Biosynthesis and Molecular Genetics of Polyketides in Marine Dinoflagellates

Abstract
:1. Introduction
2. Dinoflagellates Have Peculiar Cellular and Genomic Features and an Evolutionary History of Multiple Endosymbioses
3. Precursor Incorporation Studies Reveal a Novel Mode of Polyketide Synthesis in Dinoflagellate
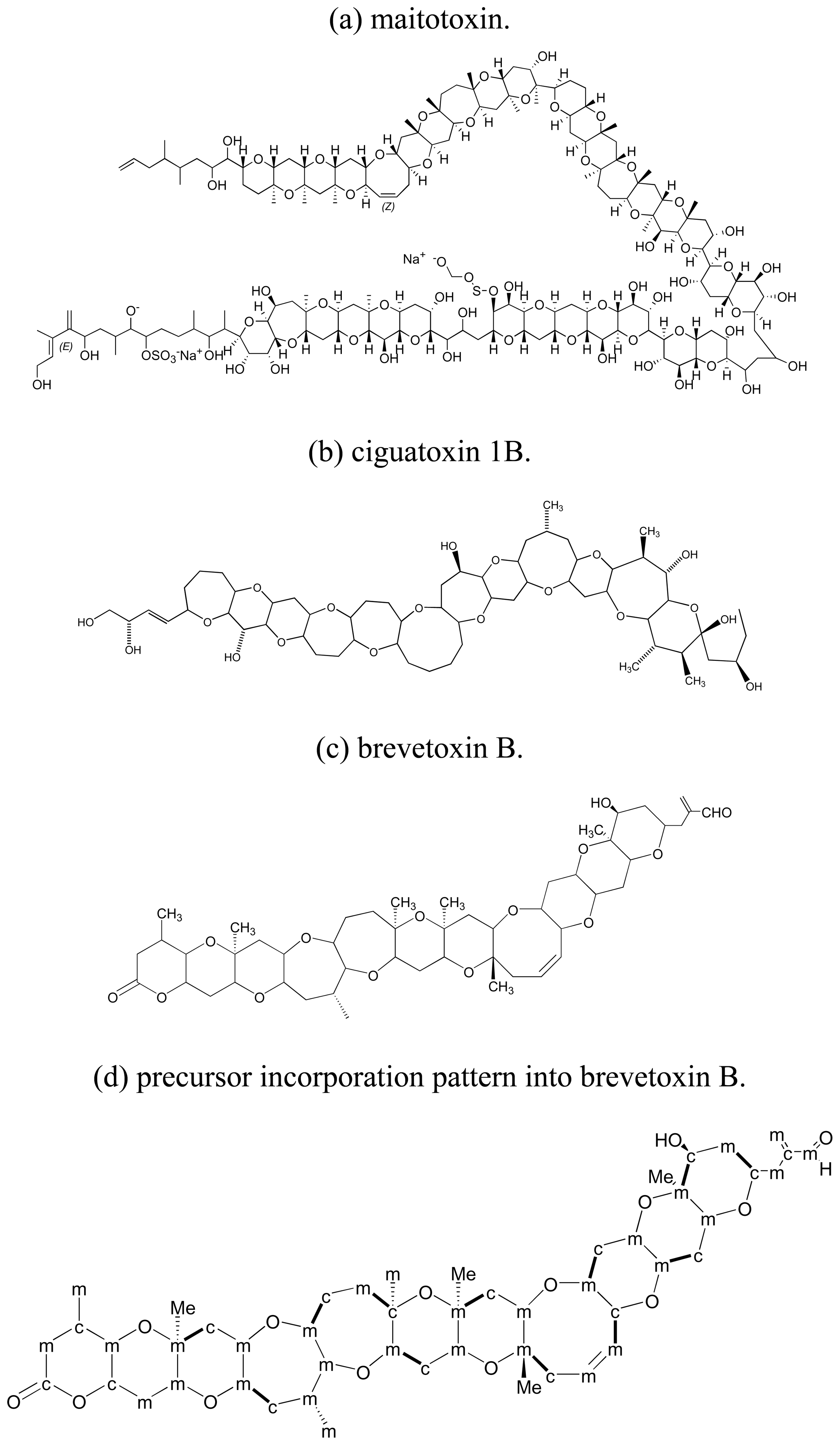
3.1. Polyether ladder toxins
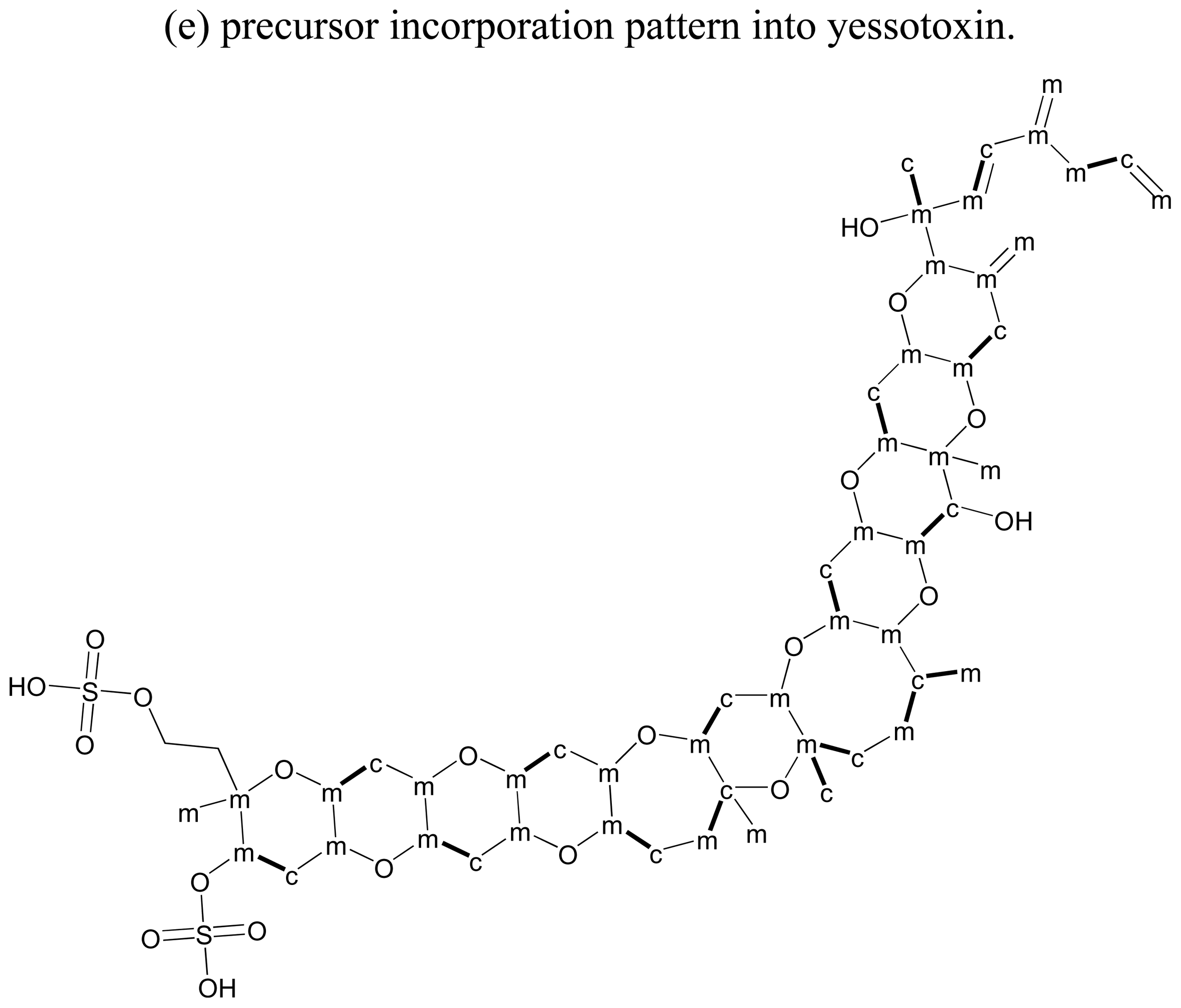
3.2. Precursor incorporation studies on polyether ladder toxins
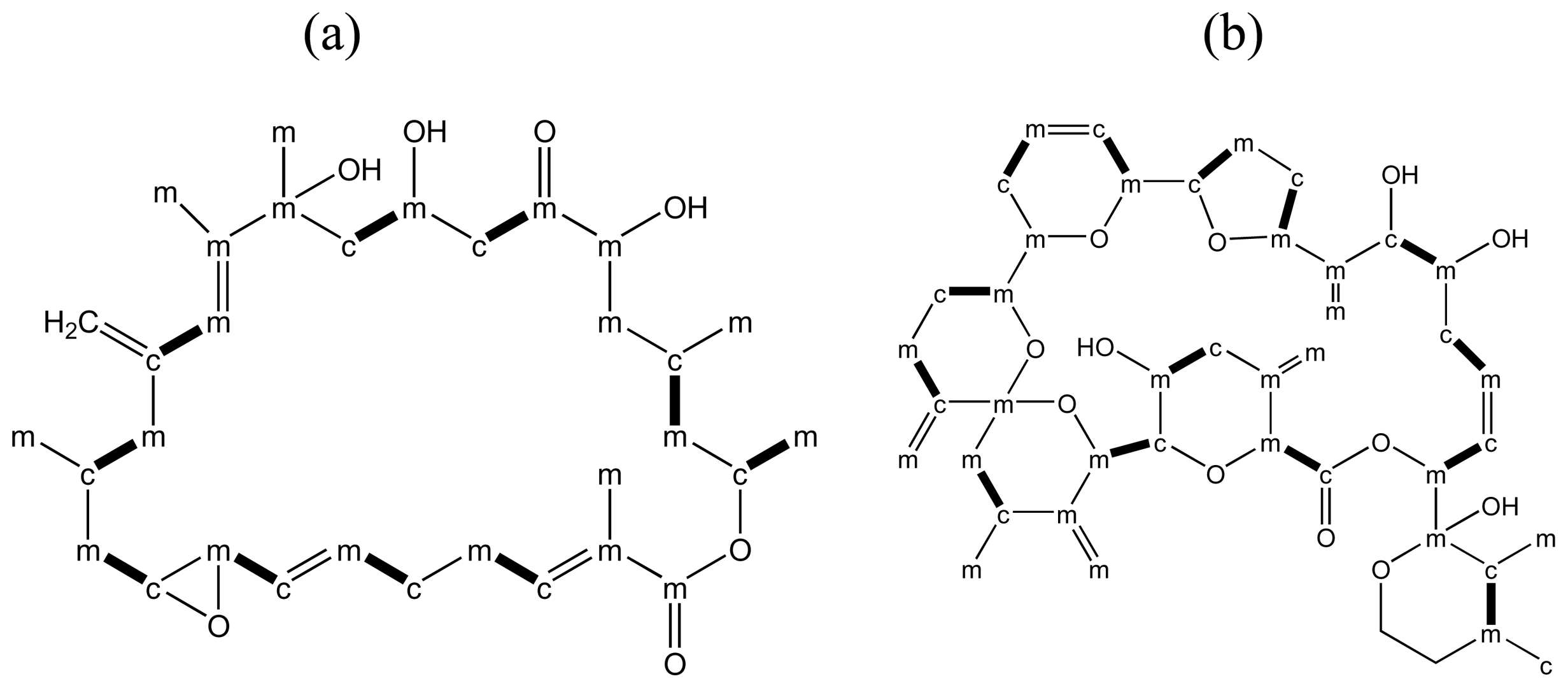
3.3. Linear polyether toxins
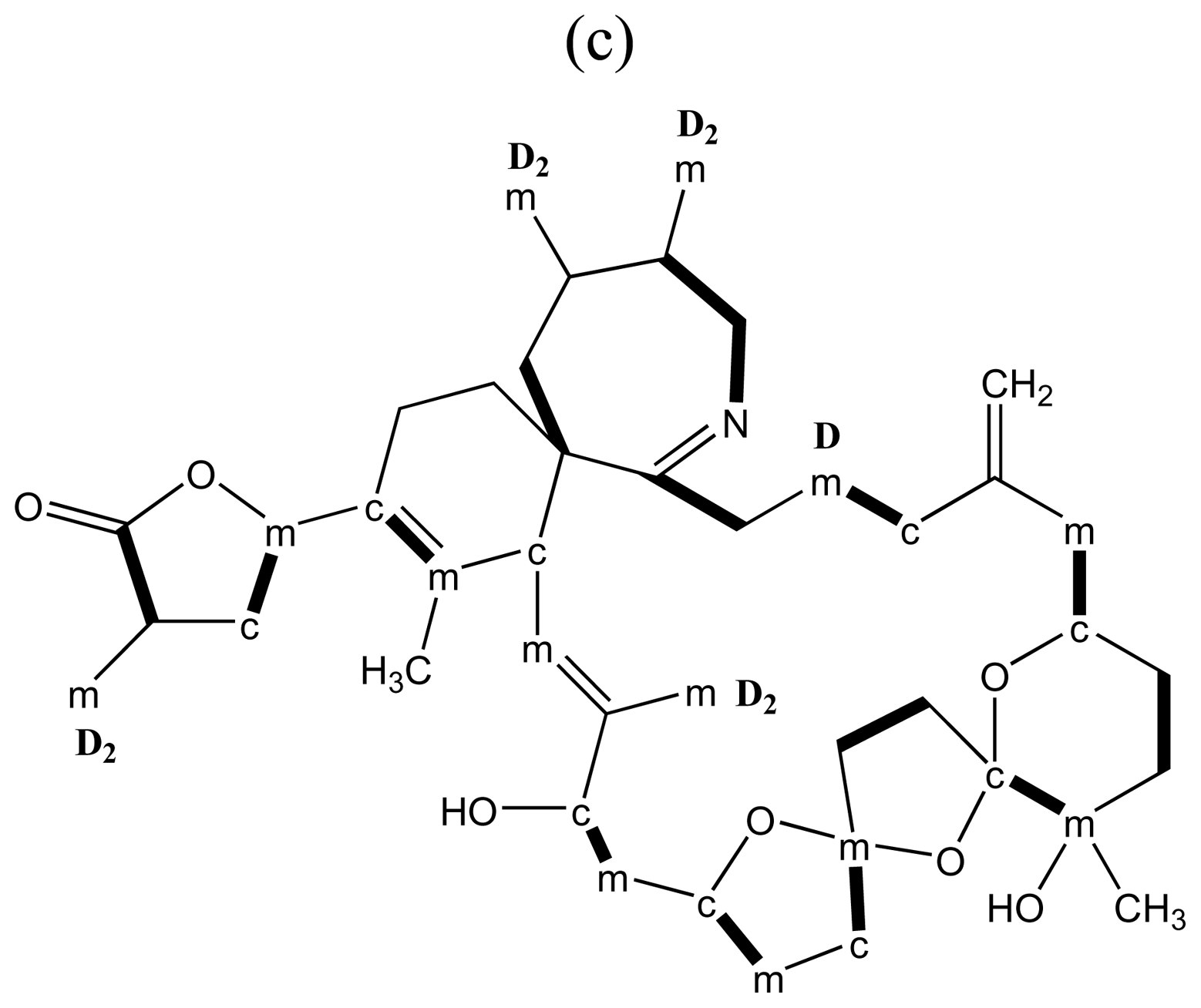
3.4. Macrolides
3.5. Dinoflagellates produce polyethers by a distinct polyketide pathway
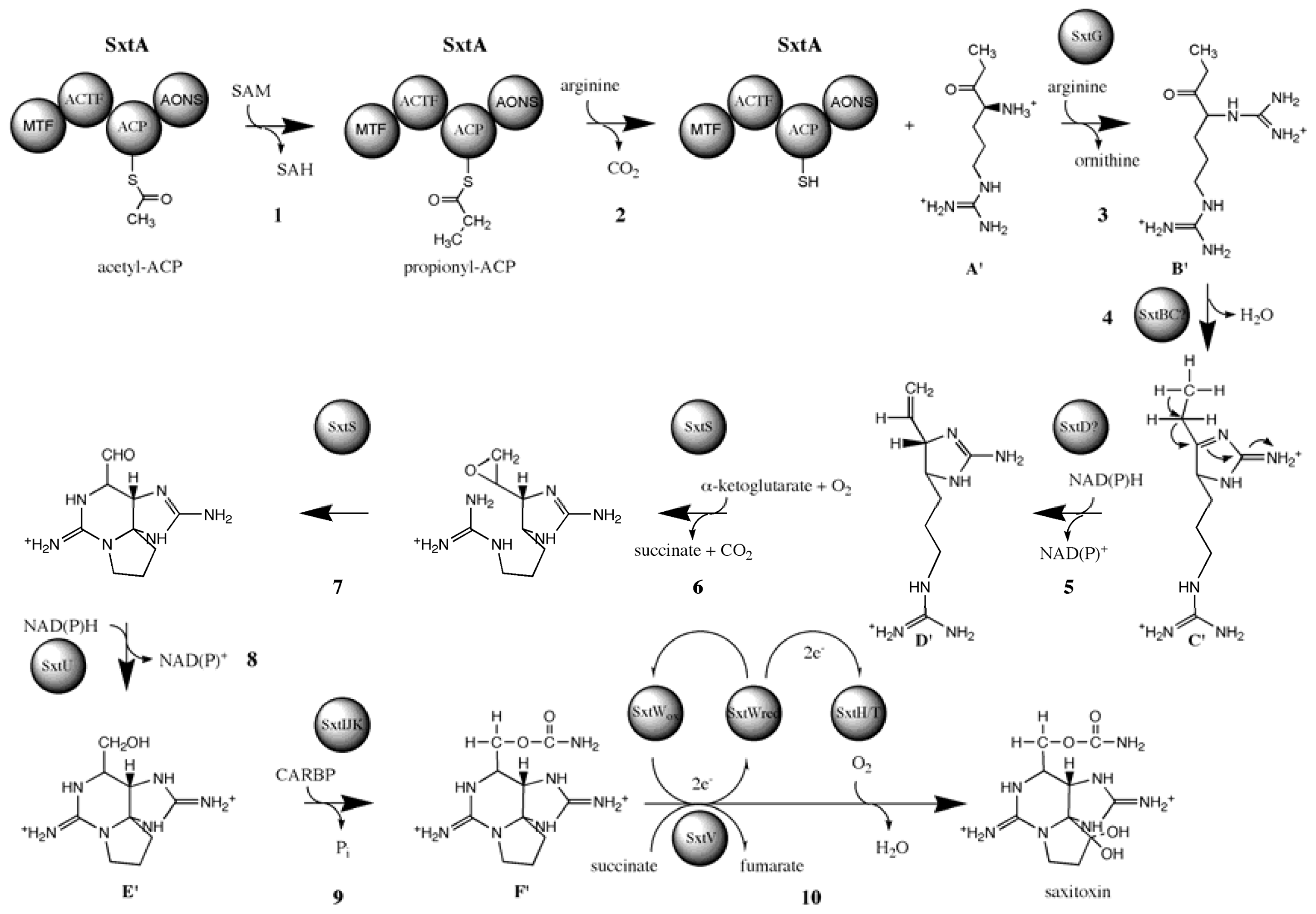
3.6. Biosynthesis of paralytic shellfish toxins
4. Phycotoxin Biosynthesis Genes
4.1. An attempt to identify putative amphidinolide biosynthesis genes on the genomic level
4.2. Brevetoxins biosynthesis genes
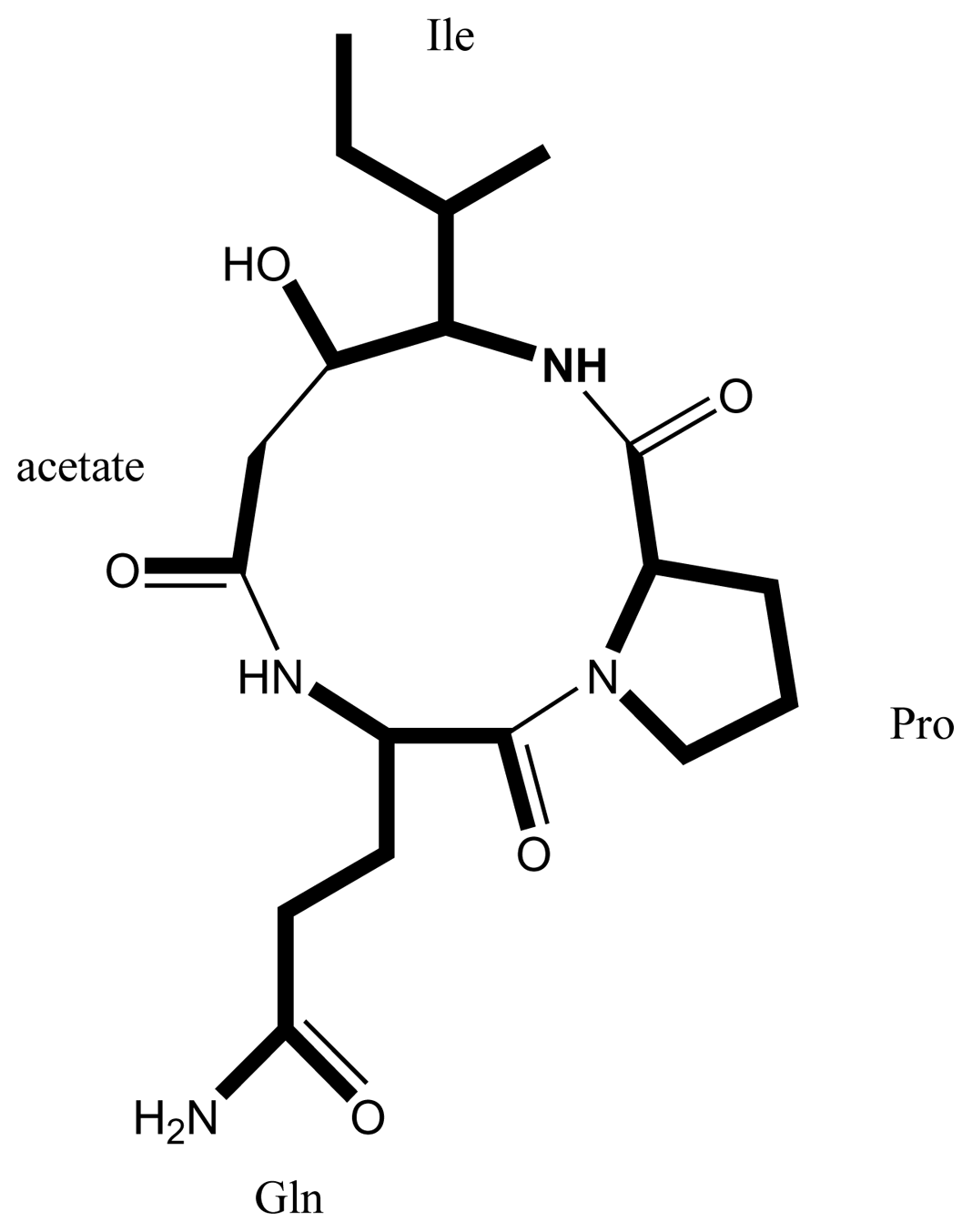
4.3. Identification of a hybrid NRPS/PKS gene cluster from Karenia brevis
4.4. Polyketide synthase genes from DSP-toxin producing dinoflagellates
4.5. Phylogenetic analysis of protist KS sequences
4.6. Biosynthesis genes and enzymes of paralytic shellfish toxins
5. A Search for sxt Genes in the Oceans
6. Possible Applications and Future Outlook
Acknowledgments
- Samples Availability: Available from the authors.
References and Notes
- Taylor, FJR. The Biology of Dinoflagellates; Blackwell Scientific Publications: Oxford, UK, 1987; p. 785. [Google Scholar]
- Hallegraeff, GM. A review of harmful algal blooms and their apparent global increase. Phycologia 1993, 32, 79–99. [Google Scholar]
- Anderson, DM. Red tides. Sci Am 1994, 271, 52–58. [Google Scholar]
- Hallegraeff, GM. Hallegraeff, GM, Anderson, DM, Cembella, AD, Eds.; Harmful algal blooms: A global overview. In Manual on Harmful Marine Microalgae; UNESCO: Paris, France, 1995; pp. 1–22. [Google Scholar]
- Wang, DZ. Neurotoxins from marine dinoflagellates: a brief review. Mar Drugs 2008, 6, 349–731. [Google Scholar]
- Rein, KS; Borrone, J. Polyketides from dinoflagellates: Origins, pharmacology and biosynthesis. Comp Biochem Physiol B Biochem Mol Biol 1999, 124, 117–131. [Google Scholar]
- Satake, M; Murata, M; Yasumoto, T; Fujita, T; Naoki, H. Amphidinol, a polyhydroxypolyene antifungal agent with an unprecedented structure, from a marine dinoflagellate Amphidinium klebsii. J Am Chem Soc 1991, 113, 9859–9861. [Google Scholar]
- Berry, JP; Reece, KS; Rein, KS; Baden, DG; Haas, LW; Ribeiro, WL; Shields, JD; Snyder, RV; Vogelbein, WK; Gawley, RE. Are Pfiesteria species toxicogenic? Evidence against production of ichthyotoxins by Pfiesteria shumwayae. Proc Natl Acad Sci USA 2002, 99, 10970–10975. [Google Scholar]
- Steidinger, KA; Burkholder, JM; Glasgow, HB, Jr; Hobbs, CW; Garrett, JK; Truby, EW; Noga, EJ; Smith, SA. Pfiesteria piscicida gen. et sp. nov. (Pfiesteriaceae fam. nov.), a new toxic dinoflagellate with a complex life cycle and behavior. J Phycol 1996, 32, 157–164. [Google Scholar]
- Moeller, PD; Beauchesne, KR; Huncik, KM; Davis, WC; Christopher, SJ; Riggs-Gelasco, P; Gelasco, AK. Metal complexes and free radical toxins produced by Pfiesteria piscicida. Environ Sci Technol 2007, 41, 1166–1172. [Google Scholar]
- Rein, KS; Snyder, RV. The biosynthesis of polyketide metabolites by dinoflagellates. Adv Appl Microbiol 2006, 59, 93–125. [Google Scholar]
- Blunt, JW; Copp, BR; Hu, WP; Munro, MH; Northcote, PT; Prinsep, MR. Marine natural products. Nat Prod Rep 2008, 25, 35–94. [Google Scholar]
- Kalaitzis, JA; Chau, R; Kohli, GS; Murray, SA; Neilan, BA. Biosynthesis of toxic naturally-occurring seafood contaminants. Toxicon 2009. [Google Scholar] [CrossRef]
- Robinson, JA. Polyketide synthase complexes: their structure and function in antibiotic biosynthesis. Philos Trans R Soc Lond B Biol Sci 1991, 332, 107–114. [Google Scholar]
- Moffitt, MC; Neilan, BA. Evolutionary affiliations within the superfamily of ketosynthases reflect complex pathway associations. J Mol Evol 2003, 56, 446–457. [Google Scholar]
- Silakowski, B; Nordsiek, G; Kunze, B; Blocker, H; Muller, R. Novel features in a combined polyketide synthase/non-ribosomal peptide synthetase: the myxalamid biosynthetic gene cluster of the myxobacterium Stigmatella aurantiaca Sga15. Chem Biol 2001, 8, 59–69. [Google Scholar]
- Van Lanen, SG; Shen, B. Advances in polyketide synthase structure and function. Curr Opin Drug Discov Devel 2008, 11, 186–195. [Google Scholar]
- Müller, R. Don’t classify polyketide synthases. Chem Biol 2004, 11, 4–6. [Google Scholar]
- Sattely, ES; Fischbach, MA; Walsh, CT. Total biosynthesis: in vitro reconstitution of polyketide and nonribosomal peptide pathways. Nat Prod Rep 2008, 25, 757–793. [Google Scholar]
- Callahan, B; Thattai, M; Shraiman, BI. Emergent gene order in a model of modular polyketide synthases. Proc Natl Acad Sci USA 2009, 106, 19410–19415. [Google Scholar]
- Shen, B. Polyketide biosynthesis beyond the type I, II and III polyketide synthase paradigms. Curr Opin Chem Biol 2003, 7, 285–295. [Google Scholar]
- Weissman, KJ; Muller, R. A brief tour of myxobacterial secondary metabolism. Bioorg Med Chem 2009, 17, 2121–2136. [Google Scholar]
- Kalaitzis, JA; Lauro, FM; Neilan, BA. Mining cyanobacterial genomes for genes encoding complex biosynthetic pathways. Nat Prod Rep 2009, 26, 1447–1465. [Google Scholar]
- Tengs, T; Dahlberg, OJ; Shalchian-Tabrizi, K; Klaveness, D; Rudi, K; Delwiche, CF; Jakobsen, KS. Phylogenetic analyses indicate that the 19′Hexanoyloxy-fucoxanthin-containing dinoflagellates have tertiary plastids of haptophyte origin. Mol Biol Evol 2000, 17, 718–729. [Google Scholar]
- Shalchian-Tabrizi, K; Minge, MA; Cavalier-Smith, T; Nedreklepp, J; Klaveness, D; Jakobsen, KS. Combined Hsp90 and rRNA sequence phylogeny supports multiple replacements of dinoflagellate plastids. J Eukaryot Microbiol 2006, 53, 217–224. [Google Scholar]
- Koike, K; Sekiguchi, H; Kobiyama, A; Takishita, K; Kawachi, M; Ogata, T. A novel type of kleptoplastidy in Dinophysis (Dinophyceae): presence of haptophyte-type plastid in Dinophysis mitra. Protist 2005, 156, 225–237. [Google Scholar]
- Minnhagen, S; Janson, S. Genetic analyses of Dinophysis spp. support kleptoplastidy. FEMS Microbiol Ecol 2006, 57, 47–54. [Google Scholar]
- Burki, F; Inagaki, Y; Bråte, J; Archibald, JM; Keeling, PJ; Cavalier-Smith, T; Sakaguchi, M; Hashimoto, T; Horak, A; Kumar, S; Klaveness, D; Jakobsen, KJ; Pawlowski, J; Shalchian-Tabrizi, K. Large-scale phylogenomic analyses reveal that two enigmatic protist lineages, Telonemia and Centroheliozoa, are related to photosynthetic chromalveolates. Genome Biol Evol 2009, 231–238. [Google Scholar]
- Burki, F; Shalchian-Tabrizi, K; Minge, M; Skjaeveland, A; Nikolaev, SI; Jakobsen, KS; Pawlowski, J. Phylogenomics reshuffles the eukaryotic supergroups. PLoS One 2007, 2, e790. [Google Scholar]
- Archibald, JM. Genomics: Green evolution, green revolution. Science 2009, 324, 191–192. [Google Scholar]
- Hampla, V; Huga, L; Leigha, JW; Dacksd, JB; Lang, F; Simpson, A; Rogera, AJ. Phylogenomic analyses support the monophyly of Excavata and resolve relationships among eukaryotic “supergroups”. Proc Natl Acad Sci USA 2009, 106, 3859–3864. [Google Scholar]
- Shalchian-Tabrizi, K; Skanseng, M; Ronquist, F; Klaveness, D; Bachvaroff, TR; Delwiche, CF; Botnen, A; Tengs, T; Jakobsen, KS. Heterotachy processes in rhodophyte-derived secondhand plastid genes: Implications for addressing the origin and evolution of dinoflagellate plastids. Mol Biol Evol 2006, 23, 1504–1515. [Google Scholar]
- Patron, NJ; Waller, RF; Keeling, PJ. A tertiary plastid uses genes from two endosymbionts. J Mol Biol 2006, 357, 1373–1382. [Google Scholar]
- Nosenko, T; Lidie, KL; Van Dolah, FM; Lindquist, E; Cheng, JF; Bhattacharya, D. Chimeric plastid proteome in the Florida “red tide” dinoflagellate Karenia brevis. Mol Biol Evol 2006, 23, 2026–2038. [Google Scholar]
- Minge, MA; Shalchian-Tabrizi, K; Tørresen, OK; Takishita, K; Probert, I; Inagaki, Y; Klaveness, D. A phylogenetic mosaic plastid proteome and unusual plastid-targeting signals in the green-colored dinoflagellate Lepidodinium chlorophorum. BMC Evol Biol 2010, in press. [Google Scholar]
- Keeling, PJ; Palmer, JD. Horizontal gene transfer in eukaryotic evolution. Nat Rev Genet 2008, 9, 605–618. [Google Scholar]
- Koumandou, VL; Nisbet, RE; Barbrook, AC; Howe, CJ. Dinoflagellate chloroplasts--where have all the genes gone. Trends Genet 2004, 20, 261–267. [Google Scholar]
- Dodge, JD. The Dinophyceae: The Chromosomes of the Algae; St Martins Press: New York, NY, USA, 1966; pp. 96–115. [Google Scholar]
- Veldhuis, MJW; Cucci, TL; Sieracki, ME. Cellular DNA content of marine phytoplankton using two new fluorochromes: Taxonomic and ecological implications. J Phycol 1997, 33, 527–541. [Google Scholar]
- Rae, PM. Hydroxymethyluracil in eukaryote DNA: a natural feature of the pyrrophyta (dinoflagellates). Science 1976, 194, 1062–1064. [Google Scholar]
- Rae, PMM; Steele, RE. Modified Bases in DNAs of Unicellular Eukaryotes-Examination of Distributions and Possible Roles, with Emphasis on Hydroxy-Methyl-Uracil in Dinoflagellates. Biosystems 1978, 10, 37–53. [Google Scholar]
- Davies, W; Jakobsen, KS; Nordby, O. Characterization of DNA from the Dinoflagellate Woloszynskia-Bostoniensis. J Protozool 1988, 35, 418–422. [Google Scholar]
- Boorstein, RJ; Cummings, AJ; Marenstein, DR; Chan, MK; Ma, Y; Neubert, TA; Brown, SM; Teebor, GW. Definitive identification of mammalian 5-hydroxymethyluracil DNA N-glycosylase activity as SMUG1. J Biol Chem 2001, 276, 41991–41997. [Google Scholar]
- Zhang, Z; Green, BR; Cavalier-Smith, T. Single gene circles in dinoflagellate chloroplast genomes. Nature 1999, 400, 155–159. [Google Scholar]
- Nisbet, RE; Hiller, RG; Barry, ER; Skene, P; Barbrook, AC; Howe, CJ. Transcript analysis of dinoflagellate plastid gene minicircles. Protist 2008, 159, 31–39. [Google Scholar]
- Bachvaroff, TR; Place, AR. From stop to start: tandem gene arrangement, copy number and trans-splicing sites in the dinoflagellate Amphidinium carterae. PLoS One 2008, 3, e2929. [Google Scholar]
- Zhang, H; Hou, Y; Miranda, L; Campbell, DA; Sturm, NR; Gaasterland, T; Lin, S. Spliced leader RNA trans-splicing in dinoflagellates. Proc Natl Acad Sci USA 2007, 104, 4618–4623. [Google Scholar]
- Lidie, KB; van Dolah, FM. Spliced leader RNA-mediated trans-splicing in a dinoflagellate, Karenia brevis. J Eukaryot Microbiol 2007, 54, 427–435. [Google Scholar]
- Douris, V; Telford, MJ; Averof, M. Evidence for multiple independent origins of trans-splicing in Metazoa. Mol Biol Evol 2009. [Google Scholar] [CrossRef]
- Zhang, H; Lin, S. Retrieval of missing spliced leader in dinoflagellates. PLoS One 2009, 4, e4129. [Google Scholar]
- Slamovits, CH; Keeling, PJ. Widespread recycling of processed cDNAs in dinoflagellates. Curr Biol 2008, 18, R550–552. [Google Scholar]
- Hou, Y; Lin, S. Distinct gene number-genome size relationships for eukaryotes and non-eukaryotes: gene content estimation for dinoflagellate genomes. PLoS One 2009, 4, e6978. [Google Scholar]
- Lin, S; Zhang, H; Gray, MW. Smith, HC, Ed.; RNA editing in dinoflagellates and its implications for the evolutionary history of the editing machinery. In RNA and DNA Editing: Molecular Mechanisms and Their Integration into Biological Systems; John Wiley & Sons, Inc: Hoboken, NJ, USA, 2008; pp. 280–309. [Google Scholar]
- Lin, SJ; Zhang, HA; Spencer, DF; Norman, JE; Gray, MW. Widespread and extensive editing of mitochondrial mRNAs in dinoflagellates. J Mol Biol 2002, 320, 727–739. [Google Scholar]
- Ting, JY; Brown, AF. Ciguatera poisoning: a global issue with common management problems. Eur J Emerg Med 2001, 8, 295–300. [Google Scholar]
- Swift, AE; Swift, TR. Ciguatera. J Toxicol Clin Toxicol 1993, 31, 1–29. [Google Scholar]
- Satake, M; Murata, M; Yasumoto, T. The structure of CTX3C, a ciguatoxin congener isolated from cultured Gambierdiscus toxicus. Tetrahedron Lett 1993, 34, 1975–1978. [Google Scholar]
- Gillespie, NC; Lewis, RJ; Pearn, JH; Bourke, AT; Holmes, MJ; Bourke, J. Ciguatera in Australia. Occurrence, clinicalfeatures, pathophysiology and management. Med J Aust 1986, 145, 584–590. [Google Scholar]
- Zheng, WJ; DeMattei, JA; Wu, JP; Duan, JJW; Cook, LR; Oinuma, H; Kishi, Y. Complete relative stereochemistry of maitotoxin. J Am Chem Soc 1996, 118, 7946–7968. [Google Scholar]
- Gusovsky, F; Daly, JW. Maitotoxin: a unique pharmacological tool for research on calcium-dependent mechanisms. Biochem Pharmacol 1990, 39, 1633–1639. [Google Scholar]
- Soergel, DG; Yasumoto, T; Daly, JW; Gusovsky, F. Maitotoxin effects are blocked by SK-and-F-96365, an inhibitor of receptor-mediated calcium entry. Mol Pharmacol 1992, 41, 487–493. [Google Scholar]
- Takahashi, M; Ohizumi, Y; Yasumoto, T. Maitotoxin, a Ca2+ channel activator candidate. J Biol Chem 1982, 257, 7287–7289. [Google Scholar]
- Holmes, MJ; Lewis, RJ; Poli, MA; Gillespie, NC. Strain dependent production of ciguatoxin precursors (gambiertoxins) by Gambierdiscus toxicus (Dinophyceae) in culture. Mem Queensl Mus 1994, 34, 447–453. [Google Scholar]
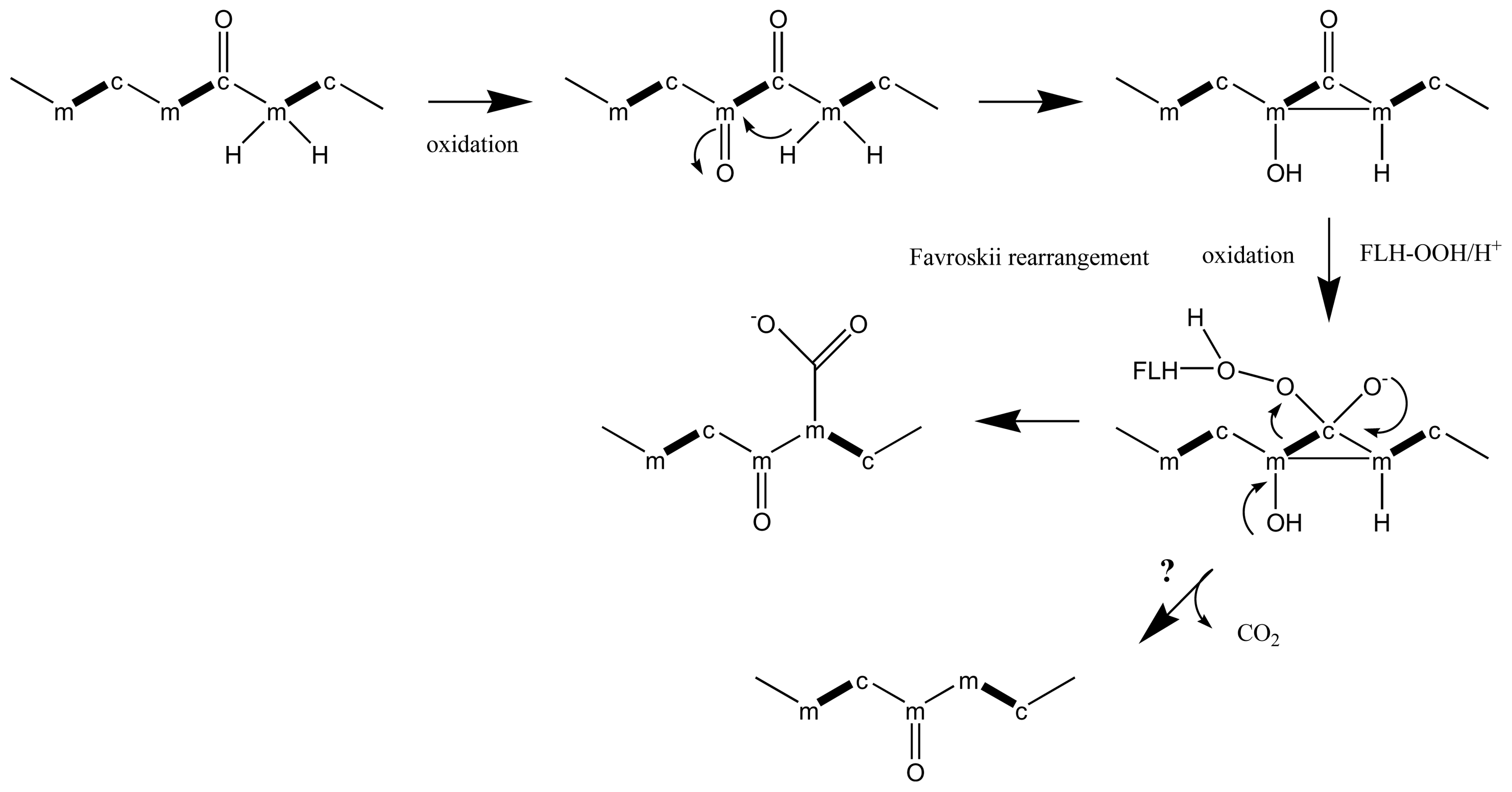
- Chou, HN; Shimizu, Y. Biosynthesis of brevetoxins. Evidence for the mixed origin of the backbone carbon chain and possible involvement of dicarboxylic acids. J Am Chem Soc 1987, 109, 2184–2185. [Google Scholar]
- Satake, M. Biosynthesis of the marine polyether toxin, yessotoxin. Symp Chem Nat Prod 2000, 42, 259–264. [Google Scholar]
- Baden, DG. Brevetoxins-unique polyether dinoflagellate toxins. FASEB J 1989, 3, 1807–1817. [Google Scholar]
- van Dolah, MF; Lidie, KB; Monroe, EA; Bhattacharya, D; Campbell, L; Doucette, GJ; Kamykowski, D. The Florida red tide dinoflagellate Karenia brevis: New insights into cellular and molecular processes underlying bloom dynamics. Harmful Algae 2009, 8, 562–572. [Google Scholar]
- Lin, YY; Risk, M. Isolation and structure of brevetoxin B from the “red tide” dinoflagellate Ptychodiscus brevis (Gymnodinium breve). J Am Chem Soc 1981, 103, 6773–6775. [Google Scholar]
- Ogino, H; Kumagai, M; Yasumoto, T. Toxicologic evaluation of yessotoxin. Nat Toxins 1997, 5, 255–259. [Google Scholar]
- Terao, K; Ito, E; Oarada, M; Murata, M; Yasumoto, T. Histopathological studies on experimental marine toxin poisoning--5. The effects in mice of yessotoxin isolated from Patinopecten yessoensis and of a desulfated derivative. Toxicon 1990, 28, 1095–1104. [Google Scholar]
- Bowden, BF. Yessotoxins-polycyclic ethers from dinoflagellates: Relationships to diarrhetic shellfish toxins. Toxin Rev 2006, 25, 137–157. [Google Scholar]
- Eiki, K; Satake, M; Koike, K; Ogata, T; Mitsuya, T; Oshima, Y. Confirmation of yessotoxin production by the dinoflagellate Protoceratium reticulatum in Mutsu Bay. Fish Sci 2005, 71, 633–638. [Google Scholar]
- Draisci, R; Ferretti, E; Palleschi, L; Marchiafava, C; Poletti, R; Milandri, A; Ceredi, A; Pompei, M. High levels of yessotoxin in mussels and presence of yessotoxin and homoyessotoxin in dinoflagellates of the Adriatic Sea. Toxicon 1999, 37, 1187–1193. [Google Scholar]
- Rhodes, L; McNabb, P; de Salas, M; Briggs, L; Beuzenberg, V; Gladstone, M. Yessotoxin production by Gonyaulax spinifera. Harmful Algae 2006, 5, 148–155. [Google Scholar]
- Chou, HN; Shimizu, Y; Duyne, GV; Clardy, J. Isolation and structures of two new polycyclic ethers from Gymnodinium breve Davis (Ptychodiscus brevis). Tetrahedron Lett 1985, 26, 2865–2868. [Google Scholar]
- Golik, J; James, JC; Nakanishi, K; Lin, YY. The structure of brevetoxin C. Tetrahedron Lett 1982, 23, 2535–2538. [Google Scholar]
- Lee, MS; Qin, GW; Nakanishi, K; Zagorski, MG. Biosynthesis Studies of Brevetoxins, Potent Neurotoxins Produced by the Dinoflagellate Gymnodinium breve. J Am Chem Soc 1989, 111, 6234–6241. [Google Scholar]
- Nakanishi, K. The chemistry of brevetoxins - a review. Toxicon 1985, 23, 473–479. [Google Scholar]
- Xiang, L; Kalaitzis, JA; Moore, BS. EncM, a versatile enterocin biosynthetic enzyme involved in Favorskii oxidative rearrangement, aldol condensation, and heterocycle-forming reactions. Proc Natl Acad Sci USA 2004, 101, 15609–5614. [Google Scholar]
- Julien, B; Tian, ZQ; Reid, R; Reeves, CD. Analysis of the ambruticin and jerangolid gene clusters of Sorangium cellulosum reveals unusual mechanisms of polyketide biosynthesis. Chem Biol 2006, 13, 1277–1286. [Google Scholar]
- Tsuda, M; Kubota, T; Sakuma, Y; Kobayashi, J. Biosynthetic study of amphidinolide B. Chem Pharm Bull 2001, 49, 1366–1367. [Google Scholar]
- Murakami, M; Okita, Y; Matsuda, H; Okino, T; Yamaguchi, K. From the dinoflagellate Alexandrium hiranoi. Phytochemistry 1998, 48, 85–88. [Google Scholar]
- MacKinnon, SL; Cembella, AD; Burton, IW; Lewis, N; LeBlanc, P; Walter, JA. Biosynthesis of 13-desmethyl spirolide C by the dinoflagellate Alexandrium ostenfeldii. J Org Chem 2006, 71, 8724–8731. [Google Scholar]
- Satake, M; Bourdelais, AJ; Van Wagoner, RM; Baden, DG; Wright, JL. Brevisamide: an unprecedented monocyclic ether alkaloid from the dinoflagellate Karenia brevis that provides a potential model for ladder-frame initiation. Org Lett 2008, 10, 3465–3468. [Google Scholar]
- Vilotijevic, I; Jamison, TF. Epoxide-opening cascades promoted by water. Science 2007, 317, 1189–1192. [Google Scholar]
- Leadlay, PF; Staunton, J; Oliynyk, M; Bisang, C; Cortes, J; Frost, E; Hughes-Thomas, ZA; Jones, MA; Kendrew, SG; Lester, JB; Long, PF; McArthur, HA; McCormick, EL; Oliynyk, Z; Stark, CB; Wilkinson, CJ. Engineering of complex polyketide biosynthesis--insights from sequencing of the monensin biosynthetic gene cluster. J Ind Microbiol Biotechnol 2001, 27, 360–367. [Google Scholar]
- Vilotijevic, I; Jamison, TF. Synthesis of marine polycyclic polyethers via epoxide-opening cascades. Mar Drugs 2010, 8, 763–809. [Google Scholar]
- Lopez-Legentil, S; Song, B; Deture, M; Baden, DG. Characterization and Localization of a Hybrid Non-ribosomal Peptide Synthetase and Polyketide Synthase Gene from the Toxic Dinoflagellate Karenia brevis. Mar Biotechnol (NY) 2010, 12, 32–41. [Google Scholar]
- Shimizu, Y. Microalgal metabolites. Curr Opin Microbiol 2003, 6, 236–243. [Google Scholar]
- Bialojan, C; Takai, A. Inhibitory effect of a marine-sponge toxin, okadaic acid, on protein phosphatases - specificity and kinetics. Biochem J 1988, 256, 283–290. [Google Scholar]
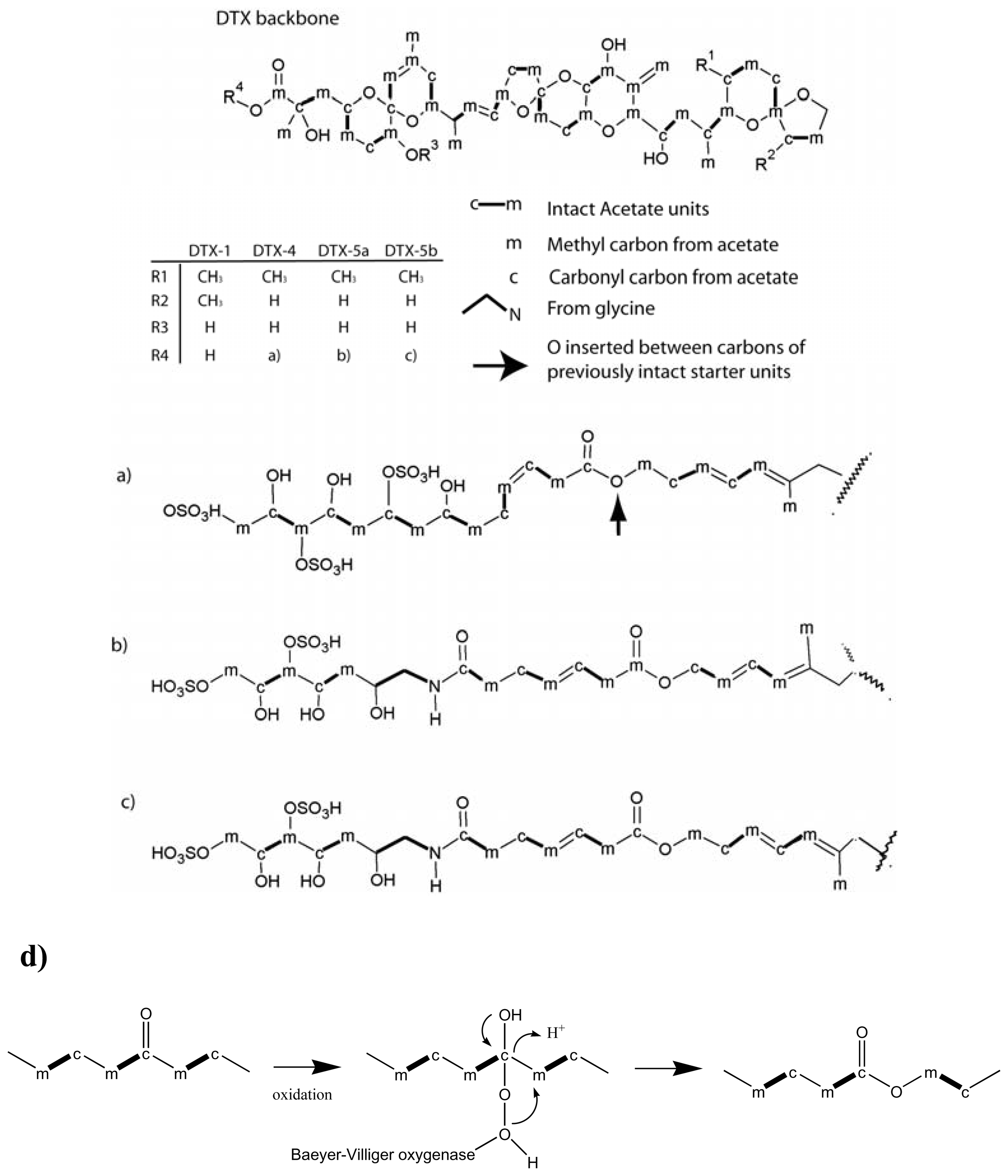
- Norte, M; Padilla, A; Fernandez, JJ. Studies on the biosynthesis of the polyether marine toxin dinophysistoxin-1 (DTX-1). Tetrahedron Lett 1994, 35, 1441–1444. [Google Scholar]
- Macpherson, GR; Burton, IW; LeBlanc, P; Walter, JA; Wright, JL. Studies of the biosynthesis of DTX-5a and DTX-5b by the dinoflagellate Prorocentrum maculosum: regiospecificity of the putative Baeyer-Villigerase and insertion of a single amino acid in a polyketide chain. J Org Chem 2003, 68, 1659–1664. [Google Scholar]
- Daranas, AH; Fernandez, JJ; Norte, M; Gavin, JA; Suarez-Gomez, B; Souto, ML. Biosynthetic studies of the DSP toxin skeleton. Chem Rec 2004, 4, 1–9. [Google Scholar]
- Needham, J; Hu, T; McLachlan, JL; Walter, JA; Wright, JLC. Biosynthetic studies of the DSP toxin DTX-4 and an okadaic acid diol ester. J Chem Soc, Chem Commun 1995, 16, 1623–1624. [Google Scholar]
- Wright, JLC; Hu, T; McLachlan, JL; Needham, J; Walter, JA. Biosynthesis of DTX-4: Confirmation of a Polyketide Pathway, Proof of a Baeyer-Villiger Oxidation Step, and Evidence for an Unusual Carbon Deletion Process. J Am Chem Soc 1996, 118, 8757–8758. [Google Scholar]
- Doekel, S; Marahiel, MA. Biosynthesis of natural products on modular peptide synthetases. Metab Eng 2001, 3, 64–77. [Google Scholar]
- Murata, M; Izumikawa, M; Tachibana, K; Fujita, T; Naoki, H. Labelling pattern of okadaic acid from 18O2 and [18O2]acetate elucidated by collision-induced dissociation tandem mass spectrometry. J Am Chem Soc 1998, 120, 147–151. [Google Scholar]
- Izumikawa, M; Murata, M; Tachibana, K; Fujita, T; Naoki, H. 18O-Labelling pattern of okadaic acid from H218O in dinoflagellate Prorocentrum lima elucidated by tandem mass spectrometry. Eur J Biochem 2000, 267, 5179–5183. [Google Scholar]
- Oliynyk, M; Stark, CB; Bhatt, A; Jones, MA; Hughes-Thomas, ZA; Wilkinson, C; Oliynyk, Z; Demydchuk, Y; Staunton, J; Leadlay, PF. Analysis of the biosynthetic gene cluster for the polyether antibiotic monensin in Streptomyces cinnamonensis and evidence for the role of monB and monC genes in oxidative cyclization. Mol Microbiol 2003, 49, 1179–1190. [Google Scholar]
- Kobayashi, J; Tsuda, M. Amphidinolides, bioactive macrolides from symbiotic marine dinoflagellates. Nat Prod Rep 2004, 21, 77–93. [Google Scholar]
- Sato, M; Shimbo, K; Tsuda, M; Kobayashi, J. Biosynthetic studies of amphidinolides G and H: unusual labeling patterns in feeding experiments with C-13-labeled acetates. Tetrahedron Lett 2000, 41, 503–506. [Google Scholar]
- Cembella, AD; Lewis, NI; Quilliam, MA. The marine dinoflagellate Alexandrium ostenfeldii (Dinophyceae) as the causative organism of spirolide shellfish toxins. Phycologia 2000, 39, 67–74. [Google Scholar]
- Ciminiello, P; Catalanotti, B; Dell’Aversano, C; Fattorusso, C; Fattorusso, E; Forino, M; Grauso, L; Leo, A; Tartaglione, L. Full relative stereochemistry assignment and conformational analysis of 13,19-didesmethyl spirolide C via NMR- and molecular modeling-based techniques. A step towards understanding spirolide’s mechanism of action. Org Biomol Chem 2009, 7, 3674–3681. [Google Scholar]
- Guéret, SM; Brimble, MA. The Spirolide Family of Shellfish Toxins: Isolation, Structure, Biological Activity and Synthesis. Mar Drugs 2010. submitted. [Google Scholar]
- Kao, CY; Levinson, SR. Tetrodotoxin, Saxitoxin, and the Molecular Biology of the Sodium Channel; The New York Academy of Science: New York, NY, USA, 1986; Volume 479, pp. 1–445. [Google Scholar]
- Wang, J; Salata, JJ; Bennett, PB. Saxitoxin is a gating modifier of HERG K+ channels. J Gen Physiol 2003, 121, 583–598. [Google Scholar]
- Su, Z; Sheets, M; Ishida, H; Li, F; Barry, WH. Saxitoxin blocks L-type ICa. J Pharmacol Exp Ther 2004, 308, 324–329. [Google Scholar]
- Shimizu, Y; Norte, M; Hori, A; Genenah, A; Kobayashi, M. Biosynthesis of saxitoxin analogues: The unexpected pathway. J Am Chem Soc 1984, 106, 6433–6434. [Google Scholar]
- Shimizu, Y. Microalgal metabolites. Chem Rev 1993, 93, 1685–1698. [Google Scholar]
- Gupta, S; Norte, M; Shimizu, Y. Biosynthesis of saxitoxin analogues: the origin and introduction mechanism of the side-chain carbon. J Chem Soc Chem Commun 1989, 1421–1424. [Google Scholar]
- Shimizu, Y. Microalgal metabolites: a new perspective. Annu Rev Microbiol 1996, 50, 431–465. [Google Scholar]
- Kellmann, R; Neilan, BA. Biochemical characterisation of paralytic shellfish toxin biosynthesis in vitro. J Phycol 2007, 43, 497–508. [Google Scholar]
- Kellmann, R; Mihali, TK; Jeon, YJ; Pickford, R; Pomati, F; Neilan, BA. Biosynthetic intermediate analysis and functional homology reveal a saxitoxin gene cluster in cyanobacteria. Appl Environ Microbiol 2008, 74, 4044–4053. [Google Scholar]
- Mahmood, NA; Carmichael, WW. Paralytic shellfish poisons produced by the freshwater cyanobacterium Aphanizomenon flos-aquae NH-5. Toxicon 1986, 24, 175–186. [Google Scholar]
- Kubota, T; Iinuma, Y; Kobayashi, J. Cloning of polyketide synthase genes from amphidinolide-producing dinoflagellate Amphidinium sp. Biol Pharm Bull 2006, 29, 1314–1318. [Google Scholar]
- Snyder, RV; Gibbs, PD; Palacios, A; Abiy, L; Dickey, R; Lopez, JV; Rein, KS. Polyketide synthase genes from marine dinoflagellates. Mar Biotechnol (NY) 2003, 5, 1–12. [Google Scholar]
- Nakamura, H; Asari, T; Fujimaki, K; Maruyama, K; Murai, A; Ohizumi, Y; Kan, Y. Zooxanthellatoxin-B, vasoconstrictive congener of zooxanthellatoxin-a from a symbiotic dinoflagellate Symbiodinium sp. Tetrahedron Lett 1995, 36, 7255–7258. [Google Scholar]
- Kobayashi, J; Ishibashi, M. Bioactive metabolites of symbiotic marine microorganisms. Chem Rev 1993, 93, 1753–1769. [Google Scholar]
- Snyder, RV; Guerrero, MA; Sinigalliano, CD; Winshell, J; Perez, R; Lopez, JV; Rein, KS. Localization of polyketide synthase encoding genes to the toxic dinoflagellate Karenia brevis. Phytochemistry 2005, 66, 1767–1780. [Google Scholar]
- Monroe, EA; Van Dolah, FM. The toxic dinoflagellate Karenia brevis encodes novel type I-like polyketide synthases containing discrete catalytic domains. Protist 2008, 159, 471–482. [Google Scholar]
- John, U; Beszteri, B; Derelle, E; de Peer, YV; Read, B; Moreau, H; Cembella, A. Novel insights into evolution of protistan polyketide synthases through phylogenomic analysis. Protist 2008, 159, 21–30. [Google Scholar]
- Stachelhaus, T; Mootz, HD; Marahiel, MA. The specificity-conferring code of adenylation domains in nonribosomal peptide synthetases. Chem Biol 1999, 6, 493–505. [Google Scholar]
- Perez, R; Liu, L; Lopez, J; An, T; Rein, KS. Diverse bacterial PKS sequences derived from okadaic acid-producing dinoflagellates. Mar Drugs 2008, 6, 164–179. [Google Scholar]
- de Traubenberg, CR. Interactions between a dinoflagellate and its associated bacterial microflora: Role of bacteria in the toxicity of Prorocentrum lima Ehrenberg (Dodge). Ph.D. Dissertation, University of Nantes, Nantes, France, 1993. [Google Scholar]
- Zhu, G; LaGier, MJ; Stejskal, F; Millership, JJ; Cai, X; Keithly, JS. Cryptosporidium parvum: the first protist known to encode a putative polyketide synthase. Gene 2002, 298, 79–89. [Google Scholar]
- Taroncher-Oldenburg, G; Anderson, DM. Identification and characterization of three differentially expressed genes, encoding S-adenosylhomocysteine hydrolase, methionine aminopeptidase, and a histone-like protein, in the toxic dinoflagellate Alexandrium fundyense. Appl Environ Microbiol 2000, 66, 2105–2112. [Google Scholar]
- Sako, Y; Yoshida, T; Uchida, A; Arakawa, O; Noguchi, T; Ishida, Y. Purification and characterization of a sulfotransferase specific to N-21 of saxitoxin and gonyautoxin 2+3 from the toxic dinoflagellate Gymnodinium catenatum (Dinophyceae). J Phycol 2001, 37, 1044–1051. [Google Scholar]
- Yoshida, T; Sako, Y; Uchida, A; Kakutani, T; Arakawa, O; Noguchi, T; Ishida, Y. Purification and characterization of sulfotransferase specific to O-22 of 11-hydroxy saxitoxin from the toxic dinoflagellate Gymnodinium catenatum (Dinophyceae). Fish Sci 2002, 68, 634–642. [Google Scholar]
- Hackett, JD; Scheetz, TE; Yoon, HS; Soares, MB; Bonaldo, MF; Casavant, TL; Bhattacharya, D. Insights into a dinoflagellate genome through expressed sequence tag analysis. BMC Genomics 2005, 6, 80. [Google Scholar]
- Taroncher Oldenburg, G; Kulis, DM; Anderson, DM. Toxin variability during the cell cycle of the dinoflagellate Alexandrium fundyense. Limnol Oceanogr 1997, 42, 1178–1188. [Google Scholar]
- Taroncher-Oldenburg, G; Kulis, DM; Anderson, DM. Coupling of saxitoxin biosynthesis to the G1 phase of the cell cycle in the dinoflagellate Alexandrin fundyense: temperature and nutrient effects. Nat Toxins 1999, 7, 207–219. [Google Scholar]
- Leighfield, TA; Barbier, M; Van Dolah, FM. Evidence for cAMP-dependent protein kinase in the dinoflagellate, Amphidinium operculatum. Comp Biochem Physiol B Biochem Mol Biol 2002, 133, 317–324. [Google Scholar]
- Leighfield, TA; Van Dolah, FM. Identification of a cyclic AMP-dependent protein kinase in the dinoflagellate Amphidinium operculatum. J Phycol 2000, 36(suppl). [Google Scholar] [CrossRef]
- Salois, P; Morse, D. Characterization and molecular phylogeny of a protein kinase cDNA from the dinoflagellate Gonyaulax (Dinophyceae). J Phycol 1997, 33, 1063–1072. [Google Scholar]
- Lin, S; Zhang, H. Mitogen-activated protein kinase in Pfiesteria piscicida and its growth rate-related expression. Appl Environ Microbiol 2003, 69, 343–349. [Google Scholar]
- Yoshida, T; Sako, Y; Fujii, A; Uchida, A; Ishida, Y; Arakawa, O; Noguchi, T. Comparative study on two sulfotransferases involved with sulfation to N-21 of PSP toxins from Gymnodinium catenatum and Alexandrium catenella. VIII International conference on Harmful algae- Abstracts and Posters Classification, Vigo (Spain), 25–29 Jun 1997; Reguera, B, Ed.; Instituto Espanol de Oceanografia, Centro Oceanografico de Vigo, Vigo, Espana: Vigo, Spain, 1997. [Google Scholar]
- Wang, D; Zhang, S; Hong, HZ. A sulfotransferase specific to N-21 of gonyautoxin 2/3 from crude enzyme extraction of toxic dinoflagellate Alexandrium tamarense CI01. Chin J Oceanol Limnol 2007, 25, 227–234. [Google Scholar]
- Matsui, M; Homma, H. Biochemistry and molecular biology of drug-metabolizing sulfotransferase. Int J Biochem 1994, 26, 1237–1247. [Google Scholar]
- Barnes, S; Buchina, ES; King, RJ; McBurnett, T; Taylor, KB. Bile acid sulfotransferase I from rat liver sulfates bile acids and 3-hydroxy steroids: purification, N-terminal amino acid sequence, and kinetic properties. J Lipid Res 1989, 30, 529–540. [Google Scholar]
- Saidha, T; Schiff, JA. Purification and properties of a phenol sulphotransferase from Euglena using L-tyrosine as substrate. Biochem J 1994, 298 Pt 1, 45–50. [Google Scholar]
- Uribe, P; Fuentes, D; Valdes, J; Shmaryahu, A; Zuniga, A; Holmes, D; Valenzuela, PD. Preparation and analysis of an expressed sequence tag library from the toxic dinoflagellate Alexandrium catenella. Mar Biotechnol (NY) 2008, 10, 692–700. [Google Scholar]
- Humpage, AR; Rositano, J; Bretag, AH; Brown, R; Baker, PD; Nicholson, BC; Steffensen, DA. Paralytic shellfish poisons from Australian cyanobacterial blooms. Aust J Mar Freshw Res 1994, 45, 761–771. [Google Scholar]
- Cembella, AD. Anderson, DM, Cembella, AD, Hallegraeff, GM, Eds.; Ecophysiology and metabolism of paralytic shellfish toxins in marine microalgae. In Physiological Ecology of Harmful Algal Blooms; Springer-Verlag: Berlin, Germany, 1998; Volume G41, pp. 381–403. [Google Scholar]
- Silva, SE. Intracellular bacteria: The origin of dinoflagellate toxicity. J Environ Pathol, Toxicol Oncol 1990, 10, 124–128. [Google Scholar]
- Kodoma, M; Ogata, T; Sato, S. Bacterial production of saxitoxin. Agric Biol Chem 1988, 52, 1075–1077. [Google Scholar]
- Plumley, FG. Purification of an enzyme involved in saxitoxin synthesis. J Phycol 2001, 37, 926–928. [Google Scholar]
- Kellmann, R; Mihali, TK; Neilan, BA. Identification of a saxitoxin biosynthesis gene that has an evolutionary history with frequent horizontal gene transfer events. J Mol Evol 2008, 67, 526–538. [Google Scholar]
- Mihali, TK; Kellmann, R; Neilan, BA. Characterisation of the paralytic shellfish toxin biosynthesis gene clusters in Anabaena circinalis AWQC131C and Aphanizomenon sp. NH-5. BMC Biochem 2009, 10, 8. [Google Scholar]
- Onodera, H; Satake, M; Oshima, Y; Yasumoto, T; Carmichael Wayne, W. New saxitoxin analogues from the freshwater filamentous cyanobacterium Lyngbya wollei. Nat Toxins 1997, 5, 146–151. [Google Scholar]
- Moustafa, A; Loram, JE; Hackett, JD; Anderson, DM; Plumley, FG; Bhattacharya, D. Origin of saxitoxin biosynthetic genes in cyanobacteria. PLoS One 2009, 4, e5758. [Google Scholar]
- Gallacher, S; Flynn, KJ; Franco, JM; Brueggemann, EE; Hines, HB. Evidence for production of paralytic shellfish toxins by bacteria associated with Alexandrium spp. (Dinophyta) in culture. Appl Environ Microbiol 1997, 63, 239–245. [Google Scholar]
- Baker, TR; Doucette, GJ; Powell, CL; Boyer, GL; Plumley, FG. GTX(4) imposters: characterization of fluorescent compounds synthesized by Pseudomonas stutzeri SF/PS and Pseudomonas/Alteromonas PTB-1, symbionts of saxitoxin-producing Alexandrium spp. Toxicon 2003, 41, 339–347. [Google Scholar]
- Li, Y; Muller, R. Non-modular polyketide synthases in myxobacteria. Phytochemistry 2009, 70, 1850–1857. [Google Scholar]
- Venter, JC; Remington, K; Heidelberg, JF; Halpern, AL; Rusch, D; Eisen, JA; Wu, D; Paulsen, I; Nelson, KE; Nelson, W; Fouts, DE; Levy, S; Knap, AH; Lomas, MW; Nealson, K; White, O; Peterson, J; Hoffman, J; Parsons, R; Baden-Tillson, H; Pfannkoch, C; Rogers, YH; Smith, HO. Environmental genome shotgun sequencing of the Sargasso Sea. Science 2004, 304, 66–74. [Google Scholar]
- Rusch, DB; Halpern, AL; Sutton, G; Heidelberg, KB; Williamson, S; Yooseph, S; Wu, D; Eisen, JA; Hoffman, JM; Remington, K; Beeson, K; Tran, B; Smith, H; Baden-Tillson, H; Stewart, C; Thorpe, J; Freeman, J; Andrews-Pfannkoch, C; Venter, JE; Li, K; Kravitz, S; Heidelberg, JF; Utterback, T; Rogers, YH; Falcon, LI; Souza, V; Bonilla-Rosso, G; Eguiarte, LE; Karl, DM; Sathyendranath, S; Platt, T; Bermingham, E; Gallardo, V; Tamayo-Castillo, G; Ferrari, MR; Strausberg, RL; Nealson, K; Friedman, R; Frazier, M; Venter, JC. The Sorcerer II Global Ocean Sampling expedition: northwest Atlantic through eastern tropical Pacific. PLoS Biol 2007, 5, e77. [Google Scholar]
- Yooseph, S; Sutton, G; Rusch, DB; Halpern, AL; Williamson, SJ; Remington, K; Eisen, JA; Heidelberg, KB; Manning, G; Li, W; Jaroszewski, L; Cieplak, P; Miller, CS; Li, H; Mashiyama, ST; Joachimiak, MP; van Belle, C; Chandonia, JM; Soergel, DA; Zhai, Y; Natarajan, K; Lee, S; Raphael, BJ; Bafna, V; Friedman, R; Brenner, SE; Godzik, A; Eisenberg, D; Dixon, JE; Taylor, SS; Strausberg, RL; Frazier, M; Venter, JC. The Sorcerer II Global Ocean Sampling expedition: expanding the universe of protein families. PLoS Biol 2007, 5, e16. [Google Scholar]
- Seshadri, R; Kravitz, SA; Smarr, L; Gilna, P; Frazier, M. CAMERA: a community resource for metagenomics. PLoS Biol 2007, 5, e75. [Google Scholar]
- Huson, DH; Auch, AF; Qi, J; Schuster, SC. MEGAN analysis of metagenomic data. Genome Res 2007, 17, 377–386. [Google Scholar]
- Udwary, DW; Zeigler, L; Asolkar, RN; Singan, V; Lapidus, A; Fenical, W; Jensen, PR; Moore, BS. Genome sequencing reveals complex secondary metabolome in the marine actinomycete Salinispora tropica. Proc Natl Acad Sci USA 2007, 104, 10376–10381. [Google Scholar]
- Coll, JM. Methodologies for transferring DNA into eukaryotic microalgae. Span J Agric Res 2006, 4, 316–330. [Google Scholar]
- Lohuis, MRt; Miller, DJ. Genetic transformation of dinoflagellates (Amphidinium and Symbiodinium): expression of GUS in microalgae using heterologous promoter constructs. Plant J 2002, 13, 427–435. [Google Scholar]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Strain | type I PKS | type II PKS | polyketide |
|---|---|---|---|---|
| Prorocentrum lima | + | + | okadaic acid | |
| Prorocentrum hoffmanianum | + | − | okadaic acid | |
| Karenia brevis | CCMP718 | + | − | brevetoxin |
| Symbiodinium sp. | CCMP831 | + | − | ND |
| Amphidinium operculatum | CCMP1342 | + | ND | ND |
| Amphidinium operculatum | CCMP120 | + | ND | ND |
| Amphidinium operculatum | CCMP121 | − | ND | ND |
| Amphidinium carterae | CCMP1314 | − | ND | ND |
| Gymnodinium catenatum | + | ND | ND |
| Name | Catalytic Residues | Known Substrates | Predicted Substrates |
|---|---|---|---|
| tyroc003 | DAWQFGLIDK | GLN | |
| McnA | DAWQTGLIDK | GLN | |
| NRPS-1 | DAWQFGLIDK | GLN | |
| Syp-M2 | DVQYIAHVTK | PRO | |
| ituri002 | DVQFIAHVXK | PRO | |
| NRPS-2 | DVQFIAXXXK | PRO | |
| bacit001 | DGFFLGVVYK | ILE | |
| NRPS-3 | DAFFLGVTYK | ILE | |
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kellmann, R.; Stüken, A.; Orr, R.J.S.; Svendsen, H.M.; Jakobsen, K.S. Biosynthesis and Molecular Genetics of Polyketides in Marine Dinoflagellates. Mar. Drugs 2010, 8, 1011-1048. https://doi.org/10.3390/md8041011
Kellmann R, Stüken A, Orr RJS, Svendsen HM, Jakobsen KS. Biosynthesis and Molecular Genetics of Polyketides in Marine Dinoflagellates. Marine Drugs. 2010; 8(4):1011-1048. https://doi.org/10.3390/md8041011
Chicago/Turabian StyleKellmann, Ralf, Anke Stüken, Russell J. S. Orr, Helene M. Svendsen, and Kjetill S. Jakobsen. 2010. "Biosynthesis and Molecular Genetics of Polyketides in Marine Dinoflagellates" Marine Drugs 8, no. 4: 1011-1048. https://doi.org/10.3390/md8041011
APA StyleKellmann, R., Stüken, A., Orr, R. J. S., Svendsen, H. M., & Jakobsen, K. S. (2010). Biosynthesis and Molecular Genetics of Polyketides in Marine Dinoflagellates. Marine Drugs, 8(4), 1011-1048. https://doi.org/10.3390/md8041011