ± SD µM | PfDd2 b IC50 ± SD µM | HEK293 c IC50 ± SD µM | SI for 3D7 d | SI for Dd2 d |
| 1 | 0.061 ± 0.009 | 0.008 ± 0.001 | 1.32 ± 0.092 | 22 | 150 |
| 2 | 0.227 ± 0.044 | 0.086 ± 0.012 | 5.53 ± 0.021 | 24 | 64 |
| 3 | 3.515 ± 0.350 | 0.583 ± 0.094 | >80 | >23 | >137 |
| 4 | 1.373 ± 0.117 | 2.060 ± 0.192 | >80 | >58 | >39 |
| 5 | 0.559 ± 0.145 | 0.058 ± 0.016 | >80 | >143 | >1379 |
| 6 | 1.653 ± 0.194 | 0.487 ± 0.098 | >80 | >48 | >164 |
| 7 | 5.623 ± 0.383 | 1.194 ± 0.270 | >80 | >14 | >67 |
| 8 | 1.623 ± 0.154 | 0.659 ± 0.119 | >80 | >49 | >121 |
| 9 | 2.223 ± 0.248 | 0.317 ± 0.008 | >80 | >36 | >252 |
| 10 | 1.685 ± 0.316 | 0.725 ± 0.244 | >80 | >47 | >110 |
| 11 | 6.800 ± 2.444 | 4.550 ± 1.154 | >80 | >12 | >18 |
| Controls | Pf3D7 a IC50 ± SD nM | PfDd2 b IC50 ± SD nM | HEK293 c IC50 ± SD nM | SI for 3D7 d | SI for Dd2 d |
| Pyrimethamine | 3.75 ± 0.30 | IA e | >40,000 | >10,666 | - |
| Artesunate | 0.49 ± 0.20 | 0.38 ± 0.01 | >4000 | >8163 | >10,526 |
| Puromycin | 31.95 ± 16.2 | 17.00 ± 3.96 | 1044 ± 66 | 33 | 61 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Egbewande, F.A.; Schwartz, B.D.; Duffy, S.; Avery, V.M.; Davis, R.A. Synthesis and Antimalarial Evaluation of Halogenated Analogues of Thiaplakortone A. Mar. Drugs 2023, 21, 317. https://doi.org/10.3390/md21050317
Egbewande FA, Schwartz BD, Duffy S, Avery VM, Davis RA. Synthesis and Antimalarial Evaluation of Halogenated Analogues of Thiaplakortone A. Marine Drugs. 2023; 21(5):317. https://doi.org/10.3390/md21050317
Chicago/Turabian StyleEgbewande, Folake A., Brett D. Schwartz, Sandra Duffy, Vicky M. Avery, and Rohan A. Davis. 2023. "Synthesis and Antimalarial Evaluation of Halogenated Analogues of Thiaplakortone A" Marine Drugs 21, no. 5: 317. https://doi.org/10.3390/md21050317
APA StyleEgbewande, F. A., Schwartz, B. D., Duffy, S., Avery, V. M., & Davis, R. A. (2023). Synthesis and Antimalarial Evaluation of Halogenated Analogues of Thiaplakortone A. Marine Drugs, 21(5), 317. https://doi.org/10.3390/md21050317