
Discovery of a Novel Chromone Enantiomer and the Precursors of Nonactic Acid from the Coral-Reef-Derived Streptomyces sp. SCSIO 66814
 , , ,
, , ,
Abstract
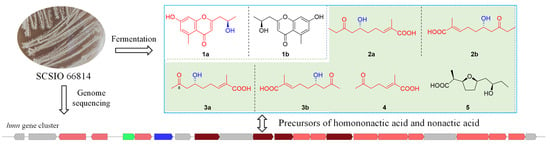
:
1. Introduction
2. Results and Discussion
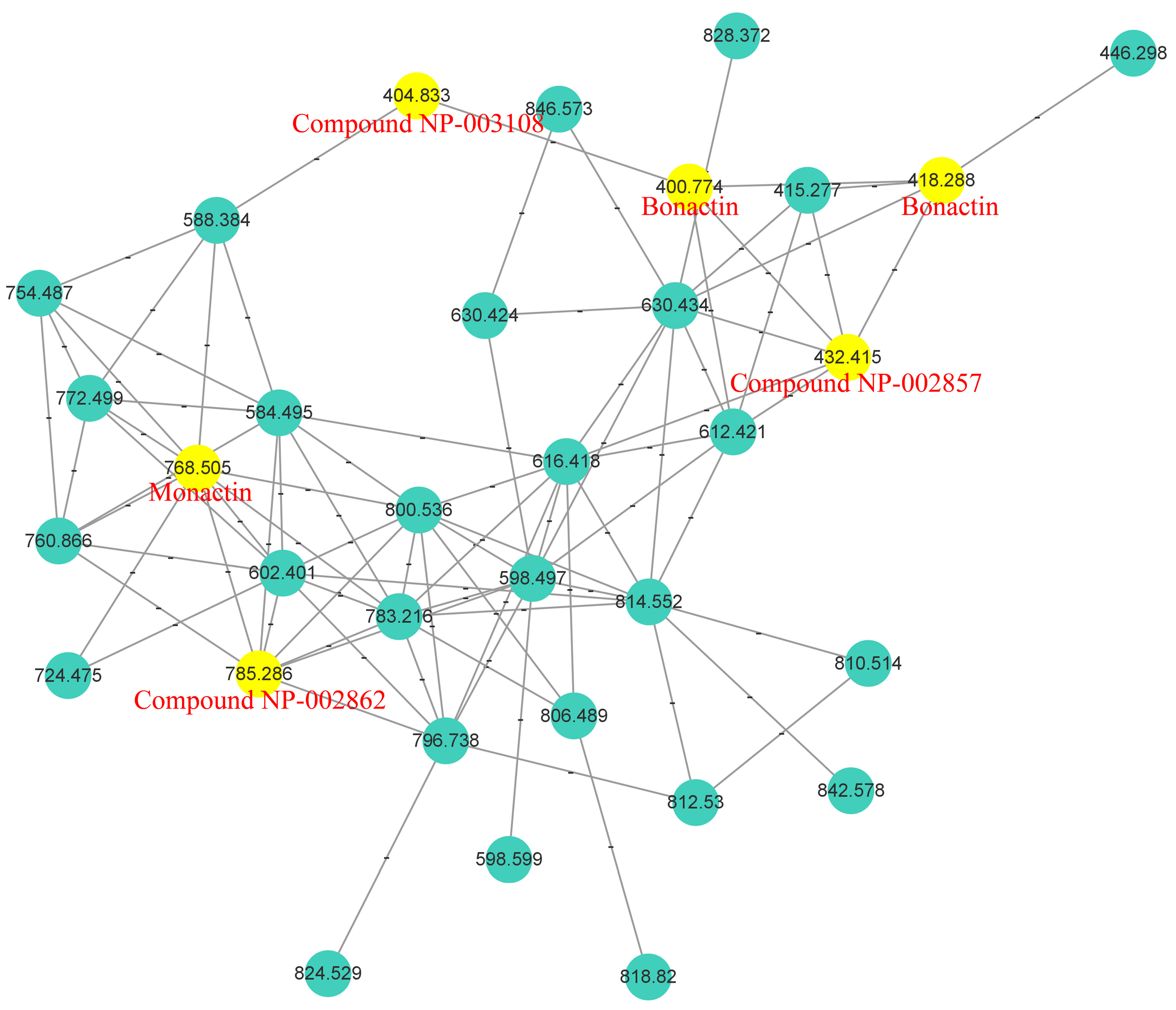
2.1. Analysis of Molecular Networking
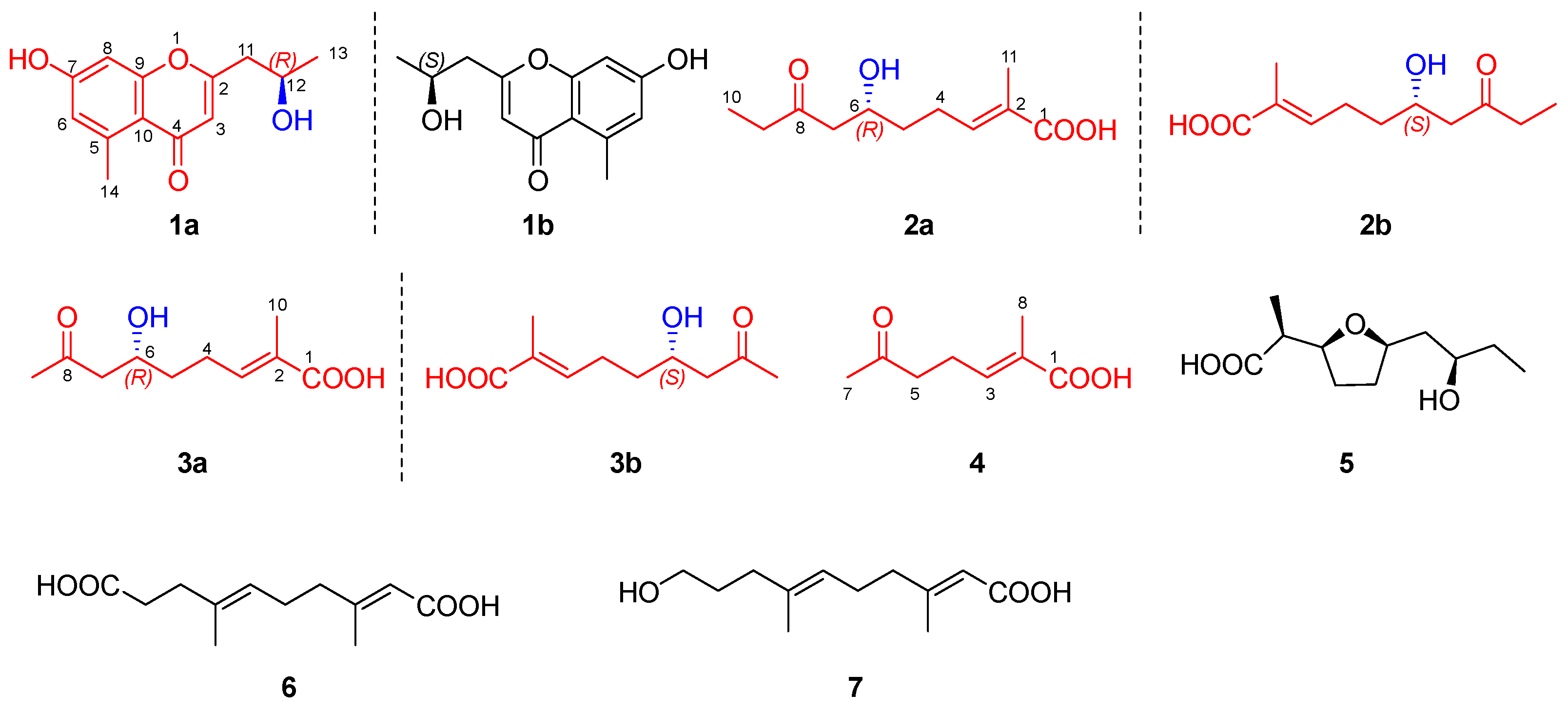
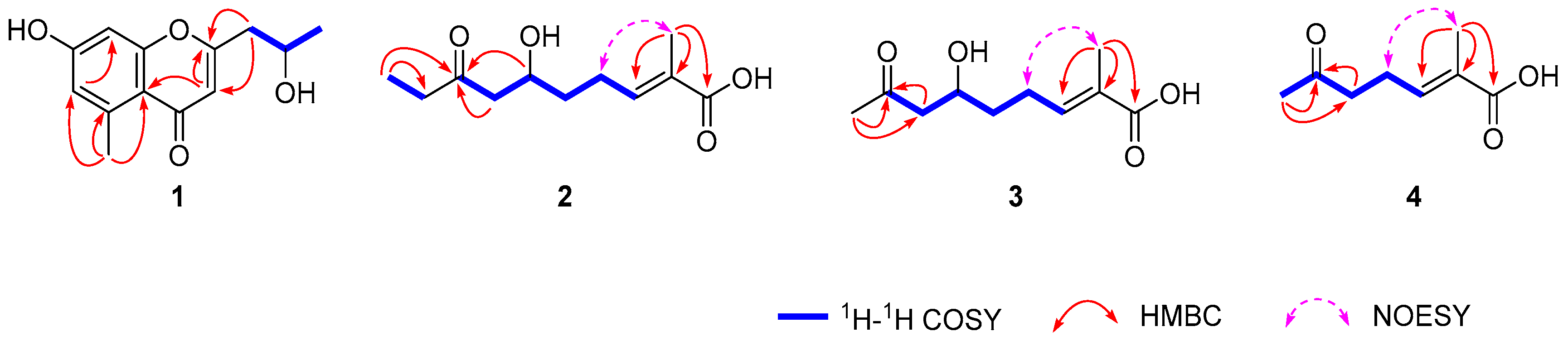
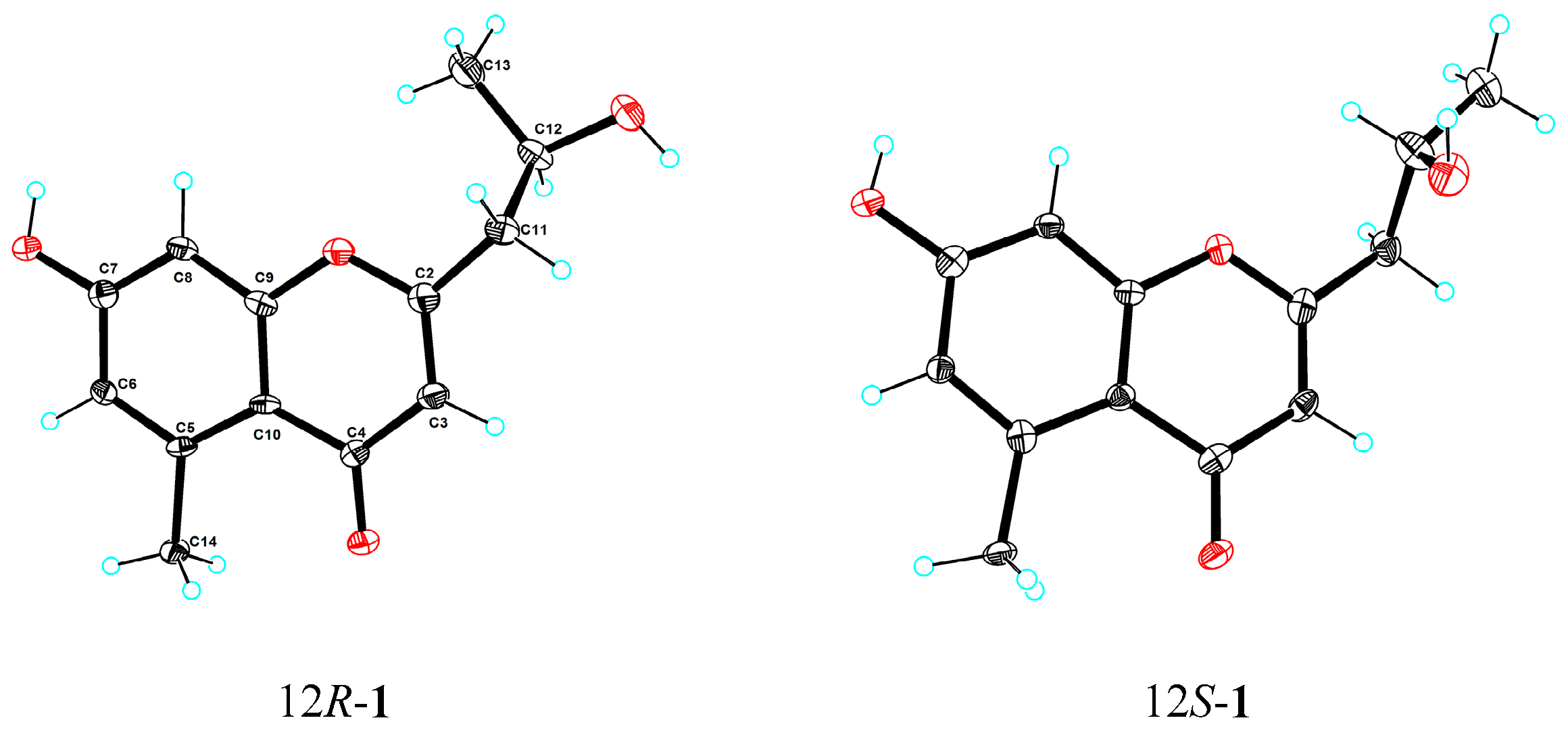
2.2. Structural Elucidation
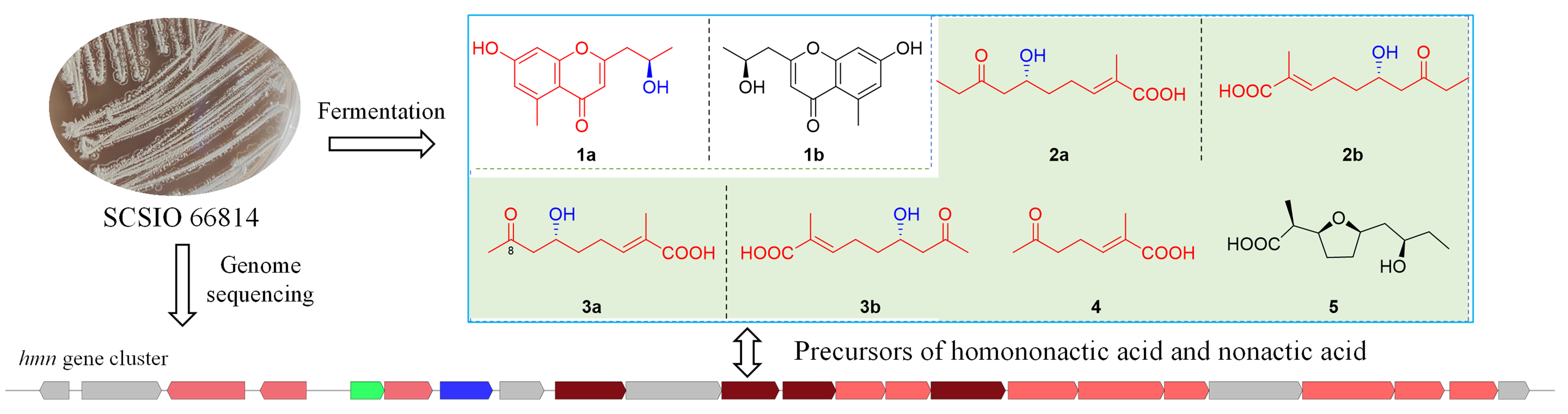
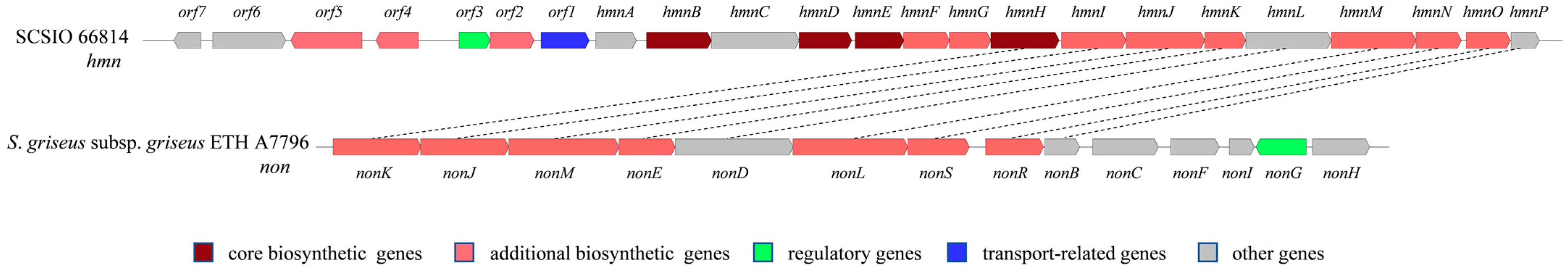
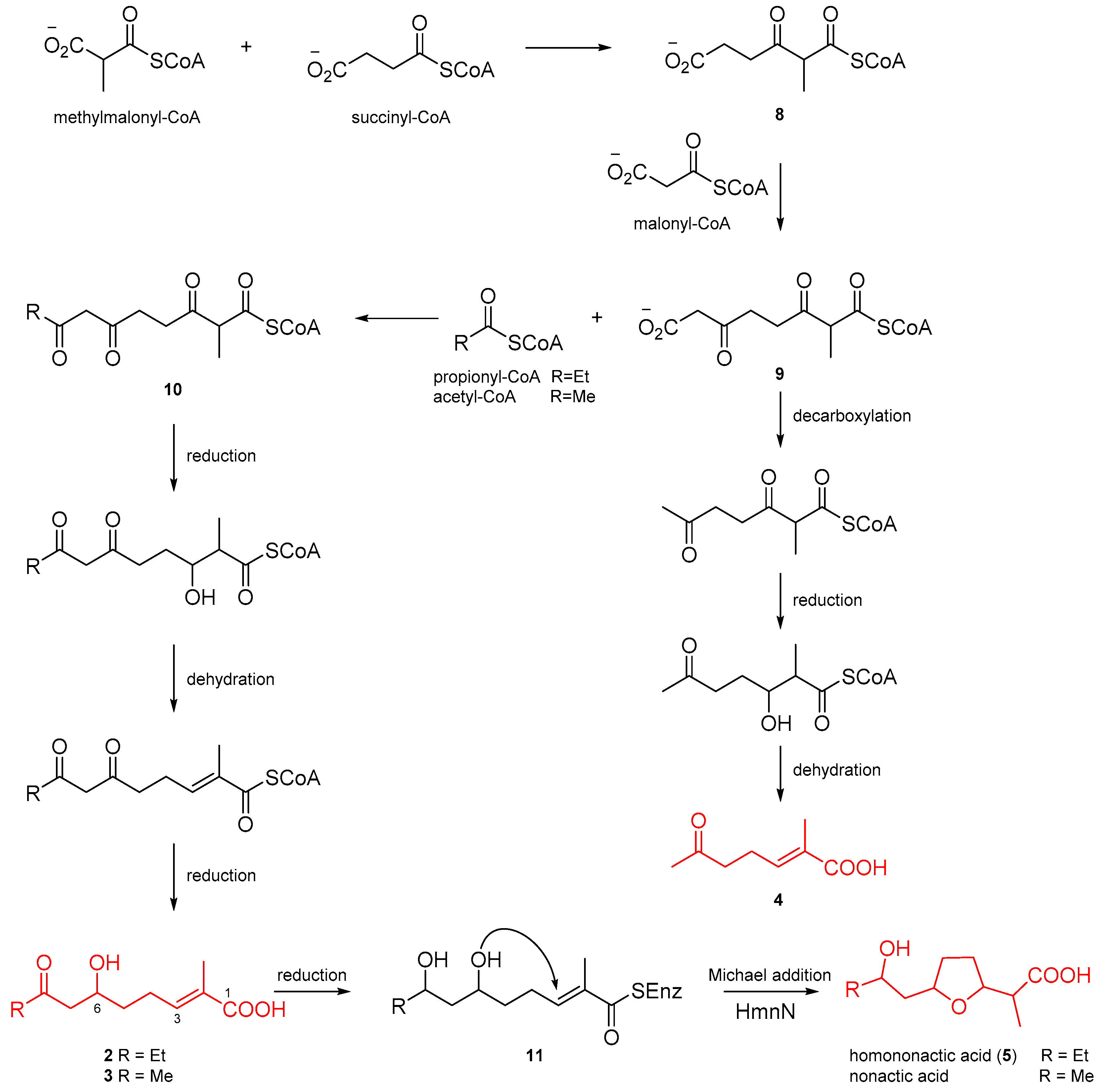
2.3. Putative Biosynthetic Pathway for Homononactic Acid and Nonactic Acid
2.4. Biological Activities
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Microorganism and Growth Conditions
3.3. Bioinformatic Analysis
3.4. Extraction and Isolation
3.5. Antibacterial Assay
3.6. DPPH Free Radical Scavenging Assay
3.7. LC-MS/MS Assay and Molecular Networking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Keller-Schierlein, W.; Gerlach, H. Macrotetrolides. Fortschr. Chem. Org. Naturst. 1968, 26, 161–189. [Google Scholar] [PubMed]
- Borrel, M.N.; Pereira, E.; Fiallo, M.; Garnier-Suillerot, A. Mobile ionophores are a novel class of P-glycoprotein inhibitors. Eur. J. Biochem. 1994, 223, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Rong, J.; Nelson, M.E.; Kusche, B.; Priestley, N.D. Nonactin biosynthesis: Unexpected patterns of label incorporation from 4, 6-dioxoheptanoate show evidence of a degradation pathway for levulinate through propionate in Streptomyces griseus. J. Nat. Prod. 2010, 73, 2009–2012. [Google Scholar] [CrossRef] [PubMed]
- Walczak, R.J.; Woo, A.J.; Strohl, W.R.; Priestley, N.D. Nonactin biosynthesis: The potential nonactin biosynthesis gene cluster contains type II polyketide synthase-like genes. FEMS Microbiol. Lett. 2000, 183, 171–175. [Google Scholar] [CrossRef] [PubMed]
- Spavold, Z.M.; Robinson, J.A. Nonactin biosynthesis: On the role of (6R, 8R)- and (6S, 8S)-2-methyl-6, 8-dihydroxynon-2E-enoic acids in the formation of nonactic acid. J. Chem. Soc. Chem. Commun. 1988, 1, 4–6. [Google Scholar] [CrossRef]
- Kashiwada, Y.; Nonaka, G.I.; Nishioka, I. Studies on Rhubarb (Rhei Rhizoma). V. Isolation and characterization of chromone and chromanone derivatives. Chem. Pharm. Bull. 1984, 32, 3493–3500. [Google Scholar] [CrossRef]
- Chen, X.; Cao, Y.G.; Ren, Y.J.; Liu, Y.L.; Fan, X.L.; He, C.; Li, X.D.; Ma, X.Y.; Zheng, X.K.; Feng, W.S. Ionones and lignans from the fresh roots of Rehmannia glutinosa. Phytochemistry 2022, 203, 113423. [Google Scholar] [CrossRef] [PubMed]
- Banwell, M.G.; Jury, J.C. Stereoselective syntheses of the methyl esters of (E)-and (Z)-2-methyl-6-oxohept-2-enoic acid. Org. Prep. Proced. Int. 2004, 36, 87–91. [Google Scholar] [CrossRef]
- Yokoyama, Y.; Arai, M.A.; Hara, Y.; Ishibashi, M. Nonactic acid derivatives isolated from Streptomyces werraensis IFM12104 in a screening program for BMI1 promoter inhibitory activity. Nat. Prod. Commun. 2019, 14, 1934578X19866583. [Google Scholar] [CrossRef]
- Meinwald, J.; Chalmers, A.M.; Pliske, T.E.; Eisner, T. Identification and synthesis of trans, trans-3, 7-dimethyl-2, 6-decadien-1, 10-dioic acid, a component of the pheromonal secretion of the male monarch butterfly. J. Chem. Soc. Chem. Commun. 1969, 86–87. [Google Scholar] [CrossRef]
- Meinwald, J.; Chalmers, A.M.; Pliske, T.E.; Eisner, T. Pheromones. III. Identification of trans, trans-10-hydroxy-3, 7-dimethyl-2, 6-decadienoic acid as a major component in “hairpencil” secretion of the male monarch butterfly. Tetrahedron Lett. 1968, 9, 4893–4896. [Google Scholar] [CrossRef]
- Zhou, S.Y.; Zou, Y.L.; Wang, G.W.; Liao, Z.H.; Chen, M. Two new compounds from a marine-derived Streptomyces sp. J. Asian Nat. Prod. Res. 2017, 19, 1172–1176. [Google Scholar] [CrossRef] [PubMed]
- Smith, W.C.; Xiang, L.; Shen, B. Genetic localization and molecular characterization of the nonS gene required for macrotetrolide biosynthesis in Streptomyces griseus DSM40695. Antimicrob. Agents Chemother. 2000, 44, 1809–1817. [Google Scholar] [CrossRef] [PubMed]
- Woo, A.J.; Strohl, W.R.; Priestley, N.D. Nonactin biosynthesis: The product of nonS catalyzes the formation of the furan ring of nonactic acid. Antimicrob. Agents Chemother. 1999, 43, 1662–1668. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.; Li, Y.; Tian, X.; Xiao, Z.; Li, R.; Zhang, S.; Yin, H. Investigation on metabolites in structure and biosynthesis from the deep-sea sediment-derived actinomycete Janibacter sp. SCSIO 52865. Molecules 2023, 28, 2133. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.; Li, Y.; Tian, X.; Chen, M.; Xiao, Z.; Chen, R.; Yin, H.; Zhang, S. Investigation on metabolites in structural diversity from the deep-sea sediment-derived bacterium Agrococcus sp. SCSIO 52902 and their biosynthesis. Mar. Drugs 2022, 20, 431. [Google Scholar] [CrossRef] [PubMed]
- Tian, S.Z.; Pu, X.; Luo, G.; Zhao, L.X.; Xu, L.H.; Li, W.J.; Luo, Y. Isolation and characterization of new p-terphenyls with antifungal, antibacterial, and antioxidant activities from halophilic actinomycete Nocardiopsis gilva YIM 90087. J. Agric. Food Chem. 2013, 61, 3006–3012. [Google Scholar] [CrossRef] [PubMed]
- Alfattani, A.; Marcourt, L.; Hofstetter, V.; Queiroz, E.F.; Leoni, S.; Allard, P.M.; Gindro, K.; Stien, D.; Perron, K.; Wolfender, J.L. Combination of pseudo-LC-NMR and HRMS/MS-based molecular networking for the rapid identification of antimicrobial metabolites from Fusarium petroliphilum. Front. Mol. Biosci. 2021, 8, 725691. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, R.W.; Talmage, S.C.; Miller, S.A.; Sarris, K.E.; Davidson, B.S.; Goldberg, A. Isolation and structure determination of an antimicrobial ester from a marine sediment-derived bacterium. J. Nat. Prod. 2003, 66, 1291–1293. [Google Scholar] [CrossRef] [PubMed]
- Řezanka, T.; Spížek, J.; Přikrylová, V.; Prell, A.; Dembitsky, V.M. Five new derivatives of nonactic and homo-nonactic acids from Streptomyces globisporus. Tetrahedron 2004, 60, 4781–4787. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 a | 2 a | ||||
|---|---|---|---|---|---|
| Position | δH | δC | Position | δH | δC |
| 2 | 167.1, s | 1 | 172.4, s | ||
| 3 | 6.06 (s) | 112.5, d | 2 | 130.0, s | |
| 4 | 182.0, s | 3 | 6.75 (tq, 7.6, 1.6) | 142.5, d | |
| 5 | 143.7, s | 4 | 2.27 (m), 2.32 (m) | 25.9, t | |
| 6 | 6.63 (d, 2.4) | 118.2, d | 5 | 1.56 (m) | 37.2, t |
| 7 | 163.5, s | 6 | 4.04 (tt, 8.4, 4.4) | 68.5, d | |
| 8 | 6.65 (d, 2.4) | 101.8, d | 7 | 2.58 (m) | 50.7, t |
| 9 | 161.6, s | 8 | 212.9, s | ||
| 10 | 115.8, s | 9 | 2.51 (m) | 37.6, t | |
| 11 | 2.65 (dd, 14.4, 7.9), 2.71 (dd, 14.4, 4.9) | 44.3, t | 10 | 1.01 (t, 7.3) | 7.8, q |
| 12 | 4.18 (dqd, 7.9, 6.2, 4.9) | 66.4, d | 11 | 1.82 (br s) | 12.6, q |
| 13 | 1.27 (d, 6.2) | 23.2, q | |||
| 14 | 2.72 (s) | 23.5, q | |||
| 3 a | 4 b | ||||
|---|---|---|---|---|---|
| Position | δH | δC | Position | δH | δC |
| 1 | 173.0, s | 1 | 171.6, s | ||
| 2 | 130.5, s | 2 | 129.8, s | ||
| 3 | 6.72 (tq,7.5, 1.5) | 142.0, d | 3 | 6.74 (tq, 7.4, 1.5) | 142.2, d |
| 4 | 2.27 (m), 2.31 (m) | 25.8, t | 4 | 2.45 (m) | 23.8, t |
| 5, | 1.56 (m) | 37.2, t | 5 | 2.68 (t, 7.2) | 42.7, t |
| 6 | 4.04 (m) | 68.3, d | 6 | 210.3, s | |
| 7 | 2.60 (d, 5.9) | 51.8, t | 7 | 2.18 (s) | 29.8, q |
| 8 | 210.6, s | 8 | 1.86 (br s) | 12.5, q | |
| 9 | 2.17 (s) | 30.7, q | |||
| 10 | 1.82 (br s) | 12.7, q | |||
| Compounds a | Scavenging Rate% | Compounds a | Scavenging Rate% |
|---|---|---|---|
| (+)-1 | 34.42% | 4 | 6.54% |
| (−)-1 | 16.04% | 5 | 3.04% |
| (+)-2 | 7.01% | 6 | 6.11% |
| (−)-2 | 16.20% | 7 | 8.87% |
| (+)-3 | 10.51% | Ascorbic acid b | 96.89% |
| (−)-3 | 15.58% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ding, W.; Li, Y.; Li, X.; Yin, J.; Shi, S.; Tian, X.; Zhang, S.; Yin, H. Discovery of a Novel Chromone Enantiomer and the Precursors of Nonactic Acid from the Coral-Reef-Derived Streptomyces sp. SCSIO 66814. Mar. Drugs 2024, 22, 181. https://doi.org/10.3390/md22040181
Ding W, Li Y, Li X, Yin J, Shi S, Tian X, Zhang S, Yin H. Discovery of a Novel Chromone Enantiomer and the Precursors of Nonactic Acid from the Coral-Reef-Derived Streptomyces sp. SCSIO 66814. Marine Drugs. 2024; 22(4):181. https://doi.org/10.3390/md22040181
Chicago/Turabian StyleDing, Wenping, Yanqun Li, Xingyu Li, Jiajia Yin, Songbiao Shi, Xinpeng Tian, Si Zhang, and Hao Yin. 2024. "Discovery of a Novel Chromone Enantiomer and the Precursors of Nonactic Acid from the Coral-Reef-Derived Streptomyces sp. SCSIO 66814" Marine Drugs 22, no. 4: 181. https://doi.org/10.3390/md22040181
APA StyleDing, W., Li, Y., Li, X., Yin, J., Shi, S., Tian, X., Zhang, S., & Yin, H. (2024). Discovery of a Novel Chromone Enantiomer and the Precursors of Nonactic Acid from the Coral-Reef-Derived Streptomyces sp. SCSIO 66814. Marine Drugs, 22(4), 181. https://doi.org/10.3390/md22040181