Marine Pyrrolocarbazoles and Analogues: Synthesis and Kinase Inhibition

Abstract
:
1. Introduction
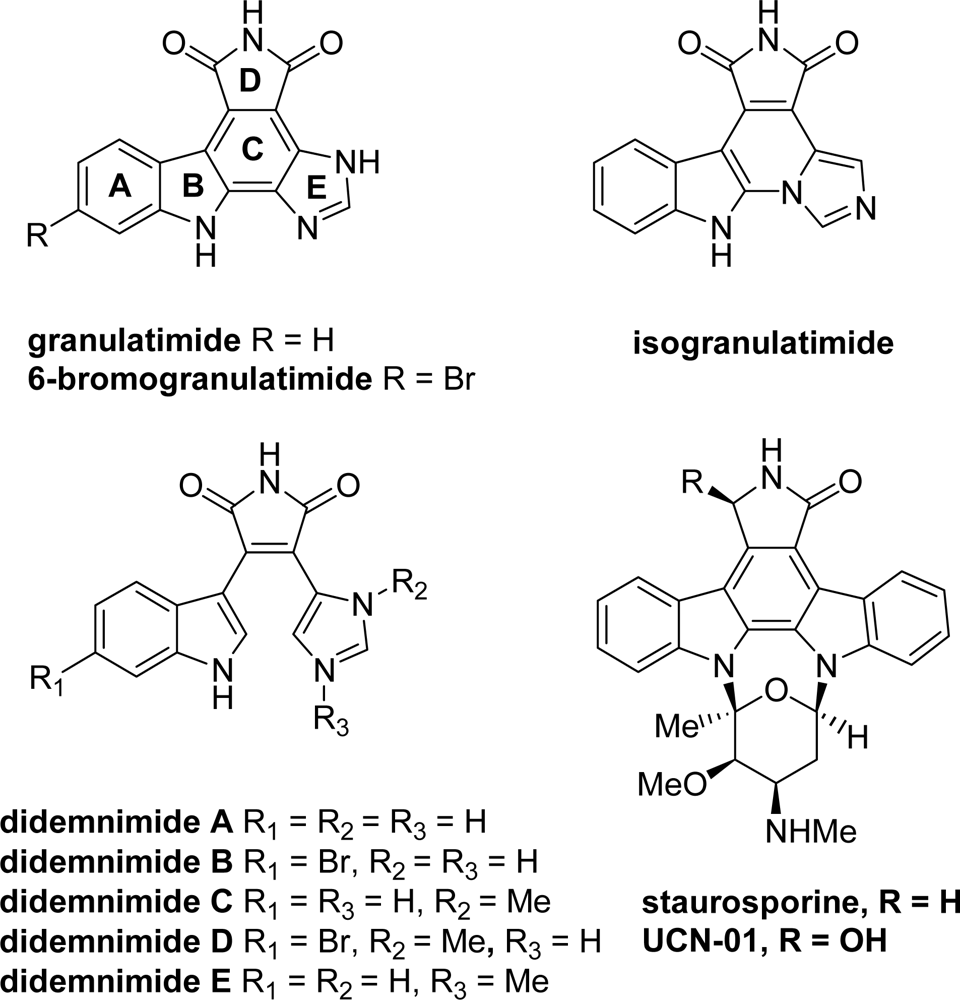
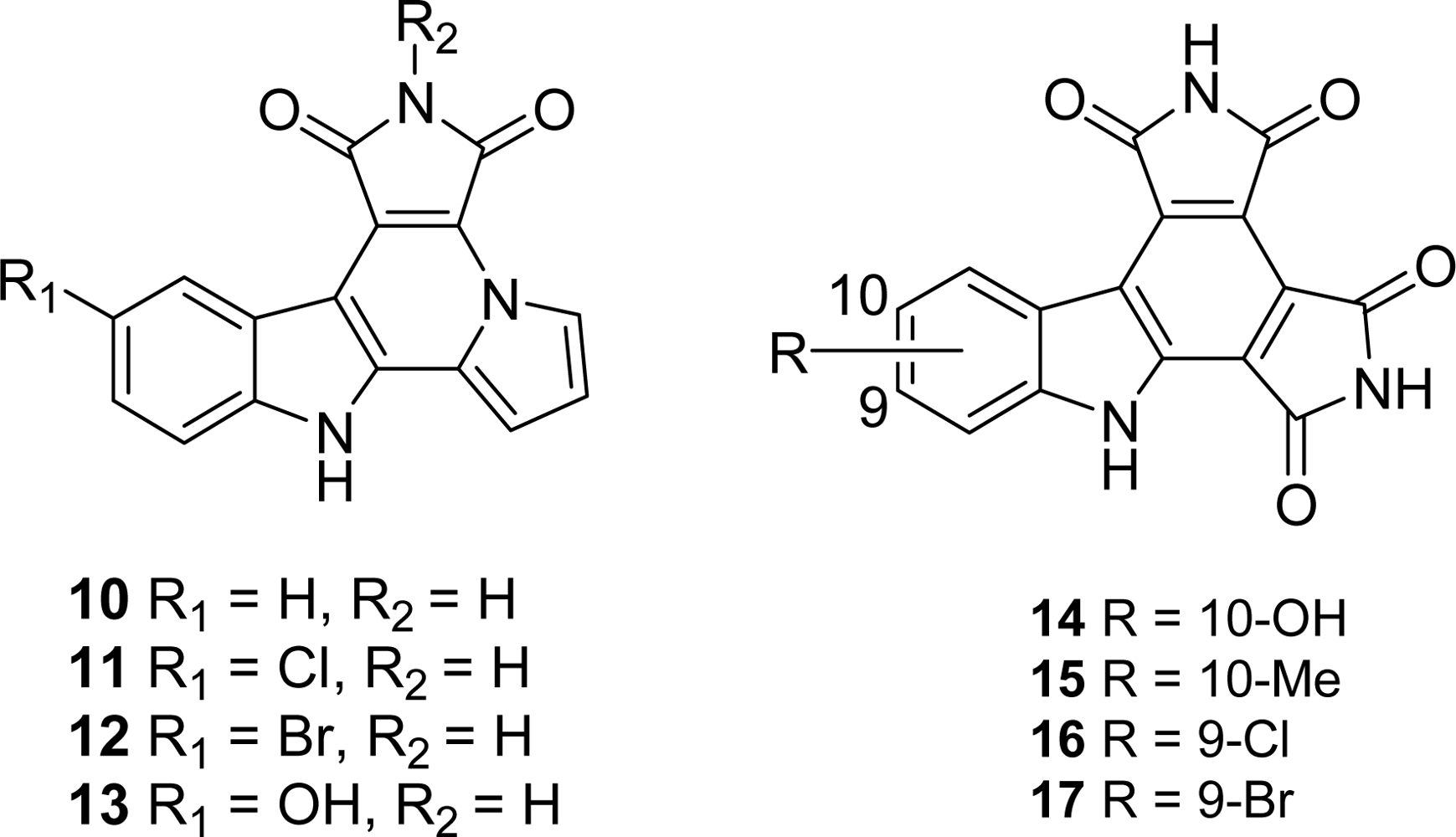
2. Marine-Related Metabolites and Positional Analogues
3. Modification of the Indole Subunit (Units A and B)
4. Replacement of the Imidazole Ring (Unit E)
4.1. By a pyrrole or by a second maleimide moiety
4.2. By a non aromatic five or six-membered-ring moiety
4.3. By another aryl or heteroaryl ring moiety
4.4. By a second indole ring
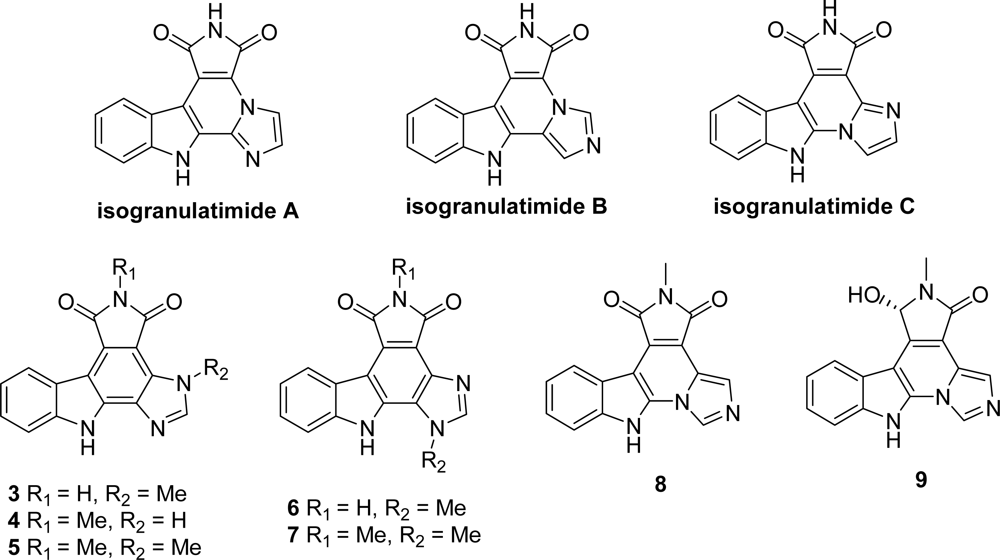
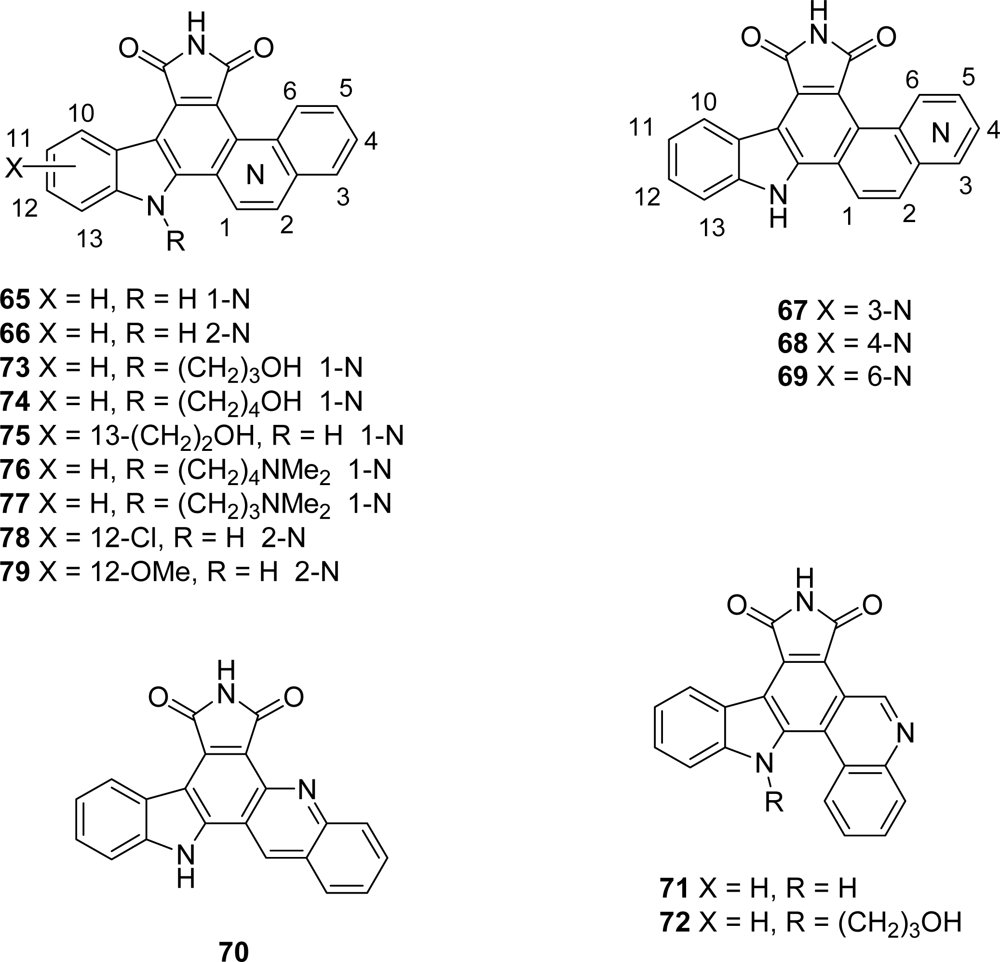
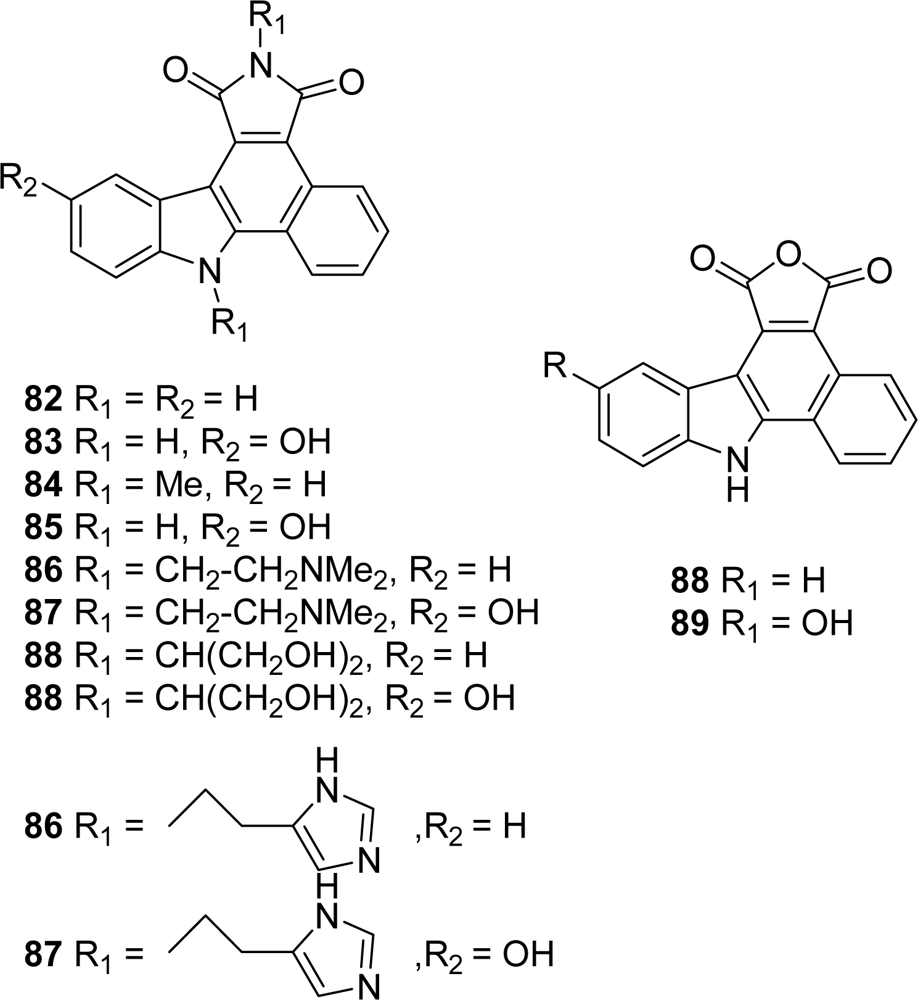
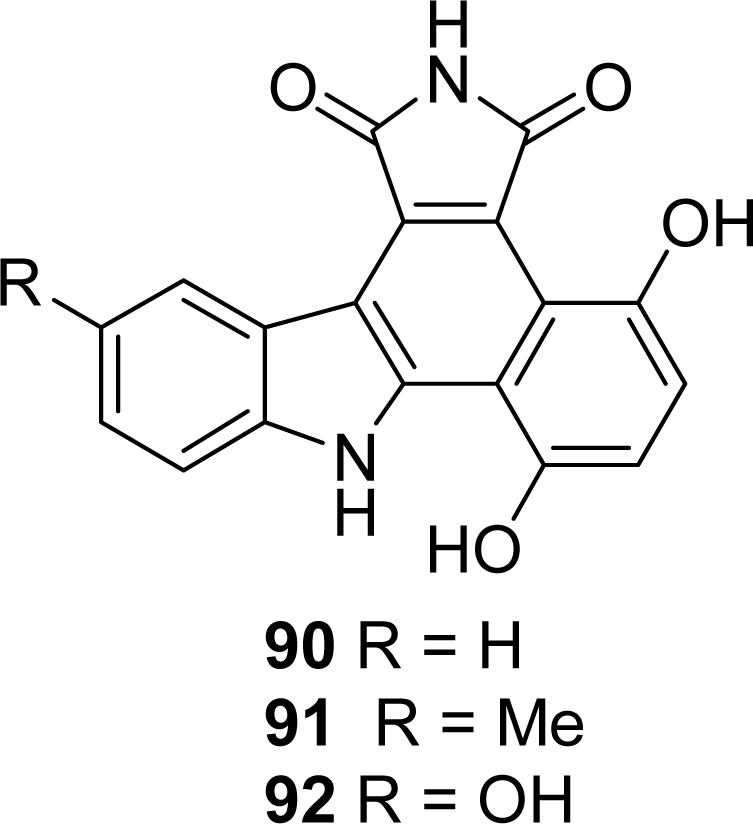
5. Granulatimide Analogues in Which the D and E Rings Are Modified or Absent
6. Conclusions
References
- Manning, G; Whyte, DB; Martinez, R; Hunter; Sudarsanam, TS. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [Google Scholar]
- Garcia-Echeverria, C; Traxler, P; Evans, DB. ATP site-directed competitive and irreversible inhibitors of protein kinases. Med Res Rev 2000, 20, 28–57. [Google Scholar]
- Sielecki, TM; Boylan, JF; Benfield, PA; Trainor, GL. Cyclin-dependent kinase inhibitors: useful targets in cell cycle regulation. J Med Chem 2000, 43, 1–18. [Google Scholar]
- Traxler, P; Bold, G; Buchdunger, E; Caravatti, G; Furet, P; Manley, P; O’Reilly, T; Wood, J; Zimmermann, J. Tyrosine kinase inhibitors: from rational design to clinical trials. Med Res Rev 2001, 21, 499–512. [Google Scholar]
- Toogood, PL. Cyclin-dependent kinase inhibitors for treating cancer. Chem Rev 2001, 101, 2541–2572. [Google Scholar]
- Bridges, AJ. Chemical inhibitors of protein kinases. Chem Rev 2001, 101, 2541–2572. [Google Scholar]
- Scapin, G. Structural biology in drug design: selective protein kinase inhibitors. Drug Discov 2001, 7, 601–611. [Google Scholar]
- Sridhar, R; Hanson-Painton, O; Cooper, DR. Protein kinase as therapeutic targets. Pharm Res 2000, 17, 1345–135. [Google Scholar]
- Zhang, J; Yang, PL; Gray, NS. Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer 2009, 9, 28–39. [Google Scholar]
- Berlinck, RGS; Britton, R; Piers, E; Lim, L; Roberge, M; Moreira da Rocha, R; Andersen, RJ. Granulatimide and isogranulatimide, aromatic alkaloids with G2 checkpoint inhibition activity isolated from the brazilian ascidian Didemnun granulatum: structure elucidation and synthesis. J Org Chem 1998, 63, 9850–9856. [Google Scholar]
- Jiang, X; Zhao, B; Britton, R; Lim, LY; Leong, D; Sanghera, JS; Zhou, BBS; Piers, E; Andersen, RJ; Roberge, M. Inhibition of Chk1 by the G2 DNA damage checkpoint inhibitor isogranulatimide. Mol Cancer Ther 2004, 3, 1221–1227. [Google Scholar]
- Zhao, S; Weng, Y-C; Yuan, S-SF; Lin, Y-T; Hsu, H-C; Lin, S-CJ; Gerbino, E; Song, M-H; Zdzienicka, P. Functional link between ataxia-telangiectasia and nijmegen breakage syndrome gene products. Nature 2000, 405, 473–476. [Google Scholar]
- Curman, D; Cinel, B; Williams, DE; Rundle, N; Block, WD; Goodarzi, AA; Hutchins, JR; Clarke, PR; Zhou, B-B; Lees-Miller, SP; Andersen, RJ; Roberge, M. Inhibition of the G2 DNA damage checkpoint and of protein kinases Chk1 and Chk2 by the marine sponge alkaloid debromohymenialdisine. J Biol Chem 2001, 276, 17914–17919. [Google Scholar]
- Zhao, B; Bower, MJ; McDevitt, PJ; Zhao, H; Davies, ST; Johanson, KO; Green, SM; Concha, NO; Zhou, BBS. Structural basis for Chk1 inhibition by UCN-01. J Biol Chem 2002, 277, 46609–46615. [Google Scholar]
- Hénon, H; Conchon, E; Hugon, B; Messaoudi, S; Golsteyn, RM. Prudhomme. Pyrrolocarbazoles as checkpoint 1 kinase inhibitors. Anti-Cancer Agents Med Chem 2008, 8, 577–597. [Google Scholar]
- Roberge, M; Berlinck, RGS; Xu, L; Anderson, HJ; Lim, LY; Curman, D; Stringer, CM; Friend, SH; Davies, P; Vincent, I; Haggarty, SJ; Kelly, MT; Britton, R; Piers, E; Andersen, RJ. High-throughput assay for G2 checkpoint inhibitors and identification of the structurally novel compound isogranulatimide. J Cancer Res 1998, 58, 5701–5706. [Google Scholar]
- Vervoort, HC; Fenical, W; Keifer, PA. A cyclised didemnimide alkaloid from the Caribbean ascidian Didemnum conchyliatum. J Nat Prod 1999, 62, 389–391. [Google Scholar]
- Vervoort, HC; Richards-Gross, SE; Fenical, W; Didemnimides, A-D. Novel predator-deterrent alkaloids from the carribean mangrove ascidian Didemnum conchyliatum. J Org Chem 1997, 62, 1486–1490. [Google Scholar]
- Britton, R; de Oliveiras, JHHL; Andersen, RJ; Berlinck, RGS. Granulatimide and 6-bromogranulatimide, minor alkaloids of the brazilian ascidian. Didemnum granulatum J Nat Prod 2001, 64, 254–255. [Google Scholar]
- Seleghim, MHR; De Lira, SP; Campana, PT; Berlink, RGS; Custodio, MR. Localization of granulatimide alkaloids in the tissues of the ascidian. Didemnum granulatum Ma Biol 2007, 150, 967–975. [Google Scholar]
- Piers, E; Britton, R; Andersen, RJ. Improved synthesis of isogranulatimide, a G2 checkpoint inhibitor synthesis of didemnimide C, isodidemnimide A, neodidemnimide A, 17-methylgranulatimide, and isogranulatimide A-C. J Org Chem 2000, 65, 530–535. [Google Scholar]
- Yoshida, T; Nishiyachi, M; Nakashima, N; Murase, M; Kotani, E. New synthetic route to granulatimide and its structural analogues. Chem Pharm Bull 2002, 50, 872–876. [Google Scholar]
- Yoshida, T; Nishiyachi, M; Nakashima, N; Murase, M; Kotani, E. Synthesis of granulatimide positional analogues. Chem Pharm Bull 2003, 51, 209–214. [Google Scholar]
- Hugon, B; Pfeiffer, B; Renard, P; Prudhomme, M. Synthesis of isogranulatimides A and B analogues possessing a 7-azaindole unit instead of an indole moiety. Tetrahedron Lett 2003, 44, 4607–4611. [Google Scholar]
- Hugon, B; Anizon, F; Bailly, C; Golsteyn, RM; Pierré, A; Léonce, S; Hickman, J; Pfeiffer, B; Prudhomme, M. Synthesis and biological activities of isogranulatimide analogues. Bioorg Med Chem 2007, 15, 5965–5980. [Google Scholar]
- Hénon, H; Messaoudi, S; Anizon, F; Aboab, B; Kucharczyk, N; Léonce, S; Golsteyn, RM; Pfeiffer, B; Prudhomme, M. Bis-imide granulatimide analogues as potent checkpoint 1 kinase inhibitors. Eur J Pharm 2007, 554, 106–112. [Google Scholar]
- Conchon, E; Anizon, F; Aboab, B; Prudhomme, M. Synthesis and biological activities of new checkpoint kinase 1 inhibitors structurally related to granulatimide. J Med Chem 2007, 50, 4669–4680. [Google Scholar]
- Conchon, E; Anizon, F; Aboab, B; Golsteyn, RM; Léonce, S; Pfeiffer, B; Prudhomme, M. Synthesis, checkpoint kinase 1 inhibitory properties and in vitro antiproliferative activities of new pyrrolocarbazoles. Bioorg Med Chem 2008, 16, 4419–4430. [Google Scholar]
- Tao, M; Park, CH; Bihovsky, R; Wells, GJ; Husten, J; Ator, MA; Hudkins, RL. Synthesis and structure-activity relationships of novel poly(ADP-ribose) polymerase-1 inhibitors. Bioorg Med Chem Lett 2006, 16, 938–942. [Google Scholar]
- Wells, GJ; Bihovsky, R; Hudkins, RL; Ator, MA; Husten, J. Synthesis and structure-activity relationships of novel pyrrolocarbazole lactam analogs as potent and cell-permeable inhibitors of poly(ADP-ribose)polymerase-1 (PARP-1). Bioorg Med Chem Lett 2006, 16, 1151–1155. [Google Scholar]
- Sanchez-Martinez, C; Shih, C; Faul, MM; Zhu, G; Paal, M; Somoza, C; Li, T; Kumrich, CA; Winneroski, LL; Xun, Z; Brooks, HB; Patel, BKR; Schultz, RM; DeHan, TB; Spencer, CD; Watkins, SA; Considine, E; Dempsey, JA; Ogg, CA; Campbell, RM; Anderson, BA; Wagner, J. Aryl[a]pyrrolo[3,4-c]carbazoles as selective Cyclyn D1-CDK4 inhibitors. Bioorg Med Chem Lett 2003, 13, 3835–3839. [Google Scholar]
- Faul, MM; Winneroski, LL; Kumrich, CA. A new efficient method for the synthesis of bisindolylmaleimides. J Org Chem 1998, 63, 6053–6058. [Google Scholar]
- Faul, MM; Winneroski, LL; Krumrich, CA. A new one step synthesis of maleimides by condensation of glyoxylate esters with acetamides. Tetrahedron Lett 1999, 40, 1109–1112. [Google Scholar]
- Zhu, G; Conner, S; Zhou, X; Shih, C; Brooks, HB; Considine, E; Dempsey, JA; Ogg, C; Patel, B; Schultz, RM; Spencer, CD; Teicher, B; Watkins, SA. Synthesis of quinolinyl/isoquinolinyl[a]pyrrolo[3,4-c]carbazoles as cyclin D1/CDK4 inhibitors. Bioorg Med Chem Lett 2003, 13, 1231–1235. [Google Scholar]
- Routier, S; Peixoto, P; Mérour, JY; Coudert, G; Dias, N; Bailly, C; Pierré, A; Léonce, S; Caignard, DH. Synthesis and biological evaluation of novel naphtocarbazoles as potential anticancer agents. J Med Chem 2005, 48, 1401–1413. [Google Scholar]
- Routier, S; Mérour, J-Y; Dias, N; Lansiaux, A; Bailly, C; Lozach, O; Meijer, L. Synthesis and biological evaluation of novel phenylcarbazoles as potential anticancer agents. J Med Chem 2006, 49, 789–799. [Google Scholar]
- Bregman, H; Williams, DS; Atila, GE; Carroll, PJ; Meggers, E. An organometallic inhibitor for glycogen synthase kinase 3. J Am Chem Soc 2004, 126, 13594–13595. [Google Scholar]
- Woodgett, JR. Molecular cloning and expression of glycogen synthase kinase-3/factor A. EMBO J 1990, 25, 923–929. [Google Scholar]
- Williams, DS; Atilla, GE; Bregman, H; Arzoumanian, A; Klein, PS; Meggers, E. Switching on a signalling pathway with an organoruthenium complex. Angew Chem Int Ed 2005, 44, 1984–1987. [Google Scholar]
- Bregman, H; Williams, DS; Meggers, W. Pyrido[2,3-a]pyrrolo[3,4-c]carbazole-5,7(6H)-diones: synthesis, cyclometalation and protein kinase inhibition. Synthesis 2005, 9, 1521–1527. [Google Scholar]
- Atilla-Gokcumen, GE; Williams, DS; Bregman, H; Pagano, N; Meggers, E. Organometallic compounds with biological activity: a very selective and highly potent cellular inhibitor for glycogen synthase kinase 3. ChemBioChem 2006, 7, 1443–1450. [Google Scholar]
- Bregman, H; Meggers, E. Ruthenium half-sandwich complexes as protein kinase inhibitors: an N-succinimidyl ester for rapid derivatizations of the cyclopentadienyl moiety. Org Lett 2006, 24, 5465–5468. [Google Scholar]
- Bregman, H; Carroll, PJ; Meggers, E. Rapid access to unexplored chemical space by ligand scanning around a ruthenium center: discovery of potent and selective protein kinase inhibitors. J Am Chem Soc 2006, 128, 877–884. [Google Scholar]
- Atilla-Gokcumen, GE; Pagano, N; Streu, C; Maksimoska, J; Filippakopoulos, P; Knapp, S; Meggers, E. Extremely tigh binding of a ruthenium complex to glycogen synthase kinase 3. ChemBioChem 2008, 9, 2933–2936. [Google Scholar]
- Anand, R; Maksimoska, J; Pagano, N; Wong, EY; Gimotty, PA; Diamond, SL; Meggers, E; Marmorstein, R. Toward the development of a potent and selective organoruthenium mammalian sterile 20 kinase inhibitor. J Med Chem 2009, 52, 1602–1611. [Google Scholar]
- Nettleton, DE; Doyle, TW; Kirshnan, B; Matsumoto, GK; Clardy, J. Isolation and structure of rebeccamycin a new antitumor antibiotic from nocardia aerocoligenes. Tetrahedron Lett 1985, 25, 4011–4014. [Google Scholar]
- Omura, S; Iwai, Y; Hirano, A; Nakagawa, A; Awaya, J; Tsuchiva, H; Takahashi, Y; Masuma, R. A new alkaloid AM-2282 of Streptomyces origin: Taxonomy, fermentation and preliminary characterization. J Antibiot 1977, 30, 275–282. [Google Scholar]
- Omura, S; Sasaki, Y; Iwai, Y; Takeshima, H. Staurosporine, a potentially important gift from a microorganism. J Antibiot 1995, 48, 535–548. [Google Scholar]
- Rodrigues Pereira, E; Belin, L; Sancelme, M; Prudhomme, M; Ollier, M; Rapp, D; Sevère, D; Riou, JF; Fabbro, D; Meyer, T. Structure-activity relationships in a series of substituted indolocarbazoles: topoisomerase I and protein kinase C inhibition and antitumoral and antimicrobial properties. J Med Chem 1996, 39, 4471–4477. [Google Scholar]
- Gray, N; Détivaud, L; Doerig, C; Meijer, L. ATP-Site directed inhibitors of cyclin-dependent kinases. Curr Med Chem 1999, 6, 859–875. [Google Scholar]
- Goeke, K; Hoehn, P; Ghisalba, O. Production of the staurosporine aglycon K-252C with a blocked mutant of the staurosporine producer strain Streptomyces longisporoflavus and by biotransformation of staurosporine with Streptomyces mediddicm ATCC 13279. J. Antibiot 1995, 48, 428–430. [Google Scholar]
- Rialet, V; Meijer, L. A new screening test for antimitotic compounds using the universal M phase-specific protein kinase, p34cdc2/cyclin Bcd13, affinity immobilized on p13suc1-coated microtitration plates. Anticancer Res 1991, 11, 1581. [Google Scholar]
- Al-Obeidi, FA; Lam, KS. Development of inhibitors for protein tyrosine kinases. Oncogene 2000, 19, 5690–5702. [Google Scholar]
- Prudhomme, M. Biological targets of antitumor indolocarbazoles bearing a sugar moiety. Curr Med Chem Anticancer Agents 2004, 4, 509–521. [Google Scholar]
- Prudhomme, M. Staurosporine and structurally related indolocarbazoles as antitumor agents. In Anticancer Agents from Natural Products; Cragg, GM, Gordon, M, Kingston, DGI, Newman, DJ, Eds.; CRC Press: Boca Raton, FL, USA, 2005; pp. 499–517. [Google Scholar]
- Hosoya, T; Yamamoto, Y; Uehara, Y; Hayashi, M; Komiyama, K; Ishibashi, M. New cytotoxic bisindole alkaloids with protein tyrosine kinase inhibitory activity from a myxomycete Lycogala epidendrum. Bioorg Med Chem Lett 2005, 15, 2776–2780. [Google Scholar]
- Engler, TA; Furnee, K; Malhotra, S; Sanchez-Martinez, C; Shih, C; Xie, W; Zhu, G; Zhou, X; Conner, S; Faul, MM; Sullivan, KA; Kolis, SP; Brooks, HB; Patel, B; Schultz, RM; DeHan, TB; Kirmani, K; Spencer, CD; Watkins, SA; Considine, EL; Dempsey, JA; Ogg, CA; Stamm, NB; Anderson, BD; Campbell, RM; Vasudevan, V; Lytle, ML. Novel, potent and selective cyclin D1/CDK4 inhibitors: indolo[6,7-a]pyrrolo[3,4-c]carbazoles. Bioorg Med Chem Lett 2003, 13, 2261–2267. [Google Scholar]
- Faul, MM; Engler, TA; Sullivan, KA; Grutsch, JL; Clayton, MT; Martinelli, MJ; Pawlak, JM; LeTourneau, M; Coffey, DS; Pedersen, SW; Kolis, SP; Furness, K; Malhotra, S; Al-awar, RS; Ray, JE. Synthetic approaches to indolo[6,7-α]pyrrolo[3,4-c]carbazoles: potent cyclin D1/CDK4 inhibitors. J Org Chem 2004, 69, 2967–2975. [Google Scholar]
- Zhu, G; Conner, SE; Zhou, X; Chan, HK; Shih, C; Engler, TA; Al-awar, RS; Brooks, HB; Watkins, SA; Spencer, CD; Schultz, RM; Dempsey, JA; Considine, EL; Patel, BR; Ogg, CA; Vasudevan, V; Lytle, ML. Synthesis of 1,7-annulated indoles and their applications in the studies of cyclin dependant kinase inhibitors. Bioorg Med Chem Lett 2004, 14, 3057–3061. [Google Scholar]
- Al-awar, RS; Ray, JE; Hecker, KA; Huang, J; Waid, PP; Shih, C; Brooks, HB; Spencer, CD; Watkins, SA; Patel, BR; Stamm, NB; Ogg, CA; Shultz, RM; Considine, EL; Faul, MM; Sullivan, KA; Kolis, SP; Grutsch, JL; Joseph, S. 1,7-Annulated indolocarbazoles as cyclin-dependent kinase inhibitors. Bioorg Med Chem Lett 2004, 14, 3217–3220. [Google Scholar]
- Moreau, P; Gaillard, N; Marminon, C; Anizon, F; Dias, N; Baldeyrou, B; Bailly, C; Pierré, A; Hickman, J; Pfeiffer, B; Renard, P; Prudhomme, M. Semi-synthesis, topoisomerase I and kinases inhibitory properties and antiproliferative activities of new rebeccamycin derivatives. Bioorg Med Chem 2003, 11, 4871–4879. [Google Scholar]
- Balasubramanian, BN; St Laurent, DR; Saulnier, MG; Long, BH; Bachand, C; Beaulieu, F; Clarke, W; Deshpande, M; Eummer, J; Fairchild, CR; Frennesson, DB; Kramer, R; Lee, FY; Mahler, M; Martel, A; Naidu, BN; Rose, WC; Russell, J; Ruediger, E; Solomon, C; Stoffan, KM; Wong, H; Zimmermann, K; Vyas, DM. Design and synthesis of a fluoroindolocarbazole series as selective topoisomerase I active agents. Discovery of water-soluble 3,9-difluoro-12,13H-benzo[b]-thienyl[2,3-a]pyrrolo[3,4-c]carbazole-5,7(6H)-dione (BMS-251873) with curative antitumor activity against xenograft tumor model. J Med Chem 2004, 47, 1609–1612. [Google Scholar]
- Saulnier, MG; Balasubramanian, BN; Long, BH; Frennesson, DB; Ruediger, E; Zimmermann, K; Eummer, JT; St Laurent, DR; Stoffan, KM; Naidu, BN; Mahler, M; Beaulieu, F; Bachand, C; Lee, FY; Fairchild, CR; Stadnick, LK; Rose, WC; Solomon, C; Wong, H; Martel, A; Wright, JJ; Kramer, R; Langley, DR; Vyas, DM. Discovery of a fluoroindolo[2,3-α]carbazole clinical candidate with broad spectrum antitumor activity in preclinical tumor models superior to the marketed oncology drug, CPT-11. J Med Chem 2005, 48, 2258–2261. [Google Scholar]
- Anizon, F; Moreau, P; Sancelme, M; Laine, W; Bailly, C; Prudhomme, M. Rebeccamycin analogues bearing amine substituents or other groups on the sugar moiety. Bioorg Med Chem 2003, 11, 3709–3722. [Google Scholar]
- Faul, MM; Sullivan, KA; Grutsch, JL; Winneroski, LL; Shih, C; Sanchez-Martinez, C; Cooper, JT. Synthesis of indolo[2,3-a]carbazole glycoside analogs of rebeccamycin: inhibitors of cyclin D1-CDK4. Tetrahedron Lett 2004, 45, 1095–1098. [Google Scholar]
- Zhang, G; Shen, J; Cheng, H; Fang, L; Luo, S; Muller, MT; Lee, GE; Wei, L; Du, Y; Sun, D; Wang, PG. Synthesis and biological activities of rebeccamycin analogues with uncommon sugars. J Med Chem 2005, 48, 2600–2611. [Google Scholar]
- Roy, S; Eastman, A; Gribble, GW. Synthesis of 7-keto-Gö6976 (ICP-103). Synth Commun 2005, 35, 595–601. [Google Scholar]
- Roy, S; Eastman, A; Gribble, GW. Synthesis of N-alkyl substituted bioactive indolocarbazoles related to Gö6979. Tetrahedron 2006, 62, 7838–7845. [Google Scholar]
- Faul, MM; Winneroski, LL; Kumrich, CA. A new efficient method for the synthesis of bisindolylmaleimides. J Org Chem 1998, 63, 6053–6058. [Google Scholar]
- Faul, MM; Winneroski, LL; Krumrich, CA. A new one step synthesis of maleimides by condensation of glyoxylate esters with acetamides. Tetrahedron Lett 1999, 40, 1109–1112. [Google Scholar]
- Roy, S; Eastman, A; Gribble, GW. Synthesis of bisindolylmaleimides related to GF109203x and their efficient conversion to the bioactive indolocarbazoles. Org Biomol Chem 2006, 4, 3228–3234. [Google Scholar]
- Routier, S; Ayerbe, N; Mérour, JY; Coudert, G; Bailly, C; Pierré, A; Pfeiffer, B; Caignard, DH; Renard, P. Synthesis and biological evaluation of 7-azaindolocarbazoles. Tetrahedron 2002, 58, 6621–6630. [Google Scholar]
- Popowycz, F; Routier, S; Joseph, B; Merour, J-Y. Synthesis and reactivity of 7-azaindole (1H-pyrrolo[2,3-b]pyridine). Tetrahedron 2007, 63, 1031–1064. [Google Scholar]
- Marminon, C; Pierré, A; Pfeiffer, B; Pérez, V; Léonce, S; Joubert, A; Bailly, C; Renard, P; Hickman, J; Prudhomme, M. Synthesis and antiproliferative activities of 7-azarebeccamycin analogues bearing one 7-azaindole moiety. J Med Chem 2003, 46, 609–622. [Google Scholar]
- Messaoudi, S; Anizon, F; Pfeiffer, B; Golsteyn, R; Prudhomme, M. Synthesis of a staurosporine analogue possessing a 7-azaindole unit instead of an indole moiety. Tetrahedron Lett 2004, 45, 4643–4647. [Google Scholar]
- Messaoudi, S; Anizon, F; Pfeiffer, B; Prudhomme, M. Synthesis of bridged aza-rebeccamycin analogues. Tetrahedron 2005, 61, 7304–7316. [Google Scholar]
- Messaoudi, S; Anizon, F; Léonce, S; Pierré, A; Pfeiffer, B; Prudhomme, M. Synthesis and cytotoxicities of 7-aza rebeccamycin analogues bearing various substituents on the sugar moiety, on the imide nitrogen and on the carbazole framework. Eur J Med Chem 2005, 40, 961–971. [Google Scholar]
- Messaoudi, S; Anizon, F; Peixoto, P; David-Cordonnier, MH; Golsteyn, RM; Léonce, S; Pfeiffer, B; Prudhomme, M. Synthesis and biological activities of 7-azarebeccamycin analogues bearing the sugar moiety on the nitrogen of the pyridine ring. Bioorg Med Chem 2006, 14, 7551–7562. [Google Scholar]
- Conchon, E; Anizon, F; Aboab, B; Golsteyn, RM; Léonce, S; Pfeiffer, B; Prudhomme, M. Synthesis, in vitro antiproliferative activities, and Chk1 inhibitory properties of pyrrolo[3,4-a]carbazole-1,3-diones, pyrrolo[3,4-c]carbazole-1,3-diones, and 2-aminopyridazino[3,4-a]pyrrolo[3,4-c]carbazole-1,3,4,7-tetraone. Eur J Med Chem 2008, 43, 282–292. [Google Scholar]
- Fabian, MA; Biggs, WH, III; Treiber, DK; Atteridge, CE; Azimioara, MD; Bendetti, MG; Carter, TA; Ciceri, P; Edeen, PT; Floyd, M; Ford, JM; Galvin, M; Gerlach, JL; Grotzfeld, RM; Herrgard, S; Insko, DE; Insko, MA; Lai, AG; Lelias, RM; Mehta, SA; Milanov, ZV; Velasco, AM; Wodicka, LM; Patel, HK; Zarrinkar, PP; Lockhardt, DJ. A small molecule-kinase interaction map for clinical kinase inhibitors. Nat Biotechnol 2005, 23, 329–336. [Google Scholar]
- Karaman, MW; Herrgard, S; Treiber, DK; Gallant, P; Atteridge, CE; Campbell, BT; Chan, KW; Ciceri, P; Davis, MI; Edeen, PT; Faraoni, R; Floyd, M; Hunt, JP; Lockhardt, DJ; Milanov, ZV; Morrison, MJ; Pallares, G; Patel, HK; Pritchard, S; Wodicka, LM; Zarrinkar, PP. A quantitative analysis of kinase inhibitor selectivity. Nat Biotechnol 2008, 26, 127–132. [Google Scholar]


































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Compound | A | B | R | PARP-1 IC50 (nM) |
|---|---|---|---|---|
| 29 | - | - | 36 | |
| 33 | H | H | H | 56 |
| 34 | H | H | Cl | 120 |
| 35 | H | H | Br | 30 |
| 36 | H | H | CN | 18 |
| 37 | H | H | CH2NH2 | 27 |
| 38 | H | H | Me | 200 |
| 39 | H | H | CO2H | 80 |
| 40 | H | H | CO2Me | 59 |
| 41 | H | H | CONH-(CH2)2-NMe2 | 165 |
| 42 | H | H | CONH-(CH2)2-morpholin-4-yl | 162 |
| 43 | H | H | CO-morpholin-4-yl | 83 |
| 44 | H | H | CON(Me)-CH2-pyrid-4-yl | 65 |
| 45 | H | H | CON(Me)-CH2-pyrid-2-yl | 237 |
| 46 | H | H | CON(Me)-(CH2)2-imidazol-4-yl | 161 |
| 47 | H | H | CONH-(CH2)2-triazol-1-yl | 105 |
| 48 | H | H | CH2NHCOCH-(NHBoc)[(CH2)4NHBoc] | 670 |
| 49 | H | H | CH2NHCOCH-(NH2)[(CH2)4NH2] | 80 |
| 50 | H | Me | H | 800 |
| 51 | Me | Me | H | 10,000 |
| 52 | CHO | H | H | 3,000 |
| 53 | - | - | 40 |

| Compound | X | R | Topoa | P388b | R/Sc | |
|---|---|---|---|---|---|---|
| rebeccamycin | NH | H | >500 | 0.54 | 1.26 | |
| 128 | 2,10-diF | NH | H | 2.0 | 0.26 | 8.7 |
| 129 | 3,9-diF | NH | H | 0.22 | 0.018 | 182.7 |
| 130 | 2,3,9,10-tetraF | NH | H | 0.69 | 0.007 | 67.1 |
| 131 | 2,3,9,10-tetraF | NMe2 | H | >600 | >8.72 | >1.1 |
| 132 | 3-F | NH | H | 6.6 | 1.036 | >905 |
| 133 | 2-F | NH | H | 3.1 | 0.392 | 4.6 |
| 134 | 10-F | NH | H | >200 | 0.098 | 11.4 |
| 135d | 9-F | NH | H | 1.7 | 0.101 | 31.7 |
| 136 | 3-F | S | H | 2.2 | 0.155 | >51.3 |
| 137 | 3,9-diF | S | H | 0.09 | 0.010 | 232.5 |
| 138 | 3-F | O | H | 1.5 | 0.529 | 13.6 |
| 139 | 3,9-diF | O | H | 0.27 | 0.114 | 63.7 |
| 140 | 3,9-diF | NH | NH2 | 0.22 | 0.020 | 196.9 |
| 141 | 3,9-diF | NH | OH | 0.08 | 0.035 | 19.6 |
| 142 | 3,9-diF | NH | Me | 1.0 | 0.862 | >10.8 |
| 143 | 3,9-diF | S | Me | 0.48 | 0.236 | 14.1 |
| 144e | 3,9-diF | NH | H | 0.28 | 0.326 | 28 |
| 145e | 3,9-diF | S | H | 0.46 | 0.068 | >115 |
| 146 | 3,9-diF | NH | H | 0.11 | 0.002 | 380 |
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Deslandes, S.; Chassaing, S.; Delfourne, E. Marine Pyrrolocarbazoles and Analogues: Synthesis and Kinase Inhibition. Mar. Drugs 2009, 7, 754-786. https://doi.org/10.3390/md7040754
Deslandes S, Chassaing S, Delfourne E. Marine Pyrrolocarbazoles and Analogues: Synthesis and Kinase Inhibition. Marine Drugs. 2009; 7(4):754-786. https://doi.org/10.3390/md7040754
Chicago/Turabian StyleDeslandes, Sébastien, Stefan Chassaing, and Evelyne Delfourne. 2009. "Marine Pyrrolocarbazoles and Analogues: Synthesis and Kinase Inhibition" Marine Drugs 7, no. 4: 754-786. https://doi.org/10.3390/md7040754
APA StyleDeslandes, S., Chassaing, S., & Delfourne, E. (2009). Marine Pyrrolocarbazoles and Analogues: Synthesis and Kinase Inhibition. Marine Drugs, 7(4), 754-786. https://doi.org/10.3390/md7040754