Cyanobacterial Cyclopeptides as Lead Compounds to Novel Targeted Cancer Drugs

Abstract
:
1. Introduction
2. Cyanotoxins–Microcystin
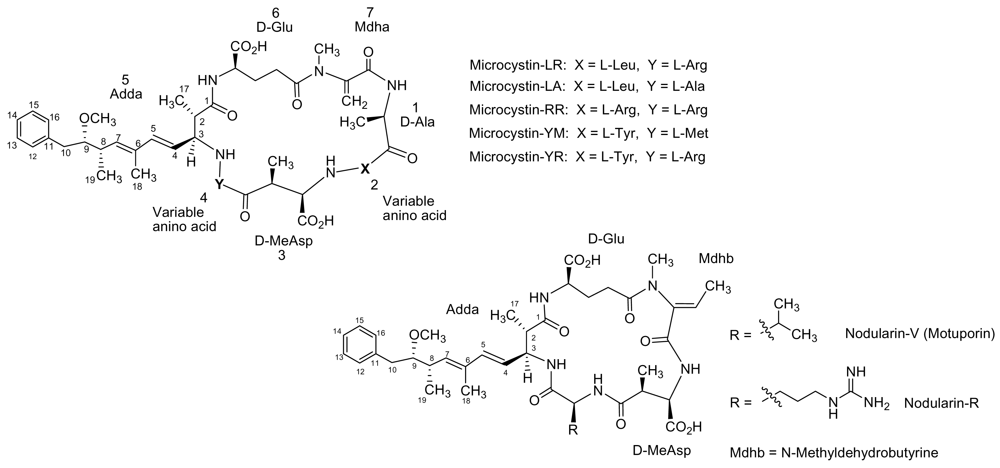
2.1. Categories
2.2. Microcystins: Physicochemical Characteristics
2.3. Microcystin Biogenesis and Ecological Role and Function
2.4. Biological Activity of Microcystins as Xenobiotics
2.4.1. In Animals
2.4.1.1. Acute Exposure
2.4.1.2. Low-dose Chronic Exposure
2.4.2. In Humans
2.4.2.1. Acute Exposure
2.4.2.2. Low-dose Chronic Exposure
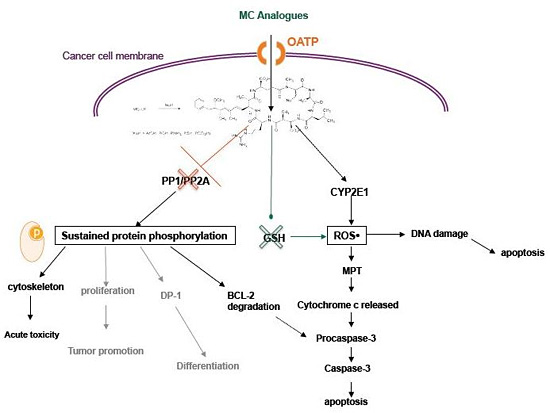
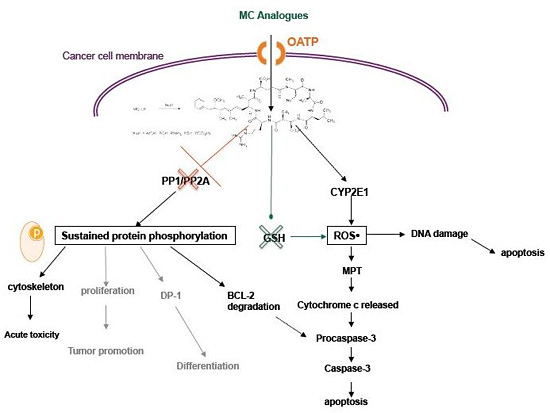
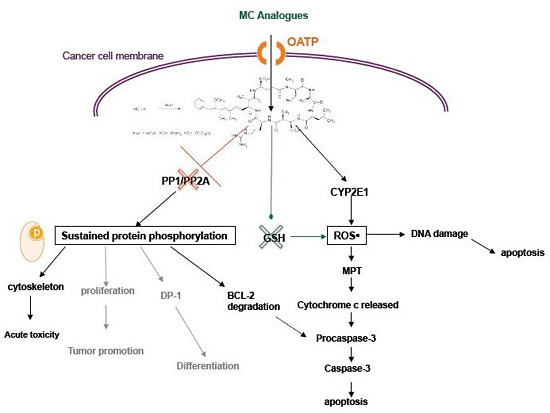
3. MC-LR Cell Molecular Targets
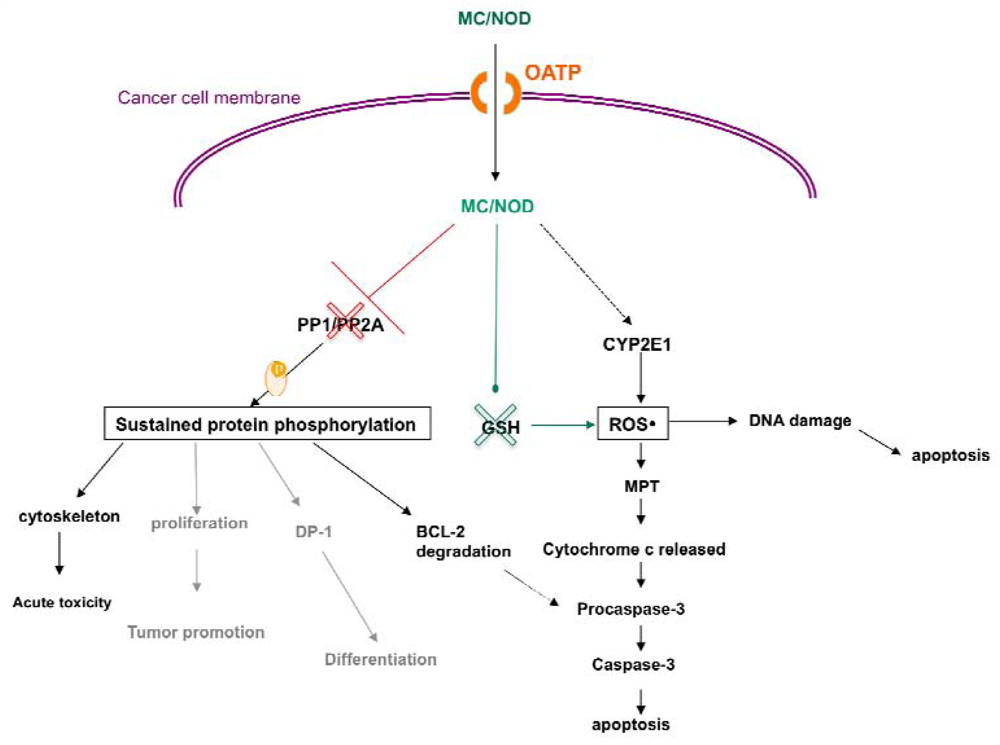
4. Cytotoxic Effects of MC-LR
4.1. Activity of MC in Normal Cell Lines and Tissues
4.2. Activity of MC in Cancer Cells
5. Organic Anion Transporting Polypeptides
5.1. OATP Substrates
5.2. OATP Expression in Normal Human Tissues
5.3. OATP Expression in Human Cancers
5.4. OATP: Cancer Trapdoors to Be Exploited
6. MC Analogues: Potentials and Perspectives
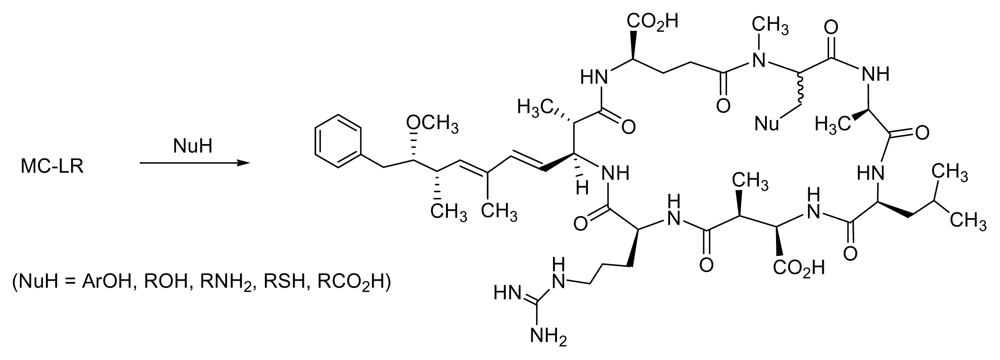
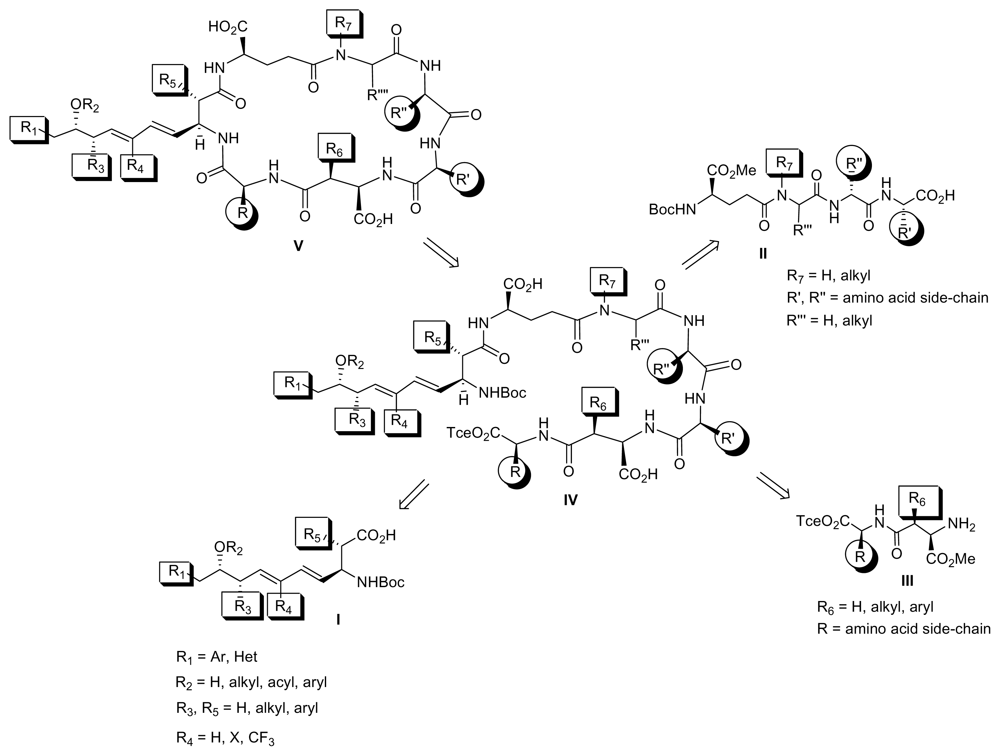
6.1. Combinatorial Chemical Synthesis
6.1.1. The Adda Issue
6.1.2. Synthetic Approaches
6.2. Combinatorial Total Biosynthesis
6.3. Selectivity and Function
7. Conclusions
Acknowledgment
- Sample Availability: Available from the authors.
References and Notes
- Awramik, SM. The oldest records of photosynthesis. Photosynth Res 1992, 33, 75–89. [Google Scholar]
- Blank, CE; Sanchez-Baracaldo, P. Timing of morphological and ecological innovations in the cyanobacteria–a key to understanding the rise in atmospheric oxygen. Geobiology 2010, 8, 1–23. [Google Scholar]
- Zehr, JP; Waterbury, JB; Turner, PJ; Montoya, JP; Omoregie, E; Steward, GF; Hansen, A; Karl, DM. Unicellular cyanobacteria fix N2 in the subtropical North Pacific Ocean. Nature 2001, 412, 635–638. [Google Scholar]
- Francis, G. Poisonous Australian lake. Nature 1878, 18, 11–12. [Google Scholar]
- Gupta, N; Pant, SC; Vijayaraghavan, R; Rao, PV. Comparative toxicity evaluation of cyanobacterial cyclic peptide toxin microcystin variants (LR, RR, YR) in mice. Toxicology 2003, 188, 285–296. [Google Scholar]
- Falconer, IR; Humpage, AR. Health risk assessment of cyanobacterial (blue-green algal) toxins in drinking water. Int J Environ Res Public Health 2005, 2, 43–50. [Google Scholar]
- Vareli, K; Briasoulis, E; Pilidis, G; Sainis, I. Molecular confirmation of Planktothrix rubescens as the cause of intense, microcystin—Synthesizing cyanobacterial bloom in Lake Ziros, Greece. Harmful Algae 2009, 8, 447–453. [Google Scholar]
- Vareli, K; Pilidis, G; Mavrogiorgou, MC; Briasoulis, E; Sainis, I. Molecular characterization of cyanobacterial diversity and yearly fluctuations of Microcystin loads in a suburban Mediterranean Lake (Lake Pamvotis, Greece). J Environ Monit 2009, 11, 1506–1512. [Google Scholar]
- Sarnelle, O; Morrison, J; Kaul, R; Horst, G; Wandell, H; Bednarz, R. Citizen monitoring: Testing hypotheses about the interactive influences of eutrophication and mussel invasion on a cyanobacterial toxin in lakes. Water Res 2009, 44, 141–150. [Google Scholar]
- Gaudin, J; Le Hegarat, L; Nesslany, F; Marzin, D; Fessard, V. In vivo genotoxic potential of microcystin-LR: a cyanobacterial toxin, investigated both by the unscheduled DNA synthesis (UDS) and the comet assays after intravenous administration. Environ Toxicol 2009, 24, 200–209. [Google Scholar]
- van Apeldoorn, ME; van Egmond, HP; Speijers, GJ; Bakker, GJ. Toxins of cyanobacteria. Mol Nutr Food Res 2007, 51, 7–60. [Google Scholar]
- Monks, NR; Liu, S; Xu, Y; Yu, H; Bendelow, AS; Moscow, JA. Potent cytotoxicity of the phosphatase inhibitor microcystin LR and microcystin analogues in OATP1B1- and OATP1B3- expressing HeLa cells. Mol Cancer Ther 2007, 6, 587–598. [Google Scholar]
- Herfindal, L; Kasprzykowski, F; Schwede, F; Lankiewicz, L; Fladmark, KE; Lukomska, J; Wahlsten, M; Sivonen, K; Grzonka, Z; Jastorff, B; Doskeland, SO. Acyloxymethyl esterification of nodularin-R and microcystin-LA produces inactive protoxins that become reactivated and produce apoptosis inside intact cells. J Med Chem 2009, 52, 5758–5762. [Google Scholar]
- Carmichael, WW. The toxins of cyanobacteria. Sci Am 1994, 270, 78–86. [Google Scholar]
- Gulledgea, BM; Aggena, JB; Huangb, HB; Nairnc, AC; Chamberlin, AR. The microcystins and nodularins: cyclic polypeptide inhibitors of PP1 and PP2A. Curr Med Chem 2002, 9, 1991–2003. [Google Scholar]
- Bouaicha, N; Maatouk, I. Microcystin-LR and nodularin induce intracellular glutathione alteration, reactive oxygen species production and lipid peroxidation in primary cultured rat hepatocytes. Toxicol Lett 2004, 148, 53–63. [Google Scholar]
- Lanaras, T; Cook, CM; Eriksson, JE; Meriluoto, JA; Hotokka, M. Computer modelling of the 3-dimensional structures of the cyanobacterial hepatotoxins microcystin-LR and nodularin. Toxicon 1991, 29, 901–906. [Google Scholar]
- Sivonen, K; Carmichael, WW; Namikoshi, M; Rinehart, KL; Dahlem, AM; Niemela, SI. Isolation and characterization of hepatotoxic microcystin homologs from the filamentous freshwater cyanobacterium Nostoc sp. strain 152. Appl Environ Microbiol 1990, 56, 2650–2657. [Google Scholar]
- Namikoshi, M; Rinehart, KL; Sakai, R; Stotts, RR; Dahlem, AM; Beasley, VR; Carmichael, WW; Evans, WR. Identification of 12 hepatotoxins from a Homer Lake bloom of the cyanobacteria Microcystis aeruginosa, Microcystis viridis, and Microcystis wesenbergii: nine new microcystins. J Org Chem 1992, 57, 866–872. [Google Scholar]
- de Figueiredo, DR; Azeiteiro, UM; Esteves, SM; Goncalves, FJ; Pereira, MJ. Microcystin-producing blooms--a serious global public health issue. Ecotoxicol Environ Saf 2004, 59, 151–163. [Google Scholar]
- Harada, K; Imanishi, S; Kato, H; Mizuno, M; Ito, E; Tsuji, K. Isolation of Adda from microcystin-LR by microbial degradation. Toxicon 2004, 44, 107–109. [Google Scholar]
- Nishiwaki-Matsushima, R; Nishiwaki, S; Ohta, T; Yoshizawa, S; Suganuma, M; Harada, K; Watanabe, MF; Fujiki, H. Structure-function relationships of microcystins, liver tumor promoters, in interaction with protein phosphatase. Jpn J Cancer Res 1991, 82, 993–996. [Google Scholar]
- Bourne, DG; Jones, GJ; Blakeley, RL; Jones, A; Negri, AP; Riddles, P. Enzymatic pathway for the bacterial degradation of the cyanobacterial cyclic peptide toxin microcystin LR. Appl Environ Microbiol 1996, 62, 4086–4094. [Google Scholar]
- Harada, K; Ogawa, K; Matsuura, K; Murata, H; Suzuki, M; Watanabe, MF; Itezono, Y; Nakayama, N. Structural determination of geometrical isomers of microcystins LR and RR from cyanobacteria by two-dimensional NMR spectroscopic techniques. Chem Res Toxicol 1990, 3, 473–481. [Google Scholar]
- Tooming-Klunderud, A; Rohrlack, T; Shalchian-Tabrizi, K; Kristensen, T; Jakobsen, KS. Structural analysis of a non-ribosomal halogenated cyclic peptide and its putative operon from Microcystis: implications for evolution of cyanopeptolins. Microbiology 2007, 153, 1382–1393. [Google Scholar]
- Hyenstrand, P; Rohrlack, T; Beattie, KA; Metcalf, JS; Codd, GA; Christoffersen, K. Laboratory studies of dissolved radiolabelled microcystin-LR in lake water. Water Res 2003, 37, 3299–3306. [Google Scholar]
- Oliveira, AC; Magalhaes, VF; Soares, RM; Azevedo, SM. Influence of drinking water composition on quantitation and biological activity of dissolved microcystin (cyanotoxin). Environ Toxicol 2005, 20, 126–130. [Google Scholar]
- Tsuji, K; Watanuki, T; Kondo, F; Watanabe, MF; Suzuki, S; Nakazawa, H; Suzuki, M; Uchida, H; Harada, KI. Stability of microcystins from cyanobacteria--II. Effect of UV light on decomposition and isomerization. Toxicon 1995, 33, 1619–1631. [Google Scholar]
- Akcaalan, R; Young, FM; Metcalf, JS; Morrison, LF; Albay, M; Codd, GA. Microcystin analysis in single filaments of Planktothrix spp. in laboratory cultures and environmental blooms. Water Res 2006, 40, 1583–1590. [Google Scholar]
- Zhang, D; Xie, P; Chen, J. Effects of Temperature on the Stability of Microcystins in Muscle of Fish and Its Consequences for Food Safety. Bull Environ Contam Toxicol 2010, 84, 202–207. [Google Scholar]
- Noguchi, T; Shinohara, A; Nishizawa, A; Asayama, M; Nakano, T; Hasegawa, M; Harada, K; Nishizawa, T; Shirai, M. Genetic analysis of the microcystin biosynthesis gene cluster in Microcystis strains from four bodies of eutrophic water in Japan. J Gen Appl Microbiol 2009, 55, 111–123. [Google Scholar]
- Sedmak, B; Elersek, T. Microcystins induce morphological and physiological changes in selected representative phytoplanktons. Microb Ecol 2006, 51, 508–515. [Google Scholar]
- Hu, ZQ; Liu, YD; Li, DH. Physiological and biochemical analyses of microcystin-RR toxicity to the cyanobacterium Synechococcus elongatus. Environ Toxicol 2004, 19, 571–577. [Google Scholar]
- DeMott, WR; Moxter, F. Foraging Cyanobacteria by Copepods: Responses to Chemical Defense and Resource Abundance. Ecology 1991, 72, 1820–1834. [Google Scholar]
- Schatz, D; Keren, Y; Vardi, A; Sukenik, A; Carmeli, S; Borner, T; Dittmann, E; Kaplan, A. Towards clarification of the biological role of microcystins, a family of cyanobacterial toxins. Environ Microbiol 2007, 9, 965–970. [Google Scholar]
- Armitage, AR; Fong, P. Upward cascading effects of nutrients: shifts in a benthic microalgal community and a negative herbivore response. Oecologia 2004, 139, 560–567. [Google Scholar]
- Jang, MH; Ha, K; Takamura, N. Microcystin production by Microcystis aeruginosa exposed to different stages of herbivorous zooplankton. Toxicon 2008, 51, 882–889. [Google Scholar]
- Utkilen, H; Gjolme, N. Iron-stimulated toxin production in Microcystis aeruginosa. Appl Environ Microbiol 1995, 61, 797–800. [Google Scholar]
- Hooser, SB. Fulminant hepatocyte apoptosis in vivo following microcystin-LR administration to rats. Toxicol Pathol 2000, 28, 726–733. [Google Scholar]
- Weng, D; Lu, Y; Wei, Y; Liu, Y; Shen, P. The role of ROS in microcystin-LR-induced hepatocyte apoptosis and liver injury in mice. Toxicology 2007, 232, 15–23. [Google Scholar]
- Oberholster, PJ; Myburgh, JG; Govender, D; Bengis, R; Botha, AM. Identification of toxigenic Microcystis strains after incidents of wild animal mortalities in the Kruger National Park, South Africa. Ecotoxicol Environ Saf 2009, 72, 1177–1182. [Google Scholar]
- Fawell, JK; Mitchell, RE; Everett, DJ; Hill, RE. The toxicity of cyanobacterial toxins in the mouse: I microcystin-LR. Hum Exp Toxicol 1999, 18, 162–167. [Google Scholar]
- Miura, GA; Robinson, NA; Lawrence, WB; Pace, JG. Hepatotoxicity of microcystin-LR in fed and fasted rats. Toxicon 1991, 29, 337–346. [Google Scholar]
- Runnegar, M; Berndt, N; Kaplowitz, N. Microcystin uptake and inhibition of protein phosphatases: effects of chemoprotectants and self-inhibition in relation to known hepatic transporters. Toxicol Appl Pharmacol 1995, 134, 264–272. [Google Scholar]
- Clark, SP; Ryan, TP; Searfoss, GH; Davis, MA; Hooser, SB. Chronic microcystin exposure induces hepatocyte proliferation with increased expression of mitotic and cyclin-associated genes in P53-deficient mice. Toxicol Pathol 2008, 36, 190–203. [Google Scholar]
- Nishiwaki-Matsushima, R; Ohta, T; Nishiwaki, S; Suganuma, M; Kohyama, K; Ishikawa, T; Carmichael, WW; Fujiki, H. Liver tumor promotion by the cyanobacterial cyclic peptide toxin microcystin-LR. J Cancer Res Clin Oncol 1992, 118, 420–424. [Google Scholar]
- Humpage, AR; Hardy, SJ; Moore, EJ; Froscio, SM; Falconer, IR. Microcystins (cyanobacterial toxins) in drinking water enhance the growth of aberrant crypt foci in the mouse colon. J Toxicol Environ Health A 2000, 61, 155–165. [Google Scholar]
- Sekijima, M; Tsutsumi, T; Yoshida, T; Harada, T; Tashiro, F; Chen, G; Yu, SZ; Ueno, Y. Enhancement of glutathione S-transferase placental-form positive liver cell foci development by microcystin-LR in aflatoxin B1-initiated rats. Carcinogenesis 1999, 20, 161–165. [Google Scholar]
- Lian, M; Liu, Y; Yu, SZ; Qian, GS; Wan, SG; Dixon, KR. Hepatitis B virus x gene and cyanobacterial toxins promote aflatoxin B1-induced hepatotumorigenesis in mice. World J Gastroenterol 2006, 12, 3065–3072. [Google Scholar]
- Carmichael, WW; Azevedo, SM; An, JS; Molica, RJ; Jochimsen, EM; Lau, S; Rinehart, KL; Shaw, GR; Eaglesham, GK. Human fatalities from cyanobacteria: chemical and biological evidence for cyanotoxins. Environ Health Perspect 2001, 109, 663–668. [Google Scholar]
- Jochimsen, EM; Carmichael, WW; An, JS; Cardo, DM; Cookson, ST; Holmes, CE; Antunes, MB; de Melo Filho, DA; Lyra, TM; Barreto, VS; Azevedo, SM; Jarvis, WR. Liver failure and death after exposure to microcystins at a hemodialysis center in Brazil. N Engl J Med 1998, 338, 873–878. [Google Scholar]
- Yuan, M; Carmichael, WW; Hilborn, ED. Microcystin analysis in human sera and liver from human fatalities in Caruaru, Brazil 1996. Toxicon 2006, 48, 627–640. [Google Scholar]
- Soares, RM; Yuan, M; Servaites, JC; Delgado, A; Magalhaes, VF; Hilborn, ED; Carmichael, WW; Azevedo, SM. Sublethal exposure from microcystins to renal insufficiency patients in Rio de Janeiro, Brazil. Environ Toxicol 2006, 21, 95–103. [Google Scholar]
- Ueno, Y; Nagata, S; Tsutsumi, T; Hasegawa, A; Watanabe, MF; Park, HD; Chen, GC; Chen, G; Yu, SZ. Detection of microcystins, a blue-green algal hepatotoxin, in drinking water sampled in Haimen and Fusui, endemic areas of primary liver cancer in China, by highly sensitive immunoassay. Carcinogenesis 1996, 17, 1317–1321. [Google Scholar]
- Svircev, Z; Krstic, S; Miladinov-Mikov, M; Baltic, V; Vidovic, M. Freshwater cyanobacterial blooms and primary liver cancer epidemiological studies in Serbia. J Environ Sci Health C Environ Carcinog Ecotoxicol Rev 2009, 27, 36–55. [Google Scholar]
- Cogliano, VJ; Baan, RA; Straif, K; Grosse, Y; Secretan, B; El Ghissassi, F. Use of mechanistic data in IARC evaluations. Environ Mol Mutagen 2008, 49, 100–109. [Google Scholar]
- Hagenbuch, B; Meier, PJ. Organic anion transporting polypeptides of the OATP/SLC21 family: phylogenetic classification as OATP/SLCO superfamily, new nomenclature and molecular/functional properties. Pflugers Arch 2004, 447, 653–665. [Google Scholar]
- Fischer, WJ; Altheimer, S; Cattori, V; Meier, PJ; Dietrich, DR; Hagenbuch, B. Organic anion transporting polypeptides expressed in liver and brain mediate uptake of microcystin. Toxicol Appl Pharmacol 2005, 203, 257–263. [Google Scholar]
- Yoshida, T; Makita, Y; Tsutsumi, T; Nagata, S; Tashiro, F; Yoshida, F; Sekijima, M; Tamura, S; Harada, T; Maita, K; Ueno, Y. Immunohistochemical localization of microcystin- LR in the liver of mice: a study on the pathogenesis of microcystin-LR-induced hepatotoxicity. Toxicol Pathol 1998, 26, 411–418. [Google Scholar]
- Runnegar, MT; Kong, S; Berndt, N. Protein phosphatase inhibition and in vivo hepatotoxicity of microcystins. Am J Physiol 1993, 265, G224–230. [Google Scholar]
- Yoshizawa, S; Matsushima, R; Watanabe, MF; Harada, K; Ichihara, A; Carmichael, WW; Fujiki, H. Inhibition of protein phosphatases by microcystins and nodularin associated with hepatotoxicity. J Cancer Res Clin Oncol 1990, 116, 609–614. [Google Scholar]
- Nong, Q; Komatsu, M; Izumo, K; Indo, HP; Xu, B; Aoyama, K; Majima, HJ; Horiuchi, M; Morimoto, K; Takeuchi, T. Involvement of reactive oxygen species in Microcystin-LR-induced cytogenotoxicity. Free Radic Res 2007, 41, 1326–1337. [Google Scholar]
- MacKintosh, RW; Dalby, KN; Campbell, DG; Cohen, PT; Cohen, P; MacKintosh, C. The cyanobacterial toxin microcystin binds covalently to cysteine-273 on protein phosphatase 1. FEBS Lett 1995, 371, 236–240. [Google Scholar]
- Fischer, WJ; Hitzfeld, BC; Tencalla, F; Eriksson, JE; Mikhailov, A; Dietrich, DR. Microcystin-LR toxicodynamics, induced pathology, and immunohistochemical localization in livers of blue-green algae exposed rainbow trout (oncorhynchus mykiss). Toxicol Sci 2000, 54, 365–373. [Google Scholar]
- Lovell, RA; Schaeffer, DJ; Hooser, SB; Haschek, WM; Dahlem, AM; Carmichael, WW; Beasley, VR. Toxicity of intraperitoneal doses of microcystin-LR in two strains of male mice. J Environ Pathol Toxicol Oncol 1989, 9, 221–237. [Google Scholar]
- Yoshida, T; Makita, Y; Nagata, S; Tsutsumi, T; Yoshida, F; Sekijima, M; Tamura, S; Ueno, Y. Acute oral toxicity of microcystin-LR, a cyanobacterial hepatotoxin, in mice. Nat Toxins 1997, 5, 91–95. [Google Scholar]
- Billam, M; Mukhi, S; Tang, L; Gao, W; Wang, JS. Toxic response indicators of microcystin- LR in F344 rats following a single-dose treatment. Toxicon 2008, 51, 1068–1080. [Google Scholar]
- Ito, E; Takai, A; Kondo, F; Masui, H; Imanishi, S; Harada, K. Comparison of protein phosphatase inhibitory activity and apparent toxicity of microcystins and related compounds. Toxicon 2002, 40, 1017–1025. [Google Scholar]
- Xing, Y; Xu, Y; Chen, Y; Jeffrey, PD; Chao, Y; Lin, Z; Li, Z; Strack, S; Stock, JB; Shi, Y. Structure of protein phosphatase 2A core enzyme bound to tumor-inducing toxins. Cell 2006, 127, 341–353. [Google Scholar]
- Gehringer, MM. Microcystin-LR and okadaic acid-induced cellular effects: a dualistic response. FEBS Lett 2004, 557, 1–8. [Google Scholar]
- Janssens, V; Goris, J. Protein phosphatase 2A: a highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem J 2001, 353, 417–439. [Google Scholar]
- Janssens, V; Goris, J; Van Hoof, C. PP2A: the expected tumor suppressor. Curr Opin Genet Dev 2005, 15, 34–41. [Google Scholar]
- Zegura, B; Zajc, I; Lah, TT; Filipic, M. Patterns of microcystin-LR induced alteration of the expression of genes involved in response to DNA damage and apoptosis. Toxicon 2008, 51, 615–623. [Google Scholar]
- Zhan, L; Sakamoto, H; Sakuraba, M; Wu, DS; Zhang, LS; Suzuki, T; Hayashi, M; Honma, M. Genotoxicity of microcystin-LR in human lymphoblastoid TK6 cells. Mutat Res 2004, 557, 1–6. [Google Scholar]
- Rao, PV; Bhattacharya, R. The cyanobacterial toxin microcystin-LR induced DNA damage in mouse liver in vivo. Toxicology 1996, 114, 29–36. [Google Scholar]
- Runnegar, MT; Andrews, J; Gerdes, RG; Falconer, IR. Injury to hepatocytes induced by a peptide toxin from the cyanobacterium Microcystis aeruginosa. Toxicon 1987, 25, 1235–1239. [Google Scholar]
- Zegura, B; Lah, TT; Filipic, M. Alteration of intracellular GSH levels and its role in microcystin-LR-induced DNA damage in human hepatoma HepG2 cells. Mutat Res 2006, 611, 25–33. [Google Scholar]
- Lankoff, A; Banasik, A; Nowak, M. Protective effect of melatonin against nodularin-induced oxidative stress. Arch Toxicol 2002, 76, 158–165. [Google Scholar]
- Mikhailov, A; Harmala-Brasken, AS; Hellman, J; Meriluoto, J; Eriksson, JE. Identification of ATP-synthase as a novel intracellular target for microcystin-LR. Chem Biol Interact 2003, 142, 223–237. [Google Scholar]
- Chen, T; Cui, J; Liang, Y; Xin, X; Owen Young, D; Chen, C; Shen, P. Identification of human liver mitochondrial aldehyde dehydrogenase as a potential target for microcystin-LR. Toxicology 2006, 220, 71–80. [Google Scholar]
- Mankiewicz, J; Tarczynska, M; Fladmark, KE; Doskeland, SO; Walter, Z; Zalewski, M. Apoptotic effect of cyanobacterial extract on rat hepatocytes and human lymphocytes. Environ Toxicol 2001, 16, 225–233. [Google Scholar]
- Ding, WX; Shen, HM; Ong, CN. Critical role of reactive oxygen species and mitochondrial permeability transition in microcystin-induced rapid apoptosis in rat hepatocytes. Hepatology 2000, 32, 547–555. [Google Scholar]
- Fladmark, KE; Brustugun, OT; Hovland, R; Boe, R; Gjertsen, BT; Zhivotovsky, B; Doskeland, SO. Ultrarapid caspase-3 dependent apoptosis induction by serine/threonine phosphatase inhibitors. Cell Death Differ 1999, 6, 1099–1108. [Google Scholar]
- Lin, SS; Bassik, MC; Suh, H; Nishino, M; Arroyo, JD; Hahn, WC; Korsmeyer, SJ; Roberts, TM. PP2A regulates BCL-2 phosphorylation and proteasome-mediated degradation at the endoplasmic reticulum. J Biol Chem 2006, 281, 23003–23012. [Google Scholar]
- Tilli, MT; Hudgins, SL; Frech, MS; Halama, ED; Renou, JP; Furth, PA. Loss of protein phosphatase 2A expression correlates with phosphorylation of DP-1 and reversal of dysplasia through differentiation in a conditional mouse model of cancer progression. Cancer Res 2003, 63, 7668–7673. [Google Scholar]
- Ding, WX; Nam Ong, C. Role of oxidative stress and mitochondrial changes in cyanobacteria-induced apoptosis and hepatotoxicity. FEMS Microbiol Lett 2003, 220, 1–7. [Google Scholar]
- Ding, WX; Shen, HM; Zhu, HG; Lee, BL; Ong, CN. Genotoxicity of microcystic cyanobacteria extract of a water source in China. Mutat Res 1999, 442, 69–77. [Google Scholar]
- Hilgendorf, C; Ahlin, G; Seithel, A; Artursson, P; Ungell, AL; Karlsson, J. Expression of thirty-six drug transporter genes in human intestine, liver, kidney, and organotypic cell lines. Drug Metab Dispos 2007, 35, 1333–1340. [Google Scholar]
- Ahlin, G; Hilgendorf, C; Karlsson, J; Szigyarto, CA; Uhlen, M; Artursson, P. Endogenous gene and protein expression of drug-transporting proteins in cell lines routinely used in drug discovery programs. Drug Metab Dispos 2009, 37, 2275–2283. [Google Scholar]
- Boaru, DA; Dragos, N; Schirmer, K. Microcystin-LR induced cellular effects in mammalian and fish primary hepatocyte cultures and cell lines: a comparative study. Toxicology 2006, 218, 134–148. [Google Scholar]
- Lee, W; Belkhiri, A; Lockhart, AC; Merchant, N; Glaeser, H; Harris, EI; Washington, MK; Brunt, EM; Zaika, A; Kim, RB; El-Rifai, W. Overexpression of OATP1B3 confers apoptotic resistance in colon cancer. Cancer Res 2008, 68, 10315–10323. [Google Scholar]
- Cui, Y; Konig, J; Nies, AT; Pfannschmidt, M; Hergt, M; Franke, WW; Alt, W; Moll, R; Keppler, D. Detection of the human organic anion transporters SLC21A6 (OATP2) and SLC21A8 (OATP8) in liver and hepatocellular carcinoma. Lab Invest 2003, 83, 527–538. [Google Scholar]
- Abe, T; Unno, M; Onogawa, T; Tokui, T; Kondo, TN; Nakagomi, R; Adachi, H; Fujiwara, K; Okabe, M; Suzuki, T; Nunoki, K; Sato, E; Kakyo, M; Nishio, T; Sugita, J; Asano, N; Tanemoto, M; Seki, M; Date, F; Ono, K; Kondo, Y; Shiiba, K; Suzuki, M; Ohtani, H; Shimosegawa, T; Iinuma, K; Nagura, H; Ito, S; Matsuno, S. LST-2, a human liver-specific organic anion transporter, determines methotrexate sensitivity in gastrointestinal cancers. Gastroenterology 2001, 120, 1689–1699. [Google Scholar]
- Konig, J; Seithel, A; Gradhand, U; Fromm, MF. Pharmacogenomics of human OATP transporters. Naunyn Schmiedebergs Arch Pharmacol 2006, 372, 432–443. [Google Scholar]
- Tamai, I; Nezu, J; Uchino, H; Sai, Y; Oku, A; Shimane, M; Tsuji, A. Molecular identification and characterization of novel members of the human organic anion transporter (OATP) family. Biochem Biophys Res Commun 2000, 273, 251–260. [Google Scholar]
- Bleasby, K; Castle, JC; Roberts, CJ; Cheng, C; Bailey, WJ; Sina, JF; Kulkarni, AV; Hafey, MJ; Evers, R; Johnson, JM; Ulrich, RG; Slatter, JG. Expression profiles of 50 xenobiotic transporter genes in humans and pre-clinical species: a resource for investigations into drug disposition. Xenobiotica 2006, 36, 963–988. [Google Scholar]
- Suzuki, T; Onogawa, T; Asano, N; Mizutamari, H; Mikkaichi, T; Tanemoto, M; Abe, M; Satoh, F; Unno, M; Nunoki, K; Suzuki, M; Hishinuma, T; Goto, J; Shimosegawa, T; Matsuno, S; Ito, S; Abe, T. Identification and characterization of novel rat and human gonad-specific organic anion transporters. Mol Endocrinol 2003, 17, 1203–1215. [Google Scholar]
- Lee, SY; Williamson, B; Caballero, OL; Chen, YT; Scanlan, MJ; Ritter, G; Jongeneel, CV; Simpson, AJ; Old, LJ. Identification of the gonad-specific anion transporter SLCO6A1 as a cancer/testis (CT) antigen expressed in human lung cancer. Cancer Immun 2004, 4, 13. [Google Scholar]
- Schiffer, R; Neis, M; Holler, D; Rodriguez, F; Geier, A; Gartung, C; Lammert, F; Dreuw, A; Zwadlo-Klarwasser, G; Merk, H; Jugert, F; Baron, JM. Active influx transport is mediated by members of the organic anion transporting polypeptide family in human epidermal keratinocytes. J Invest Dermatol 2003, 120, 285–291. [Google Scholar]
- Gao, B; Huber, RD; Wenzel, A; Vavricka, SR; Ismair, MG; Reme, C; Meier, PJ. Localization of organic anion transporting polypeptides in the rat and human ciliary body epithelium. Exp Eye Res 2005, 80, 61–72. [Google Scholar]
- Libra, A; Fernetti, C; Lorusso, V; Visigalli, M; Anelli, PL; Staud, F; Tiribelli, C; Pascolo, L. Molecular determinants in the transport of a bile acid-derived diagnostic agent in tumoral and nontumoral cell lines of human liver. J Pharmacol Exp Ther 2006, 319, 809–817. [Google Scholar]
- Bronger, H; Konig, J; Kopplow, K; Steiner, HH; Ahmadi, R; Herold-Mende, C; Keppler, D; Nies, AT. ABCC drug efflux pumps and organic anion uptake transporters in human gliomas and the blood-tumor barrier. Cancer Res 2005, 65, 11419–11428. [Google Scholar]
- Kalliokoski, A; Niemi, M. Impact of OATP transporters on pharmacokinetics. Br J Pharmacol 2009, 158, 693–705. [Google Scholar]
- Franke, RM; Scherkenbach, LA; Sparreboom, A. Pharmacogenetics of the organic anion transporting polypeptide 1A2. Pharmacogenomics 2009, 10, 339–344. [Google Scholar]
- Pizzagalli, F; Varga, Z; Huber, RD; Folkers, G; Meier, PJ; St-Pierre, MV. Identification of steroid sulfate transport processes in the human mammary gland. J Clin Endocrinol Metab 2003, 88, 3902–3912. [Google Scholar]
- Mikkaichi, T; Suzuki, T; Onogawa, T; Tanemoto, M; Mizutamari, H; Okada, M; Chaki, T; Masuda, S; Tokui, T; Eto, N; Abe, M; Satoh, F; Unno, M; Hishinuma, T; Inui, K; Ito, S; Goto, J; Abe, T. Isolation and characterization of a digoxin transporter and its rat homologue expressed in the kidney. Proc Natl Acad Sci USA 2004, 101, 3569–3574. [Google Scholar]
- Wlcek, K; Svoboda, M; Thalhammer, T; Sellner, F; Krupitza, G; Jaeger, W. Altered expression of organic anion transporter polypeptide (OATP) genes in human breast carcinoma. Cancer Biol Ther 2008, 7, 1450–1455. [Google Scholar]
- Martinez, FO; Gordon, S; Locati, M; Mantovani, A. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: new molecules and patterns of gene expression. J Immunol 2006, 177, 7303–7311. [Google Scholar]
- Adachi, H; Suzuki, T; Abe, M; Asano, N; Mizutamari, H; Tanemoto, M; Nishio, T; Onogawa, T; Toyohara, T; Kasai, S; Satoh, F; Suzuki, M; Tokui, T; Unno, M; Shimosegawa, T; Matsuno, S; Ito, S; Abe, T. Molecular characterization of human and rat organic anion transporter OATP-D. Am J Physiol Renal Physiol 2003, 285, F1188–1197. [Google Scholar]
- Umehara, K; Iwai, M; Adachi, Y; Iwatsubo, T; Usui, T; Kamimura, H. Hepatic uptake and excretion of (-)-N-{2-[(R)-3-(6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline-2-carbonyl)p iperidino]ethyl}-4-fluorobenzamide (YM758), a novel if channel inhibitor, in rats and humans. Drug Metab Dispos 2008, 36, 1030–1038. [Google Scholar]
- Huber, RD; Gao, B; Sidler Pfandler, MA; Zhang-Fu, W; Leuthold, S; Hagenbuch, B; Folkers, G; Meier, PJ; Stieger, B. Characterization of two splice variants of human organic anion transporting polypeptide 3A1 isolated from human brain. Am J Physiol Cell Physiol 2007, 292, C795–806. [Google Scholar]
- Janneh, O; Hartkoorn, RC; Jones, E; Owen, A; Ward, SA; Davey, R; Back, DJ; Khoo, SH. Cultured CD4T cells and primary human lymphocytes express hOATPs: intracellular accumulation of saquinavir and lopinavir. Br J Pharmacol 2008, 155, 875–883. [Google Scholar]
- Seki, S; Kobayashi, M; Itagaki, S; Hirano, T; Iseki, K. Contribution of organic anion transporting polypeptide OATP2B1 to amiodarone accumulation in lung epithelial cells. Biochim Biophys Acta 2009, 1788, 911–917. [Google Scholar]
- Mandery, K; Bujok, K; Schmidt, I; Wex, T; Treiber, G; Malfertheiner, P; Rau, TT; Amann, KU; Brune, K; Fromm, MF; Glaeser, H. Influence of cyclooxygenase inhibitors on the function of the prostaglandin transporter OATP2A1 expressed in human gastroduodenal mucosa. J Pharmacol Exp Ther 2010, 332, 345–351. [Google Scholar]
- Niessen, J; Jedlitschky, G; Grube, M; Bien, S; Schwertz, H; Ohtsuki, S; Kawakami, H; Kamiie, J; Oswald, S; Starke, K; Strobel, U; Siegmund, W; Rosskopf, D; Greinacher, A; Terasaki, T; Kroemer, HK. Human platelets express organic anion-transporting peptide 2B1, an uptake transporter for atorvastatin. Drug Metab Dispos 2009, 37, 1129–1137. [Google Scholar]
- Vavricka, SR; Jung, D; Fried, M; Grutzner, U; Meier, PJ; Kullak-Ublick, GA. The human organic anion transporting polypeptide 8 (SLCO1B3) gene is transcriptionally repressed by hepatocyte nuclear factor 3beta in hepatocellular carcinoma. J Hepatol 2004, 40, 212–218. [Google Scholar]
- Narita, M; Hatano, E; Arizono, S; Miyagawa-Hayashino, A; Isoda, H; Kitamura, K; Taura, K; Yasuchika, K; Nitta, T; Ikai, I; Uemoto, S. Expression of OATP1B3 determines uptake of Gd-EOB-DTPA in hepatocellular carcinoma. J Gastroenterol 2009, 44, 793–798. [Google Scholar] [Green Version]
- Muto, M; Onogawa, T; Suzuki, T; Ishida, T; Rikiyama, T; Katayose, Y; Ohuchi, N; Sasano, H; Abe, T; Unno, M. Human liver-specific organic anion transporter-2 is a potent prognostic factor for human breast carcinoma. Cancer Sci 2007, 98, 1570–1576. [Google Scholar]
- Liedauer, R; Svoboda, M; Wlcek, K; Arrich, F; Ja, W; Toma, C; Thalhammer, T. Different expression patterns of organic anion transporting polypeptides in osteosarcomas, bone metastases and aneurysmal bone cysts. Oncol Rep 2009, 22, 1485–1492. [Google Scholar]
- Oba-Shinjo, SM; Caballero, OL; Jungbluth, AA; Rosemberg, S; Old, LJ; Simpson, AJ; Marie, SK. Cancer-testis (CT) antigen expression in medulloblastoma. Cancer Immun 2008, 8, 7. [Google Scholar]
- Burris, HA, 3rd. Shortcomings of current therapies for non-small-cell lung cancer: unmet medical needs. Oncogene 2009, 28(Suppl 1), S4–13. [Google Scholar]
- Saltz, LB. Progress in cancer care: the hope, the hype, and the gap between reality and perception. J Clin Oncol 2008, 26, 5020–5021. [Google Scholar]
- Philip, PA; Mooney, M; Jaffe, D; Eckhardt, G; Moore, M; Meropol, N; Emens, L; O’Reilly, E; Korc, M; Ellis, L; Benedetti, J; Rothenberg, M; Willett, C; Tempero, M; Lowy, A; Abbruzzese, J; Simeone, D; Hingorani, S; Berlin, J; Tepper, J. Consensus report of the national cancer institute clinical trials planning meeting on pancreas cancer treatment. J Clin Oncol 2009, 27, 5660–5669. [Google Scholar]
- Hotchkiss, RS; Strasser, A; McDunn, JE; Swanson, PE. Cell death. N Engl J Med 2009, 361, 1570–1583. [Google Scholar]
- Rosen, JM; Jordan, CT. The increasing complexity of the cancer stem cell paradigm. Science 2009, 324, 1670–1673. [Google Scholar]
- Roukos, DH; Murray, S; Briasoulis, E. Molecular genetic tools shape a roadmap towards a more accurate prognostic prediction and personalized management of cancer. Cancer Biol Ther 2007, 6, 308–312. [Google Scholar]
- Gupta, PB; Onder, TT; Jiang, G; Tao, K; Kuperwasser, C; Weinberg, RA; Lander, ES. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell 2009, 138, 645–659. [Google Scholar]
- Gatenby, RA. A change of strategy in the war on cancer. Nature 2009, 459, 508–509. [Google Scholar]
- Briasoulis, E; Pavlidis, N; Terret, C; Bauer, J; Fiedler, W; Schoffski, P; Raoul, JL; Hess, D; Selvais, R; Lacombe, D; Bachmann, P; Fumoleau, P. Glufosfamide administered using a 1- hour infusion given as first-line treatment for advanced pancreatic cancer. A phase II trial of the EORTC-new drug development group. Eur J Cancer 2003, 39, 2334–2340. [Google Scholar]
- Briasoulis, E; Judson, I; Pavlidis, N; Beale, P; Wanders, J; Groot, Y; Veerman, G; Schuessler, M; Niebch, G; Siamopoulos, K; Tzamakou, E; Rammou, D; Wolf, L; Walker, R; Hanauske, A. Phase I trial of 6-hour infusion of glufosfamide, a new alkylating agent with potentially enhanced selectivity for tumors that overexpress transmembrane glucose transporters: a study of the European Organization for Research and Treatment of Cancer Early Clinical Studies Group. J Clin Oncol 2000, 18, 3535–3544. [Google Scholar]
- Laidley, CW; Cohen, E; Casida, JE. Protein phosphatase in neuroblastoma cells: [3H]cantharidin binding site in relation to cytotoxicity. J Pharmacol Exp Ther 1997, 280, 1152–1158. [Google Scholar]
- Driggers, EM; Hale, SP; Lee, J; Terrett, NK. The exploration of macrocycles for drug discovery--an underexploited structural class. Nat Rev Drug Discov 2008, 7, 608–624. [Google Scholar]
- Robinson, NA; Pace, JG; Matson, CF; Miura, GA; Lawrence, WB. Tissue distribution, excretion and hepatic biotransformation of microcystin-LR in mice. J Pharmacol Exp Ther 1991, 256, 176–182. [Google Scholar]
- Daily, A; Monks, NR; Leggas, M; Moscow, JA. Abrogation of microcystin cytotoxicity by MAP kinase inhibitors and N-acetyl cysteine is confounded by OATPIB1 uptake activity inhibition. Toxicon 2010, 55, 827–837. [Google Scholar]
- Stotts, RR; Namikoshi, M; Haschek, WM; Rinehart, KL; Carmichael, WW; Dahlem, AM; Beasley, VR. Structural modifications imparting reduced toxicity in microcystins from Microcystis spp. Toxicon 1993, 31, 783–789. [Google Scholar]
- Goldberg, J; Huang, HB; Kwon, YG; Greengard, P; Nairn, AC; Kuriyan, J. Three-dimensional structure of the catalytic subunit of protein serine/threonine phosphatase-1. Nature 1995, 376, 745–753. [Google Scholar]
- Craig, M; Luu, HA; McCready, TL; Williams, D; Andersen, RJ; Holmes, CF. Molecular mechanisms underlying he interaction of motuporin and microcystins with type-1 and type-2A protein phosphatases. Biochem Cell Biol 1996, 74, 569–578. [Google Scholar]
- Maynes, JT; Luu, HA; Cherney, MM; Andersen, RJ; Williams, D; Holmes, CF; James, MN. Crystal structures of protein phosphatase-1 bound to motuporin and dihydromicrocystin-LA: elucidation of the mechanism of enzyme inhibition by cyanobacterial toxins. J Mol Biol 2006, 356, 111–120. [Google Scholar]
- Humphrey, JM; Aggen, JB; Chamberlin, AR. Total Synthesis of the Serine-Threonine Phosphatase Inhibitor Microcystin-LA. J Am Chem Soc 1996, 118, 11759–11770. [Google Scholar]
- Aggen, JB; Humphrey, JM; Gauss, CM; Huang, HB; Nairn, AC; Chamberlin, AR. The design, synthesis, and biological evaluation of analogues of the serine-threonine protein phosphatase 1 and 2A selective inhibitor microcystin LA: rational modifications imparting PP1 selectivity. Bioorg Med Chem 1999, 7, 543–564. [Google Scholar]
- Gulledge, BM; Aggen, JB; Chamberlin, AR. Linearized and truncated microcystin analogues as inhibitors of protein phosphatases 1 and 2A. Bioorg Med Chem Lett 2003, 13, 2903–2906. [Google Scholar]
- Abdel-Rahman, S; el-Ayouty, YM; Kamael, HA. Characterization of heptapeptide toxins extracted from Microcystis aeruginosa (Egyptian isolate). Comparison with some synthesized analogs. Int J Pept Protein Res 1993, 41, 1–7. [Google Scholar]
- Taylor, C; Quinn, RJ; Suganuma, M; Fujiki, H. Inhibition of protein phosphatase 2A by cyclic peptides modeled on the microcystin ring. Bioorg Med Chem Lett 1996, 6, 2113–2116. [Google Scholar]
- Tappan, E; Chamberlin, AR. Activation of protein phosphatase 1 by a small molecule designed to bind to the enzyme’s regulatory site. Chem Biol 2008, 15, 167–174. [Google Scholar]
- Kondo, F; Matsumoto, H; Yamada, S; Ishikawa, N; Ito, E; Nagata, S; Ueno, Y; Suzuki, M; Harada, K. Detection and identification of metabolites of microcystins formed in vivo in mouse and rat livers. Chem Res Toxicol 1996, 9, 1355–1359. [Google Scholar]
- Trogen, GB; Edlund, U; Larsson, G; Sethson, I. The solution NMR structure of a blue-green algae hepatotoxin, microcystin-RR--a comparison with the structure of microcystin-LR. Eur J Biochem 1998, 258, 301–312. [Google Scholar]
- Bagu, JR; Sykes, BD; Craig, MM; Holmes, CF. A molecular basis for different interactions of marine toxins with protein phosphatase-1. Molecular models for bound motuporin, microcystins, okadaic acid, and calyculin A. J Biol Chem 1997, 272, 5087–5097. [Google Scholar]
- Bagu, JR; Sonnichsen, FD; Williams, D; Andersen, RJ; Sykes, BD; Holmes, CF. Comparison of the solution structures of microcystin-LR and motuporin. Nat Struct Biol 1995, 2, 114–116. [Google Scholar]
- Lavigne, P; Bagu, JR; Boyko, R; Willard, L; Holmes, CF; Sykes, BD. Structure-based thermodynamic analysis of the dissociation of protein phosphatase-1 catalytic subunit and microcystin-LR docked complexes. Protein Sci 2000, 9, 252–264. [Google Scholar]
- Nishizawa, T; Asayama, M; Fujii, K; Harada, K; Shirai, M. Genetic analysis of the peptide synthetase genes for a cyclic heptapeptide microcystin in Microcystis spp. J Biochem 1999, 126, 520–529. [Google Scholar]
- Nishizawa, T; Ueda, A; Asayama, M; Fujii, K; Harada, K; Ochi, K; Shirai, M. Polyketide synthase gene coupled to the peptide synthetase module involved in the biosynthesis of the cyclic heptapeptide microcystin. J Biochem 2000, 127, 779–789. [Google Scholar]
- Tillett, D; Dittmann, E; Erhard, M; von Dohren, H; Borner, T; Neilan, BA. Structural organization of microcystin biosynthesis in Microcystis aeruginosa PCC7806: an integrated peptide-polyketide synthetase system. Chem Biol 2000, 7, 753–764. [Google Scholar]
- Christiansen, G; Fastner, J; Erhard, M; Borner, T; Dittmann, E. Microcystin biosynthesis in planktothrix: genes, evolution, and manipulation. J Bacteriol 2003, 185, 564–572. [Google Scholar]
- Rouhiainen, L; Vakkilainen, T; Siemer, BL; Buikema, W; Haselkorn, R; Sivonen, K. Genes coding for hepatotoxic heptapeptides (microcystins) in the cyanobacterium Anabaena strain 90. Appl Environ Microbiol 2004, 70, 686–692. [Google Scholar]
- Marahiel, MA; Stachelhaus, T; Mootz, HD. Modular Peptide Synthetases Involved in Nonribosomal Peptide Synthesis. Chem Rev 1997, 97, 2651–2674. [Google Scholar]
- Sieber, SA; Marahiel, MA. Molecular mechanisms underlying nonribosomal peptide synthesis: approaches to new antibiotics. Chem Rev 2005, 105, 715–738. [Google Scholar]
- Mikalsen, B; Boison, G; Skulberg, OM; Fastner, J; Davies, W; Gabrielsen, TM; Rudi, K; Jakobsen, KS. Natural variation in the microcystin synthetase operon mcyABC and impact on microcystin production in Microcystis strains. J Bacteriol 2003, 185, 2774–2785. [Google Scholar]
- Tanabe, Y; Sano, T; Kasai, F; Watanabe, MM. Recombination, cryptic clades and neutral molecular divergence of the microcystin synthetase (mcy) genes of toxic cyanobacterium Microcystis aeruginosa. BMC Evol Biol 2009, 9, 115. [Google Scholar]
- Lautru, S; Challis, GL. Substrate recognition by nonribosomal peptide synthetase multienzymes. Microbiology 2004, 150, 1629–1636. [Google Scholar]
- Fewer, DP; Rouhiainen, L; Jokela, J; Wahlsten, M; Laakso, K; Wang, H; Sivonen, K. Recurrent adenylation domain replacement in the microcystin synthetase gene cluster. BMC Evol Biol 2007, 7, 183. [Google Scholar]
- Stachelhaus, T; Mootz, HD; Marahiel, MA. The specificity-conferring code of adenylation domains in nonribosomal peptide synthetases. Chem Biol 1999, 6, 493–505. [Google Scholar]
- Lautru, S; Deeth, RJ; Bailey, LM; Challis, GL. Discovery of a new peptide natural product by Streptomyces coelicolor genome mining. Nat Chem Biol 2005, 1, 265–269. [Google Scholar]
- Mootz, HD; Schwarzer, D; Marahiel, MA. Construction of hybrid peptide synthetases by module and domain fusions. Proc Natl Acad Sci USA 2000, 97, 5848–5853. [Google Scholar]
- Tanovic, A; Samel, SA; Essen, LO; Marahiel, MA. Crystal structure of the termination module of a nonribosomal peptide synthetase. Science 2008, 321, 659–663. [Google Scholar]
- Yonus, H; Neumann, P; Zimmermann, S; May, JJ; Marahiel, MA; Stubbs, MT. Crystal structure of DltA. Implications for the reaction mechanism of non-ribosomal peptide synthetase adenylation domains. J Biol Chem 2008, 283, 32484–32491. [Google Scholar]
- Koglin, A; Lohr, F; Bernhard, F; Rogov, VV; Frueh, DP; Strieter, ER; Mofid, MR; Guntert, P; Wagner, G; Walsh, CT; Marahiel, MA; Dotsch, V. Structural basis for the selectivity of the external thioesterase of the surfactin synthetase. Nature 2008, 454, 907–911. [Google Scholar]
- Frueh, DP; Arthanari, H; Koglin, A; Vosburg, DA; Bennett, AE; Walsh, CT; Wagner, G. Dynamic thiolation-thioesterase structure of a non-ribosomal peptide synthetase. Nature 2008, 454, 903–906. [Google Scholar]
- Lai, JR; Fischbach, MA; Liu, DR; Walsh, CT. A protein interaction surface in nonribosomal peptide synthesis mapped by combinatorial mutagenesis and selection. Proc Natl Acad Sci USA 2006, 103, 5314–5319. [Google Scholar]
- Watanabe, K; Hotta, K; Praseuth, AP; Koketsu, K; Migita, A; Boddy, CN; Wang, CC; Oguri, H; Oikawa, H. Total biosynthesis of antitumor nonribosomal peptides in Escherichia coli. Nat Chem Biol 2006, 2, 423–428. [Google Scholar]
- Miyahisa, I; Kaneko, M; Funa, N; Kawasaki, H; Kojima, H; Ohnishi, Y; Horinouchi, S. Efficient production of (2S)-flavanones by Escherichia coli containing an artificial biosynthetic gene cluster. Appl Microbiol Biotechnol 2005, 68, 498–504. [Google Scholar]
- Ma, SM; Li, JW; Choi, JW; Zhou, H; Lee, KK; Moorthie, VA; Xie, X; Kealey, JT; Da Silva, NA; Vederas, JC; Tang, Y. Complete reconstitution of a highly reducing iterative polyketide synthase. Science 2009, 326, 589–592. [Google Scholar]
- Santra, A; Chowdhury, A; Ghatak, S; Biswas, A; Dhali, GK. Arsenic induces apoptosis in mouse liver is mitochondria dependent and is abrogated by N-acetylcysteine. Toxicol Appl Pharmacol 2007, 220, 146–155. [Google Scholar]
- Puerto, M; Prieto, AI; Pichardo, S; Moreno, I; Jos, A; Moyano, R; Camean, AM. Effects of dietary N-acetylcysteine on the oxidative stress induced in tilapia (Oreochromis Niloticus) exposed to a microcystin-producing cyanobacterial water bloom. Environ Toxicol Chem 2009, 28, 1679–1686. [Google Scholar]
- Kortsalioudaki, C; Taylor, RM; Cheeseman, P; Bansal, S; Mieli-Vergani, G; Dhawan, A. Safety and efficacy of N-acetylcysteine in children with non-acetaminophen-induced acute liver failure. Liver Transpl 2008, 14, 25–30. [Google Scholar]
- Terneus, MV; Brown, JM; Carpenter, AB; Valentovic, MA. Comparison of S-adenosyl-L-methionine (SAMe) and N-acetylcysteine (NAC) protective effects on hepatic damage when administered after acetaminophen overdose. Toxicology 2008, 244, 25–34. [Google Scholar]
- Manov, I; Hirsh, M; Iancu, TC. N-acetylcysteine does not protect HepG2 cells against acetaminophen-induced apoptosis. Basic Clin Pharmacol Toxicol 2004, 94, 213–225. [Google Scholar]
- Campos, A; Vasconcelos, V. Molecular mechanisms of microcystin toxicity in animal cells. Int J Mol Sci 2010, 11, 268–287. [Google Scholar]
- Trachootham, D; Alexandre, J; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach. Nat Rev Drug Discov 2009, 8, 579–591. [Google Scholar]
- Pelicano, H; Carney, D; Huang, P. ROS stress in cancer cells and therapeutic implications. Drug Resist Updat 2004, 7, 97–110. [Google Scholar]
- Schumacker, PT. Reactive oxygen species in cancer cells: live by the sword, die by the sword. Cancer Cell 2006, 10, 175–176. [Google Scholar]
- Trachootham, D; Zhou, Y; Zhang, H; Demizu, Y; Chen, Z; Pelicano, H; Chiao, PJ; Achanta, G; Arlinghaus, RB; Liu, J; Huang, P. Selective killing of oncogenically transformed cells through a ROS-mediated mechanism by beta-phenylethyl isothiocyanate. Cancer Cell 2006, 10, 241–252. [Google Scholar]
- Treiber, A; Schneiter, R; Hausler, S; Stieger, B. Bosentan is a substrate of human OATP1B1 and OATP1B3: inhibition of hepatic uptake as the common mechanism of its interactions with cyclosporin A, rifampicin, and sildenafil. Drug Metab Dispos 2007, 35, 1400–1407. [Google Scholar]
- He, YJ; Zhang, W; Chen, Y; Guo, D; Tu, JH; Xu, LY; Tan, ZR; Chen, BL; Li, Z; Zhou, G; Yu, BN; Kirchheiner, J; Zhou, HH. Rifampicin alters atorvastatin plasma concentration on the basis of SLCO1B1 521T>C polymorphism. Clin Chim Acta 2009, 405, 49–52. [Google Scholar]
- Ogasawara, MA; Zhang, H. Redox regulation and its emerging roles in stem cells and stem-like cancer cells. Antioxid Redox Signal 2009, 11, 1107–1122. [Google Scholar]
- Ito, K; Oleschuk, CJ; Westlake, C; Vasa, MZ; Deeley, RG; Cole, SP. Mutation of Trp1254 in the multispecific organic anion transporter, multidrug resistance protein 2 (MRP2) (ABCC2), alters substrate specificity and results in loss of methotrexate transport activity. J Biol Chem 2001, 276, 38108–38114. [Google Scholar]
- Klaassen, CD; Lu, H. Xenobiotic transporters: ascribing function from gene knockout and mutation studies. Toxicol Sci 2008, 101, 186–196. [Google Scholar]
- Janga, SC; Tzakos, A. Structure and organization of drug-target networks: insights from genomic approaches for drug discovery. Mol Biosyst 2009, 5, 1536–1548. [Google Scholar]
- Barglow, KT; Cravatt, BF. Activity-based protein profiling for the functional annotation of enzymes. Nat Methods 2007, 4, 822–827. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Approved gene symbol | Expression in human normal tissue | Expression in human tumor tissue |
|---|---|---|
| SLCO1A2 | Liver (m; P), Brain (m), blood barrier (P), Kidney (m; P), Testis (m), Prostate (m), Breast (m), Retina (m) | Glioma (m;P); Bone tumors (m) |
| SLCO1B1 | Liver (m; P), Mononuclear cells (m) | HCC (m; P); Colorectal Cancer (m) |
| SLCO1B3 | Liver (m; P), Cervix (m), Mononuclear cells (m) | Colon Cancer (m; P); Breast cancer (P); Non Small Cell Lung Cancer (m); HCC (m; P) |
| SLCO1C1 | Brain (m); Testis (m; P), Heart (m), Retina (m), Breast (m) | Glioma (m); Bone tumors (m) |
| SLCO2A1 | Ubiquitous (protein detected only in GI tract tissue) | Colon cancer (m); Lung cancer (m); Bone tumor (m), Breast cancer (m) |
| SLCO2B1 | Ubiquitous (protein detected only in liver tissue) | Glioma (m; P); Colon cancer (m); Lung cancer (m); Bone tumors (m); Breast cancer (m) |
| SLCO3A1 | Ubiquitous and also in Peripheral Blood Mononuclear Cells (PBMC) (data available only on mRNA level) | Lung cancer (m); Colon cancer (m); Bone tumors (m); Breast cancer (m) |
| SLCO4A1 | Ubiquitous (protein detected only in brain and placenta tissues) | Glioma (m); Lung cancer (m); colon cancer(m), Bone tumor (m); Breast cancer (m) |
| SLCO4C1 | Kidney (m), Lung (m), Skin (m), PBMC (m), Kidney (m), Liver (m), Neutrophils (m), Breast (m), peripheral leukocytes (m) | Lung cancer (m); Bone tumor (m); Breast cancer (m) |
| SLCO5A1 | Prostate (m), Skeletal muscles (m), Thymus (m), Classically activated macrophages (m), Breast (m). | Bone tumors (m); Breast cancer (m) |
| SLCO6A1 | Testis (m), Spleen (m), Brain (m) (especially fetal brain), Placenta (m) | Non small cell lung cancer (m); Bladder cancer (m); Esophagus cancer (m); medulloblastoma (m) |
 | |||||||
|---|---|---|---|---|---|---|---|
| Inhibition (IC50 nm) | |||||||
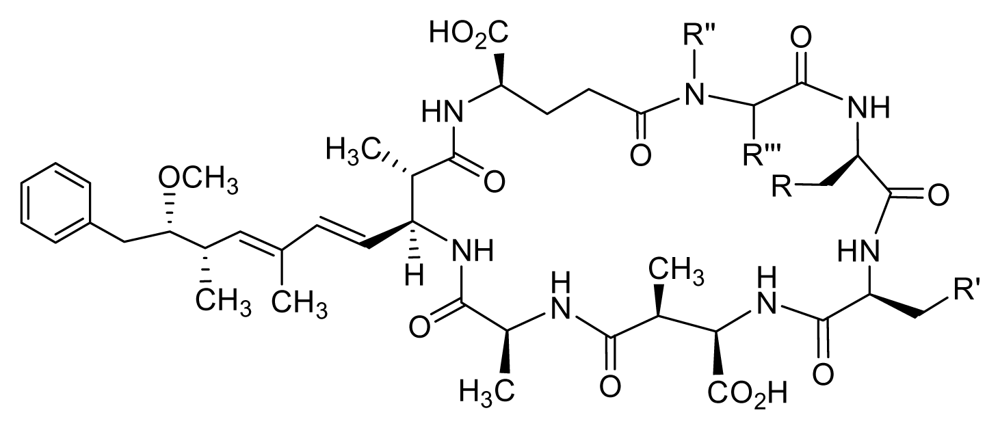
| Inhibitor | R | R′ | R″ | R‴ | PP1c | PP2Ac | PP1 selectivity |
| MC-LA (synthetic) | H | CH(CH3)2 | CH3 | =CH2 | 0.3 | 0.3 | 1 |
| 1 | H | Cyclohexyl | CH3 | CH2 | 0.52 | 3.4 | 7 |
| 2 | H | i-Propyl | Cyclohexyl | H | 0.8 | 1.5 | 2 |
| 3 | H | i-Propyl | CH3 | H | 0.8 | 1.5 | 2 |
| 4 | NH3+ | i-Propyl | CH3 | CH2 | 3 | 9 | 3 |
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sainis, I.; Fokas, D.; Vareli, K.; Tzakos, A.G.; Kounnis, V.; Briasoulis, E. Cyanobacterial Cyclopeptides as Lead Compounds to Novel Targeted Cancer Drugs. Mar. Drugs 2010, 8, 629-657. https://doi.org/10.3390/md8030629
Sainis I, Fokas D, Vareli K, Tzakos AG, Kounnis V, Briasoulis E. Cyanobacterial Cyclopeptides as Lead Compounds to Novel Targeted Cancer Drugs. Marine Drugs. 2010; 8(3):629-657. https://doi.org/10.3390/md8030629
Chicago/Turabian StyleSainis, Ioannis, Demosthenes Fokas, Katerina Vareli, Andreas G. Tzakos, Valentinos Kounnis, and Evangelos Briasoulis. 2010. "Cyanobacterial Cyclopeptides as Lead Compounds to Novel Targeted Cancer Drugs" Marine Drugs 8, no. 3: 629-657. https://doi.org/10.3390/md8030629
APA StyleSainis, I., Fokas, D., Vareli, K., Tzakos, A. G., Kounnis, V., & Briasoulis, E. (2010). Cyanobacterial Cyclopeptides as Lead Compounds to Novel Targeted Cancer Drugs. Marine Drugs, 8(3), 629-657. https://doi.org/10.3390/md8030629