Synthesis of the Marine Pyrroloiminoquinone Alkaloids, Discorhabdins

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
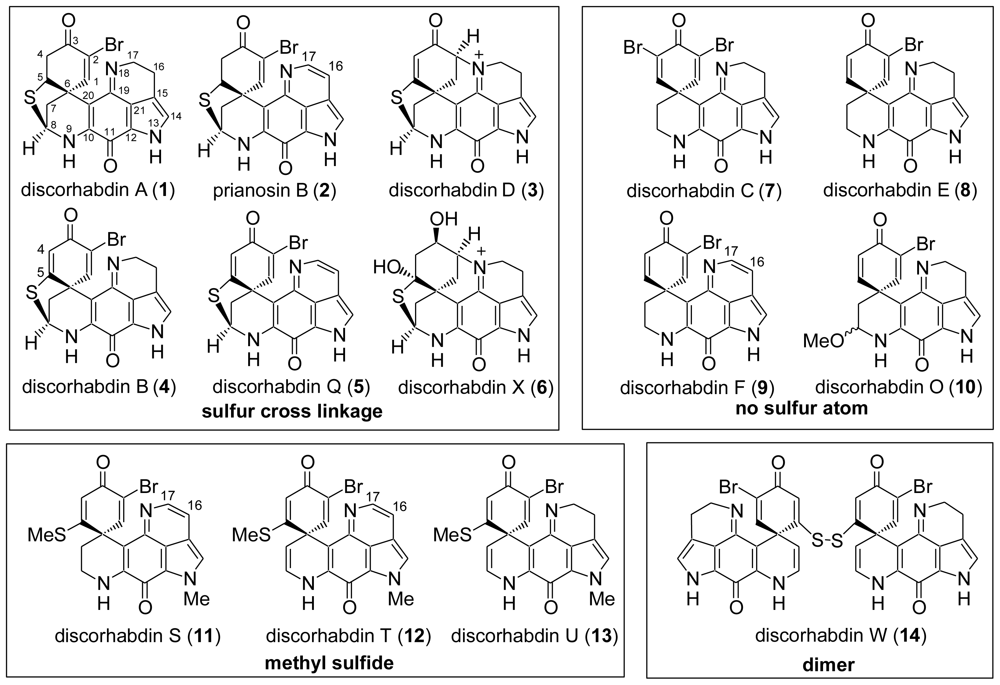
:1. Introduction
2. Studies of the Total Synthesis of Discorhabdin C
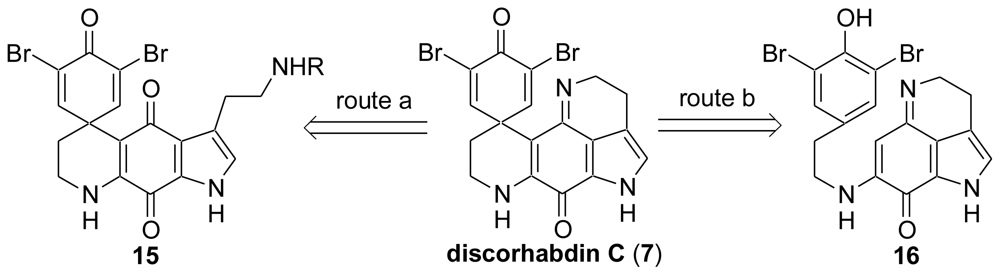
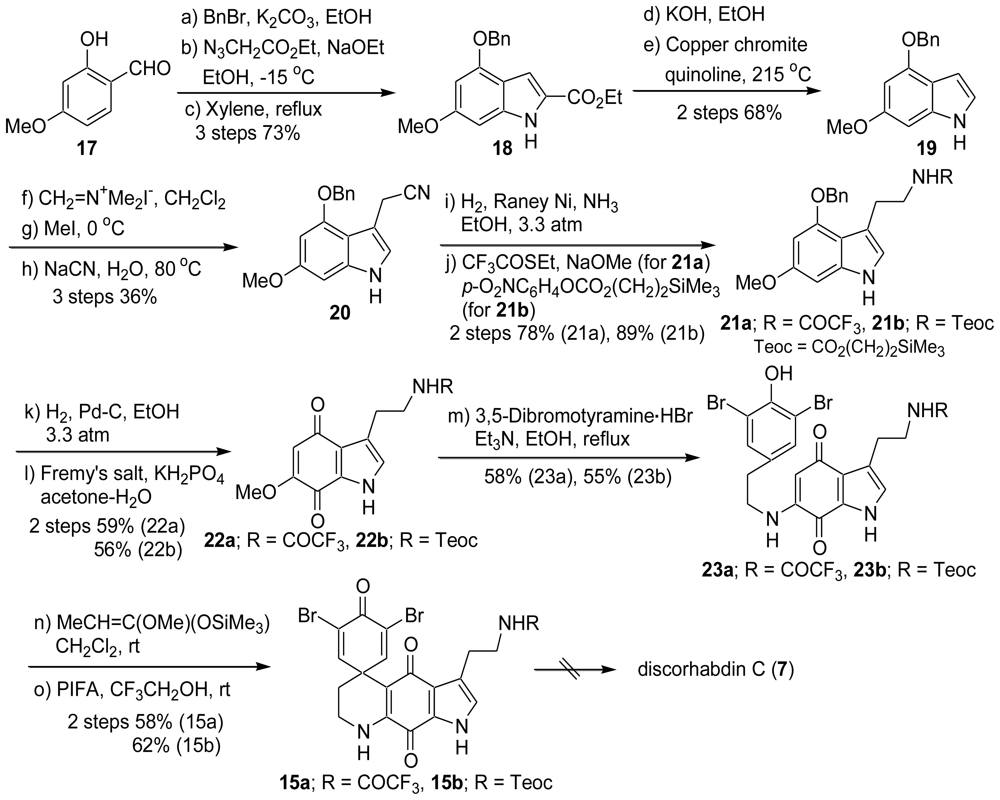
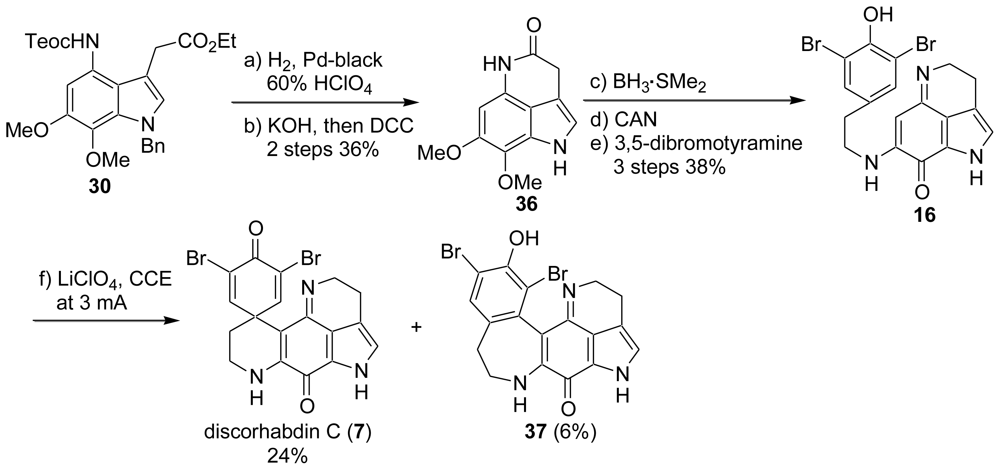
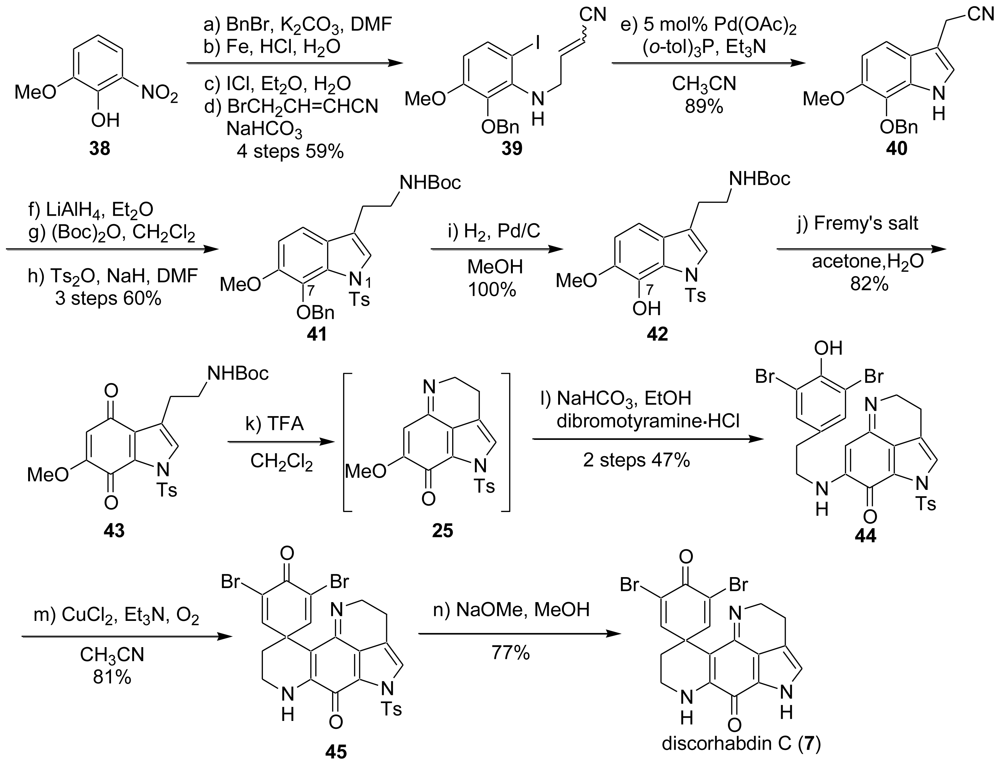
2.1. Our Synthesis [37]
2.2. Yamamura’s Synthesis [47,48]
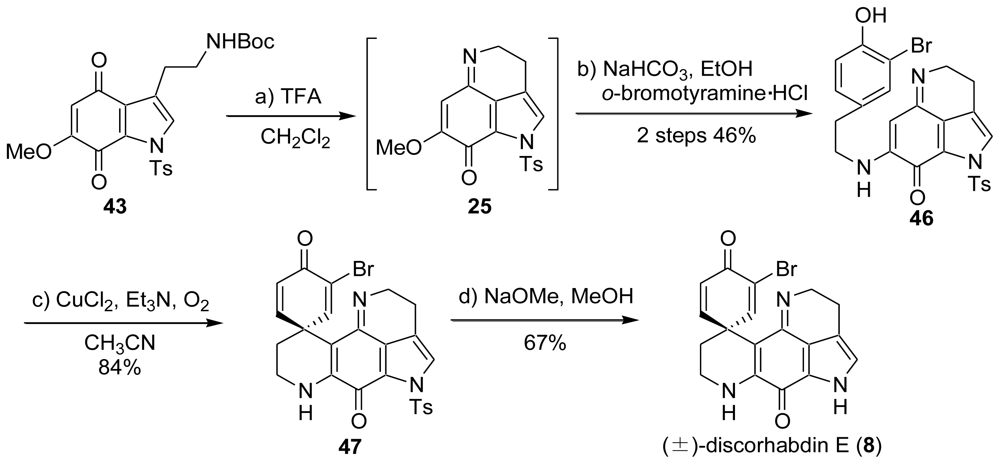
2.3. Heathcock’s Synthesis [49]
3. Studies of the Total Synthesis of Sulfur-Containing Makaluvamine, Makaluvamine F (48) [38,39]
3.1. Facile and Direct Synthesis of the Pyrroloiminoquinone Unit [50]
3.2. Synthesis of the Dihydrothiophene Units [52,53]
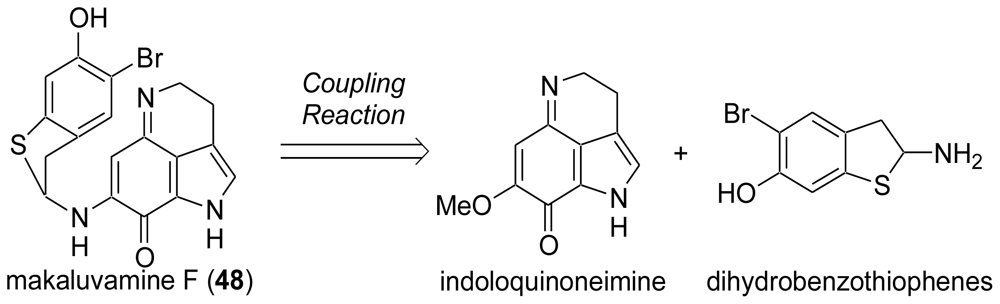
3.3. Total Synthesis of (±)-Makaluvamine F (48) [38,39]
4. Total Synthesis of Discorhabdin A [40–43]
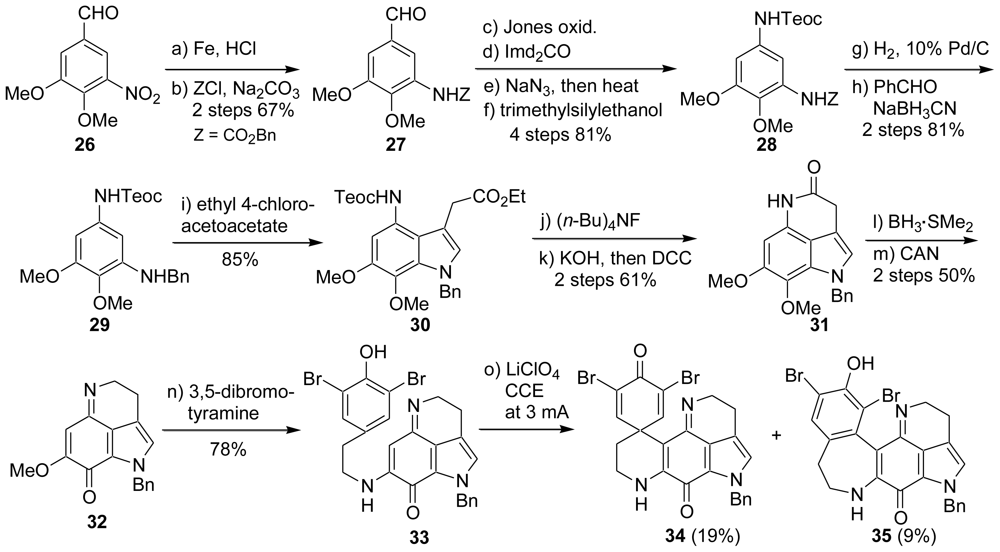
5. Semi-Synthesis of Discorhabdins P and U [57]
6. Total Synthesis of Prianosin B [44]
7. Summary
- Samples Availability: Available from the authors.
References and Notes
- Carté, BK. Biomedical potential of marine natural products. BioScience 1996, 46, 271–285. [Google Scholar]
- Faulkner, DJ. Biomedical Uses for Natural Marine Chemicals. Oceanus 1992, 35, 29–35. [Google Scholar]
- Faulkner, DJ. Marine natural products: metabolites of marine algae and herbivorous marine mollusks. Nat Prod Rep 1984, 1, 251–280. [Google Scholar]
- Barasch, D. Fishing for Drugs. Am Health 1990, 9, 20–21. [Google Scholar]
- Cohen, T. Pharmaceuticals from the sea. Technol Rev 1993, 96, 15–16. [Google Scholar]
- Liu, R; Cui, C; Duan, L; Gu, Q; Zhu, W. Potent in vitro anticancer activity of metacycloprodigiosin and undecylprodigiosin from a sponge-derived actinomycete Saccharopolyspora sp. nov. Archives of Pharm Res 2005, 28, 1341–1344. [Google Scholar]
- Jeong, SY; Ishida, K; Ito, Y; Okada, S; Murakami, M. Bacillamide, a novel algicide from the marine bacterium, Bacillus sp. SY-1, against the harmful dinoflagellate Cochlodinium polykrikoides. Tetrahedron Lett 2003, 44, 8005–8007. [Google Scholar]
- Xin, ZH; Zhu, WM; Gu, QQ; Fang, YC; Duan, L; Cui, CB. A new cytotoxic compound from Penicillium auratiogriseum, symbiotic or epiphytic fungus of sponge Mycale plumose. Chin Chem Lett 2005, 16, 1227–1229. [Google Scholar]
- Xin, Z; Tian, L; Zhu, T; Wang, W; Du, L; Fang, Y; Gu, Q; Zhu, W. Isocoumarin derivatives from the sea squirt-derived fungus Penicillium stoloniferum QY2-10 and the halotolerant fungus Penicillium notatum B-52. Arch Pharm Res 2007, 30, 816–819. [Google Scholar]
- Wang, F; Fang, Y; Zhang, M; Lin, A; Zhu, T; Gu, Q; Zhu, W. Six new ergosterols from the marine-derived fungus Rhizopus sp. Steroids 2008, 73, 19–26. [Google Scholar]
- Wang, F; Fang, Y; Zhu, T; Zhang, M; Lin, A; Gu, Q; Zhu, W. Seven new prenylated indole diketopiperazine alkaloids from holothurianderived fungus Aspergillus fumigatus. Tetrahedron 2008, 64, 7986–7991. [Google Scholar]
- Urban, S; Hickford, SJH; Blunt, JW; Munro, MHG. Bioactive Marine Alkaloids. Curr Org Chem 2000, 4, 765–805. [Google Scholar]
- Gunasekera, SP; Zuleta, IA; Longley, RE; Wright, AE; Pomponi, SA. Discorhabdins S, T, and U, New Cytotoxic Pyrroloiminoquinones from a Deep-Water Caribbean Sponge of the Genus Batzella. J Nat Prod 2003, 66, 1615–1617. [Google Scholar]
- Reyes, F; Martin, R; Rueda, A; Fernandez, R; Montalvo, D; Gomez, C; Sanchez-Puelles, JM. Discorhabdins I and L, Cytotoxic Alkaloids from the Sponge Latrunculia brevis. J Nat Prod 2004, 67, 463–465. [Google Scholar]
- Antunes, EM; Beukes, DR; Kelly, M; Samaai, T; Barrows, LR; Marshall, KM; Sincich, C; Davies-Coleman, MT. Cytotoxic Pyrroloiminoquinones from Four New Species of South African Latrunculid Sponges. J Nat Prod 2004, 67, 1268–1276. [Google Scholar]
- Harayama, Y; Kita, Y. Pyrroloiminoquinone alkaloids. Discorhabdins and makaluvamines. Curr Org Chem 2005, 9, 1567–1588. [Google Scholar]
- Cheng, J; Ohizumi, Y; Wälchli, MR; Nakamura, H; Hirata, Y; Sasaki, T; Kobayashi, J. Prianosins B, C, and D, novel sulfur-containing alkaloids with potent antineoplastic activity from the Okinawan marine sponge Prianos melanos. J Org Chem 1988, 53, 4621–4624. [Google Scholar]
- Monks, A; Scuderio, DA; Skehan, P; Shoemaker, RH; Paull, KD; Hose, VC; Langley, J; Cronise, P; Vaigro-Wolff, A; Grey-Goodrich, M; Campball, H; Boyd, MR. Feasibility of a high-flux anticancer drug screen using a diverse panel of cultured human tumor cell lines. J Natl Cancer Inst 1991, 83, 757–766. [Google Scholar]
- Carney, JR; Scheuer, PJ; Kelly-Borges, M. Makaluvamine G, a cytotoxic pigment from an an Indonesian Sponge Histodermella sp. Tetrahedron 1993, 49, 8483–8486. [Google Scholar]
- Schmidt, EW; Harper, MK; Faulkner, DJ. Makaluvamines H-M and Damirone C from the Pohnpeian Sponge Zyzzya fuliginosa. J Nat Prod 1995, 58, 1861–1867. [Google Scholar]
- Venables, DA; Concepción, GP; Matsumoto, S; Barrows, LR; Ireland, CM. Makaluvamine N: A New Pyrroloiminoquinone from Zyzzya fuliginosa. J Nat Prod 1997, 60, 408–410. [Google Scholar]
- Sun, HH; Sakemi, S; Burres, N; McCarthy, P. Isobatzellines A, B, C, and D. Cytotoxic and antifungal pyrroloquinoline alkaloids from the marine sponge Batzella sp. J Org Chem 1990, 55, 4964–4966. [Google Scholar]
- Gunasekera, SP; McCarthy, PJ; Longley, RE; Pomponi, SA; Wright, AE. Secobatzellines A and B, Two New Enzyme Inhibitors from a Deep-Water Caribbean Sponge of the Genus Batzella. J Nat Prod 1999, 62, 1208–1211. [Google Scholar]
- Copp, BR; Ireland, CM. Wakayin: A Novel Cytotoxic Pyrroloiminoquinone Alkaloid from the Ascidian Clavelina Species. J Org Chem 1991, 56, 4596–4597. [Google Scholar]
- Stierle, DB; Faulkner, DJ. Two New Pyrroloquinoline Alkaloids from the Sponge Damiria sp. J Nat Prod 1991, 54, 1131–1133. [Google Scholar]
- Schmidt, EW; Harper, MK; Faulkner, DJ. Makaluvamines H-M and Damirone C from the Pohnpeian Sponge Zyzzya Fuliginosa. J Nat Prod 1995, 58, 1861–1867. [Google Scholar]
- Delfourne, E. Analogues of Marine Pyrroloiminoquinone Alkaloids: Synthesis and Antitumore Properties. Anti-Cancer Agents Med Chem 2008, 8, 910–916. [Google Scholar]
- Kobayashi, J; Cheng, JF; Ishibashi, M; Nakamura, H; Ohizumi, Y; Hirata, Y; Sasaki, T; Lu, H; Clardy, J. Prianosin A, a novel antileukemic alkaloid from the okinawan marine sponge Prianos melanos. Tetrahedron Lett 1987, 28, 4939–4942. [Google Scholar]
- Perry, NB; Blunt, JW; Munro, MHG. Cytotoxic pigments from new zealand sponges of the genus latrunculia: discorhabdins A, B and C. Tetrahedron 1988, 44, 1727–1734. [Google Scholar]
- Perry, NB; Blunt, JW; Munro, MHG. Discorhabdin D, an antitumor alkaloid from the sponges Latrunculia brevis and Prianos sp. J Org Chem 1988, 53, 4127–4128. [Google Scholar]
- Dijoux, MG; Gamble, WR; Hallock, YF; Cardellina, JH, II; Soest, R; Boyd, MR. A New Discorhabdin from Two Sponge Genera. J Nat Prod 1999, 62, 636–637. [Google Scholar]
- Ford, J; Capon, RJ. Discorhabdin R: A New Antibacterial Pyrroloiminoquinone from Two Latrunculiid Marine Sponges, Latrunculia sp. and Negombata sp. J Nat Prod 2000, 63, 1527–1528. [Google Scholar]
- El-Naggar, M; Capon, RJ. Discorhabdins Revisited: Cytotoxic Alkaloids from Southern Australian Marine Sponges of the Genera Higginsia and Spongosorites. J Nat Prod 2009, 72, 460–464. [Google Scholar]
- Cheng, J; Ohizumi, Y; Wälchli, MR; Nakamura, H; Hirata, Y; Sasaki, T; Kobayashi, J. Prianosins B, C, and D, novel sulfur-containing alkaloids with potent antineoplastic activity from the Okinawan marine sponge Prianos melanos. J Org Chem 1988, 53, 4621–4624. [Google Scholar]
- Lang, G; Pinkert, A; Blunt, JW; Munro, MHG. Discorhabdin W, the First Dimeric Discorhabdin. J Nat Prod 2005, 68, 1796–1798. [Google Scholar]
- Blunt, JW; Munro, MHG; Battershill, CN; Copp, BR; McCombs, JD; Perry, NB; Prinsep, M; Thompson, AM. From the Antarctic to the Antipodes; 45° of marine chemistry. New J Chem 1990, 14, 761–775. [Google Scholar]
- Kita, Y; Tohma, H; Inagaki, M; Hatanaka, K; Yakura, T. Total synthesis of discorhabdin C: a general aza spiro dienone formation from O-silylated phenol derivatives using a hypervalent iodine reagent. J Am Chem Soc 1992, 114, 2175–2180. [Google Scholar]
- Kita, Y; Egi, M; Tohma, H. Total synthesis of sulfur-containing pyrroloiminoquinone marine product, (±)-makaluvamine F using hypervalent iodine(III)-induced reactions. Chem Commun 1999, 143–144. [Google Scholar]
- Kita, Y; Egi, M; Takada, T; Tohma, H. Development of novel reactions using hypervalent iodine(III) reagents. Total synthesis of sulfur-containing pyrroloiminoquinone marine product, (±)-makaluvamine F. Synthesis 1999, 885–897. [Google Scholar]
- Tohma, H; Harayama, Y; Hashizume, M; Iwata, M; Egi, M; Kita, Y. Synthetic studies on the sulfur-cross-linked core of antitumor marine alkaloid, discorhabdins: total synthesis of discorhabdin A. Angew Chem Int Ed 2002, 41, 348–350. [Google Scholar]
- Tohma, H; Harayama, Y; Hashizume, M; Iwata, M; Kiyono, Y; Egi, M; Kita, Y. The First Total Synthesis of Discorhabdin A. J Am Chem Soc 2003, 125, 11235–11240. [Google Scholar]
- Harayama, Y; Yoshida, M; Kamimura, D; Kita, Y. The novel and efficient direct synthesis of N,O-acetal compounds using a hypervalent iodine(III) reagent: an improved synthetic method for a key intermediate of discorhabdins. Chem Commun 2005, 1764–1766. [Google Scholar]
- Harayama, Y; Yoshida, M; Kamimura, D; Wada, Y; Kita, Y. The efficient direct synthesis of N,O-acetal compounds as key intermediates of discorhabdin A: oxidative fragmentation reaction of α-amino acids or β-amino alcohols by using hypervalent iodine(III) reagents. Chem Eur J 2006, 12, 4893–4899. [Google Scholar]
- Wada, Y; Otani, K; Endo, N; Harayama, Y; Kamimura, D; Yoshida, M; Fujioka, H; Kita, Y. The first total synthesis of prianosin B. Tetrahedron 2009, 65, 1059–1062. [Google Scholar]
- Perry, NB; Blunt, JW; McCombs, JD; Munro, MHG. Discorhabdin C, a highly cytotoxic pigment from a sponge of the genus Latrunculia. J Org Chem 1986, 51, 5476–5478. [Google Scholar]
- Kita, Y; Yakura, T; Tohma, H; Kikuchi, K; Tamura, Y. A synthetic approach to discorhabdin alkaloids: Hypervalent iodine oxidation of p-substituted phenol derivatives to azacarbocyclic spirodienones. Tetrahedron Lett 1989, 30, 1119–1120. [Google Scholar]
- Nishiyama, S; Cheng, JF; Tao, XL; Yamamura, S. Synthetic studies on novel sulfur-containing alkaloids, prianosins and discorhabdins: total synthesis of discorhabdin C. Tetrahedron Lett 1991, 32, 4151–4154. [Google Scholar]
- Tao, XL; Cheng, JF; Nishiyama, S; Yamamura, S. Synthetic studies on tetrahydropyrroloquinoline-containing natural products: Syntheses of discorhabdin C, batzelline C and isobatzelline C. Tetahedron 1994, 50, 2017–2028. [Google Scholar]
- Aubart, KM; Heathcock, CH. A Biomimetic Approach to the Discorhabdin Alkaloids: Total Syntheses of Discorhabdins C and E and Dethiadiscorhabdin D. J Org Chem 1999, 64, 16–22. [Google Scholar]
- Kita, Y; Egi, M; Ohtsubo, M; Saiki, T; Okajima, A; Takada, T; Tohma, H. Hypervalent iodine(III)-induced intramolecular cyclization reaction of substituted phenol ethers with an alkyl azido side-chain: a novel and efficient synthesis of quinone imine derivatives. Chem Pharm Bull 1999, 47, 241–245. [Google Scholar]
- Kita, Y; Egi, M; Okajima, A; Ohtsubo, M; Takada, T; Tohma, H. Hypervalent iodine(III) induced intramolecular cyclization of substituted phenol ethers bearing an alkyl azido sidechain-a novel synthesis of quinone imine ketals. Chem Commun 1996, 1491–1492. [Google Scholar]
- Kita, Y; Egi, M; Ohtsubo, M; Saiki, T; Takada, T; Tohma, H. Novel and efficient synthesis of sulfur-containing heterocycles using a hypervalent iodine(III) reagent. Chem Commun 1996, 2225–2226. [Google Scholar]
- Tohma, H; Egi, M; Ohtsubo, M; Watanabe, H; Takizawa, S; Kita, Y. A novel and direct α-azidation of cyclic sulfides using a hypervalent iodine(III) reagent. Chem Commun 1998, 173–174. [Google Scholar]
- Kita, Y; Takada, T; Mihara, S; Tohma, H. A novel and direct sulfenylation of phenol ethers using phenyliodine(III) bis(trifluoroacetate) (PIFA) and various thiophenols. Synlett 1995, 211–212. [Google Scholar]
- Kita, Y; Takada, T; Mihara, S; Whelan, BA; Tohma, H. Novel and Direct Nucleophilic Sulfenylation and Thiocyanation of Phenol Ethers Using a Hypervalent Iodine(III) Reagent. J Org Chem 1995, 60, 7144–7148. [Google Scholar]
- Lill, RE; Major, DA; Blunt, JW; Munro, MHG. Studies on the Biosynthesis of Discorhabdin B in the New Zealand Sponge Latrunculia sp. B. J Nat Prod 1995, 58, 306–311. [Google Scholar]
- Grkovic, T; Kaur, B; Webb, VL; Copp, BR. Semi-synthetic preparation of the rare, cytotoxic, deep-sea sourced sponge metabolites discorhabdins P and U. Bioorg Med Chem Lett 2006, 16, 1944–1946. [Google Scholar]
- Miller, K; Alvarez, B; Bettershill, C; Northcote, P; Parthasarathy, H. Genetic, morphological, and chemical divergence in the sponge genus Latrunculia (Porifera: Demospongiae) from New Zealand. Mar Biol 2001, 139, 235–250. [Google Scholar]
- Izawa, T; Nishiyama, S; Yamamura, S. Total syntheses of makaluvamines A, B, C, D and E, cytotoxic pyrroloiminoquinone alkaloids isolated from marine sponge bearing inhibitory activities against topoisomerase II. Tetrahedron 1994, 50, 13593–13600. [Google Scholar]
- White, JD; Yager, KM; Yakura, T. Synthetic Studies of the Pyrroloquinoline Nucleus of the Makaluvamine Alkaloids. Synthesis of the Topoisomerase II Inhibitor Makaluvamine D. J Am Chem Soc 1994, 116, 1831–1838. [Google Scholar]
- Patel, SP; Nadkarni, DH; Murugesan, S; King, JR; Velu, SE. Azide-Mediated Detosylation of N-Tosylpyrroloiminoquinones and N-Tosylindole-4,7-quinones. Synlett 2008, 2864–2868. [Google Scholar]
- Regitz, M. Recent synthetic methods in diazo chemistry. Synthesis 1972, 351–373. [Google Scholar]
- Evans, DA; Britton, TC. Electrophilic azide transfer to chiral enolates. A general approach to the asymmetric synthesis of .alpha.-amino acids. J Am Chem Soc 1987, 109, 6881–6883. [Google Scholar]
- Weil, T; Cais, M. A Simplified Procedure for the Preparation of Diazocyclopentadiene and Some Related Compounds. J Org Chem 1963, 28, 2472. [Google Scholar]
- Regitz, M; Liedhegener, A. Reaktionen aktiver methylenverbindungen mit aziden—XV *1: Synthese von diazocyclopentadienen durch diazogruppenübertragung und einige reaktionen. Tetrahedron 1967, 23, 2701–2708. [Google Scholar]
- Wada, Y; Otani, K; Endo, N; Harayama, Y; Kamimura, D; Yoshida, M; Fujioka, H; Kita, Y. Synthesis of Antitumor Marine Alkaloid Discorhabdin A Oxa Analogues. Org Lett 2009, 11, 4048–4050. [Google Scholar]
- Radisky, DC; Radisky, ES; Barrows, LR; Copp, BR; Kramer, RA; Ireland, CM. Novel cytotoxic topoisomerase II inhibiting pyrroloiminoquinones from Fijian sponges of the genus Zyzzya. J Am Chem Soc 1993, 115, 1632–1638. [Google Scholar]
- Dijoux, M-G; Schnabel, PC; Hallock, YF; Boswell, JL; Johnson, TR; Wilson, JA; Ireland, CM; Soest, R; Boyd, MR; Barrows, LR; Cardellina, JH. Antitumor activity and distribution of pyrroloiminoquinones in the sponge genus. Zyzzya Bioorg Med Chem 2005, 13, 6035–6044. [Google Scholar]
- Shinkre, BA; Raisch, KP; Fan, L; Velu, SE. Analogs of the marine alkaloid makaluvamines: Synthesis, topoisomerase II inhibition, and anticancer activity. Bioorg Med Chem Lett 2007, 17, 2890–2893. [Google Scholar]
- Passarella, D; Belinghieri, F; Scarpellini, M; Pratesi, G; Zunino, F; Gia, OM; Via, LD; Santoro, G; Danieli, B. Analogs of the marine alkaloid makaluvamines: Synthesis, topoisomerase II inhibition, and anticancer activity. Bioorg Med Chem 2008, 16, 2431–2438. [Google Scholar]
- Shinkre, BA; Raisch, KP; Fan, L; Velu, SE. Synthesis and antiproliferative activity of benzyl and phenethyl analogs of makaluvamines. Bioorg Med Chem 2008, 16, 2541–2549. [Google Scholar]



























© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wada, Y.; Fujioka, H.; Kita, Y. Synthesis of the Marine Pyrroloiminoquinone Alkaloids, Discorhabdins. Mar. Drugs 2010, 8, 1394-1416. https://doi.org/10.3390/md8041394
Wada Y, Fujioka H, Kita Y. Synthesis of the Marine Pyrroloiminoquinone Alkaloids, Discorhabdins. Marine Drugs. 2010; 8(4):1394-1416. https://doi.org/10.3390/md8041394
Chicago/Turabian StyleWada, Yasufumi, Hiromichi Fujioka, and Yasuyuki Kita. 2010. "Synthesis of the Marine Pyrroloiminoquinone Alkaloids, Discorhabdins" Marine Drugs 8, no. 4: 1394-1416. https://doi.org/10.3390/md8041394
APA StyleWada, Y., Fujioka, H., & Kita, Y. (2010). Synthesis of the Marine Pyrroloiminoquinone Alkaloids, Discorhabdins. Marine Drugs, 8(4), 1394-1416. https://doi.org/10.3390/md8041394