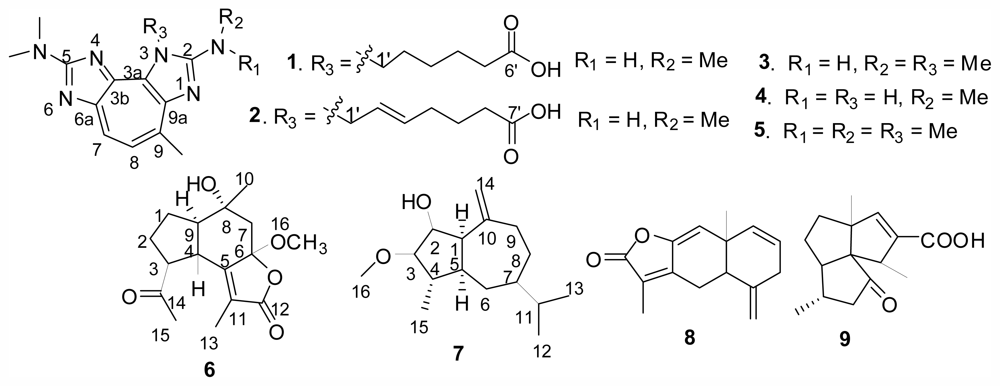
2. Results and Discussion
Compound
1 had a molecular formula of C
19H
26N
6O
2 deduced from its ESIMS and NMR data. The
1H and
13C NMR spectra of
1 were similar to those of pseudozoanthoxanthin A [
3], pseudozoanthoxanthins I and II [
6,
10], zoanthoxanthin 1 (
3) [
4], paragracine (
4) [
4] and zoanthoxanthin (
5) [
4] (
Table 1), except for the addition of five methylene units and one carboxyl group (δ
C 176.4), which suggested that
1 has the same 3
H-pseudozoanthoxanthin core as
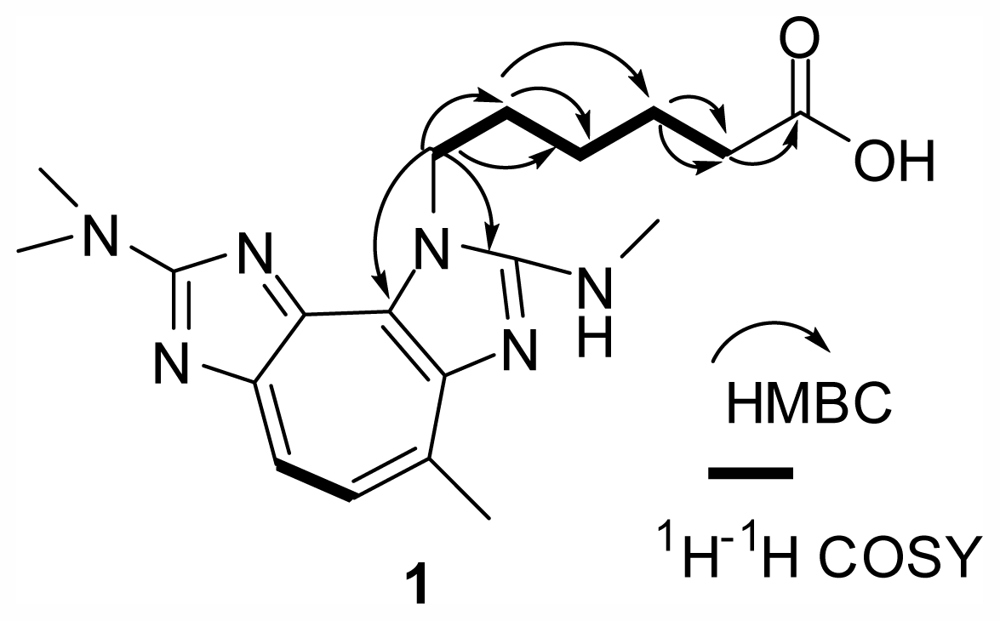
4, the difference between them existing in the side chain. The HMBC spectrum of
1 (
Figure 2) showed correlations of H-1′ (δ
H 3.20, t,
J = 6.5 Hz) with C-2′ (δ
C 29.7)/C-3′ (δ
C 26.9), H-2′ (δ
H 1.54, m) with C-1′ (δ
C 39.9)/C-3′/C-4′ (δ
C 26.3), H-3′ (δ
H 1.35, m) with C-1′ (δ
C 39.9)/C-2′/C-4′/C-5′ (δ
C 36.7), H-4′ (δ
H 1.65, m) with C-3′/C-5′/C-6′ (δ
C 176.4), H-5′ (δ
H 2.21, t,
J = 7.5 Hz ) with C-3′/C-4′/C-6′ (δ
C 176.4), which suggested the presence of an –N–CH
2–CH
2–CH
2–CH
2–CH
2–COOH unit. The suggestion was supported by the
1H–
1H COSY spectrum (
Figure 2) showing correlations of H-2′ with H-1′/H-3′, and H-4′ with H-3′/H-5′, and the ESIMS (positive) spectrum showing a main fragment ion peak at
m/z 257 {100%, [M + 2H–(CH
2–CH
2–CH
2–CH
2–CH
2–COOH)]
+}. The weak HMBC correlations of H-1′ with C-2 (δ
C 160.8, s)/C-3a (δ
C 132.3, s) and comparison of the
13C NMR data of C-3a in
1 and
4 (
Table 1) suggested that the –CH
2–CH
2–CH
2–CH
2–CH
2–COOH unit should be attached on the nitrogen atom N(3) instead of the another nitrogen atom attached at C(2). So, the structure of
1 was determined as shown and the compound was named pseudozoanthoxanthin III.
Compound
2 had a molecular formula of C
20H
26N
6O
2 deduced from its (−) ESIMS spectrum (
m/z 381 [M − H]
−) and NMR spectra. Comparison of
1H and
13C NMR spectral data (
Table 1) revealed close similarities between
2 and
1. The difference between them was the absence of one methylene group and the appearance of a 1,2-disubstituted double bond [δ
H 5.59 (1H, dd,
J = 6.5, 16.0 Hz), 5.50 (1H, m), δ
C 130.8, 131.8]. Extensive 2D NMR analyses, including HSQC, HMBC and
1H–
1H COSY spectra proved that
1 and
2 had the same skeleton. Moreover, the HMBC spectrum showed correlations of H-1′ (δ
H 4.14) with C-2′ (δ
C 130.8)/C-3′ (δ
C 131.8), H-2′ (δ
H 5.59) with C-1′ (δ
C 58.6)/C-3′/C-4′ (δ
C 27.7), H-3′ (δ
H 5.50) with C-1′ (δ
C 58.6)/C-2′/C-4′/C-5′ (δ
C 25.9), H-4′ (δ
H 2.14) with C-3′/C-5′/C-6′ (δ
C 34.3), H-5′ (δ
H 1.69) with C-2′/C-3′/C-4′/C-6′, and H-6′ (δ
H 2.31) with C-4′/C-5′/C-7′ (δ
C 177.6), which suggested the presence of an –N–CH
2–CH=CH– CH
2–CH
2–CH
2–COOH unit.
This suggestion was supported by the
1H–
1H COSY spectrum (
Figure 2) showing correlations of H-2′ with H-1′/H-3′, H-4′ with H-3′/H-5′, and H-5′ with H-6′, and the (−) ESIMS spectrum showing one main fragment ion peak at
m/z 255. In the
1H NMR spectrum of
2, the coupling constant of H-2′/H-3′ (
J = 16.0 Hz) indicated that geometric configuration of double bond H-2′/H-3′ was
E. The weak HMBC correlations of H-1′ with C-2 (δ
C 160.8, s)/C-3a (δ
C 131.9, s) and comparison of the
13C NMR data of C(3a) in
2 and
4 (
Table 1) suggested that the –CH
2–CH=CH–CH
2–CH
2–CH
2–COOH unit should be attached to the nitrogen N(3). Thus, the structure of
2 was determined as shown and named pseudozoanthoxanthin IV.
Compound
6 had the molecular formula of C
16H
22O
5 as deduced from EIMS and NMR spectra. Its
1H NMR spectrum displayed four methyls at δ
H 1.80 (3H, s), 1.36 (3H, s), 2.26 (3H, s), 3.14 (3H, s). The
13C and DEPT-135 NMR spectra showed 17 carbons consisting of four methyls (δ
C 8.7, 21.9, 28.7, 50.0), three methylenes (δ
C 23.4, 27.0, 51.2), three methines (δ
C 42.1, 52.1, 56.7), two oxygenated quaternary carbons (δ
C 71.5, 106.9), one double bond (δ
C 121.4, 157.5), one lactone group (δ
C 171.9), and one keto group (δ
C 208.2). The
1H and
13C NMR spectral data of
6 showed similarity to those of 7β-hydroxyoplop-11-enone [
11] and 7β-senecioyloxyoplopa-3(14)
Z,8(10)-dien-2-one [
12], which suggested that
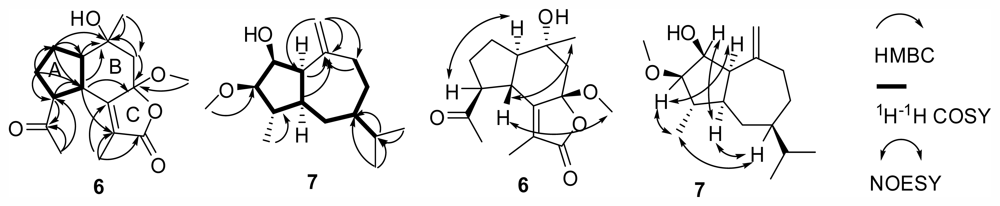
6 was an oplopane-type sesquiterpene. The suggestion was confirmed by the HMBC and
1H–
1H COSY spectra. In the HMBC spectrum (
Figure 3), correlations of H-4 (δ
H 2.65, dd,
J = 11.0, 12.5 Hz) with C-5 (δ
C 157.5)/C-6 (δ
C 106.9)/C-8 (δ
C 71.5)/C-11(δ
C 121.4, s), H-7 (δ
H 2.53, 1.77, each 1H, d,
J = 13.5 Hz) with C-6/C-8/C-9 (δ
C 56.7), H-9 (δ
H 1.84, 1H, m) with C-4 (δ
C 42.1)/C-5/C-8, and H-13 (δ
H 1.80, 3H, s) with C-5/C-11/C-12 (δ
C 171.9, s), suggested the presence of the B,C-ring substructure and Me-13 attached on C-11 to form a methyl substituted α,β-unsaturated γ-lactone unit. In addition, HMBC correlations of H-10 (δ
H 1.36, 3H, s) with C-7/C-8/C-9, and H-16 (δ
H 3.14, 3H, s) with C-6 (δ
C 106.9) indicated that Me-10 and OMe-16 were connected with C-8 and C-16, respectively. Meanwhile, the
1H–
1H COSY spectrum (
Figure 3) showed correlations of H-1 [δ
H 1.94, 1.63 (each 1H, m)] with H-9/H-2 [δ
H 2.28, 1.77 (each 1H, m)], and H-3 (δ
H 3.31, ddd,
J = 8.5, 11.0, 16.8 Hz) with H-2/H-4, suggesting the presence of A-ring unit. The suggestion was supported by HMBC correlations of H-1 with C-4/C-8/C-9, H-2 with C-1 (δ
C 23.4)/C-3 (δ
C 52.1)/C-4/C-9, and H-3 with C-2 (δ
C 27.0)/C-4. Furthermore, HMBC correlations of H-15 (δ
H 2.26, 3H, s) with C-3/C-14 (δ
C 208.2), and H-3 with C-14 indicated that an acetyl group was attached on C(3).
The relative stereochemistry of
6 was deduced from the NOESY spectrum (
Figure 3) and comparison with that of 7β-hydroxyoplop-11-enone [
11]. NOE correlations of H-3 with H-9 indicated that H-3 and H-9 were in the same α-oriented direction, and NOE correlations of H-4 with Me-10/Me-16 suggested that H-4, Me-10, and Me-16 were on the same β-oriented side. So, the structure of
6 was elucidated as shown and named 8-hydroxy-6β-methoxy-14-oxooplop-6,12-olide. Oplopanes are frequently found in terricolous plant. However this is the first report of an oplopane-type sesquiterpene isolated from a marine animal.
Compound
7 had the molecular formula of C
16H
28O
2 deduced from NMR spectra and ESIMS. The
1H NMR spectrum of
7 displayed signals for four methyls at δ
H 0.78 (3H, d,
J = 6.9 Hz), 0.95 (3H, d,
J = 6.9 Hz), 1.15 (3H, d,
J = 6.5 Hz), 3.37 (3H, s) and two oxymethines at δ
H 3.67 (1H, dd,
J = 7.0, 11.0 Hz), 4.15 (1H, dd,
J = 7.0, 10.8 Hz). The
13C NMR spectrum showed 16 carbons including four methyls (δ
C 15.8, 22.0, 30.3, 57.1), three methylenes (δ
C 24.8, 25.9, 31.5), five high-filed sp
3 methines (δ
C 29.1, 43.7, 44.5, 45.3, 53.3), two oxymethines (δ
C 78.3, 90.6), and one double bond [δ
C 109.2 (t), 147.8 (s)]. The
13C and
1H NMR data of
7 were similar to those of guaia-1(10),11-diene and guaia-9,11-diene [
13], which suggested that
7 was a guaiane-type sesquiterpene.
The suggestion was supported by
1H–
1H COSY and HMBC spectra (
Figure 3). The presence of five membered ring substructure was concluded from the
1H–
1H COSY spectrum showing correlations of H-2 (δ
H 4.15, dd,
J = 7.0, 10.8 Hz) with H-1 (δ
H 2.97, t,
J = 7.0 Hz)/H-3 (δ
H 3.67, 1H, dd,
J = 7.0, 11.0 Hz), H-4 (δ
H 1.73, m) with H-3/H-5 (δ
H 2.14, m), and H-1 with H-5. The presence of seven membered ring substructure was inferred from the
1H–
1H COSY spectrum showing correlations of H-6 (δ
H 1.97, 1.53, each 1H, m) with H-5/H-7 (δ
H 1.28, m), H-8 (δ
H 1.67, 2H, m) with H-7/H-9 (δ
H 2.04, 2.24, each 1H, m), and HMBC spectrum showing correlations C-10 (δ
C 147.8) with H-1/H-5/H-9. Furthermore, in the HMBC spectrum, correlations of H-14 [δ
H 4.64, 4.62 (each 1H, s)] with C-1 (δ
C 53.3)/C-9 (δ
C 31.5)/C-10 suggested one double bond between C-10 and C-14. HMBC correlations of H-12 (δ
H 0.78, 3H, d,
J = 6.9 Hz) and H-13 (δ
H 0.95, 3H, d,
J = 6.9 Hz) with C-7 (δ
C 43.7)/C-11 (δ
C 29.1), and H-11 (δ
H 1.73, 1H, m) with C-7/C-12 (δ
C 15.8)/C-13 (δ
C 22.0) indicated that an isopropyl unit attached on C-7 of the seven membered ring substructure. Meanwhile, HMBC correlations of H-16 (δ
H 3.37, 3H, s) with C-3 (δ
C 90.6), and H-15 (δ
H 1.15, 3H, d,
J = 6.5 Hz) with C-4 (δ
C 44.5), indicated that one methoxy group and one methyl were connected with C-3 and C-4, respectively.
The relative configuration of
7 was determined by a NOESY experiment (
Figure 3) and comparison with that of guaia-1(10),11-diene and guaia-9,11-diene [
13]. Considering the bulky isopropyl group to keep a
pseudo equatorial position and being β-oriented, H-7 had to be α-oriented. NOE correlations of H-1 with H-2/H-3/H-5, H-2 with H-5, H-3 with H-5/H-7/Me-15, and H-7 with H-5/Me-15 suggested that H-1, H-2, H-3, H-5, and Me-15 were oriented in the same direction as H-7, and should be α-orientation. Based on the above data, the structure of
7 was determined as shown and named 3β-methoxyguaian-10(14)-en-2β-ol.
In vitro antiviral activity of 1–4 against HSV-1 was evaluated using plaque reduction assay. First, the completely non-toxic concentration (CC0) of 1–4 and positive control ACV on Vero cells were tested to be 270.3, 523.6, 185.2, 195.3, >7500 μM by MTT assays, respectively, then for further antivirus studies, the concentrations of tested compounds were kept below their CC0 values. The antiviral assays displayed that 1–4 exhibited anti-HSV-1 activity with EC50 (50% effective concentration required to inhibit virus-induced cytopathicity 50%) values of 108.1, 471.2, 70.4, 117.2, 6.08 μM, respectively. The results suggested that the side chain at the nitrogen N(3) in 1–4 could affect their antiviral activity. Although 1 and 3 showed mild anti-HSV-1 activity, their activities were far lower than that of the positive control ACV.
Compound 7 was evaluated for its antilarval activity against B. amphitrite and B. neritina larvae. The results showed that 7 had significant antilarval activity towards B. amphitrite larvae with EC50 value of 17.2 μg/mL (68.2 μM), and showed 50% inhibition towards the settlement of B. neritina larvae at concentration of 25 μg/mL. The EC50 value of 7 is lower than the standard requirement of an EC50 of 25 μg/mL established by the US Navy program as an efficacy level for natural antifoulants, indicating that 7 is a potential natural antifouling agent.
3. Experimental Section
3.1. General
Optical rotations were measured with a Horiba SEAP-300 spectropolarimeter. UV spectra were measured with a Shimadzu double-beam 210A spectrophotometer in MeOH solution. 1H, 13C NMR and 2D NMR spectra were recorded on a Bruker AV-500 MHz NMR spectrometer with TMS as internal standard. MS spectral data were obtained on an LCQDECA XP HPLC/MSn spectrometer for ESIMS.
3.2. Animal Material
The South China Sea gorgonian coral E. pseudossapo (7.8 kg, wet weight) was collected in Sanya, Hainan Province, China in October 2007 and identified by Research Assistant Xiubao Li, the South China Sea Institute of Oceanology, Academia Sinica (SCSIO). A voucher specimen (No. 2007-SCSIO-3) was deposited in SCSIO, Guangzhou, China.
3.3. Extraction and Isolation
The frozen specimens of E. pseudossapo were exhaustively extracted with EtOH/CH2Cl2 (2:1) three times at room temperature, and the solvent was evaporated in vacuo. The residue was partitioned in H2O and extracted with EtOAc and n-BuOH in turn three times, respectively. The n-BuOH extract was concentrated in vacuo to afford 10.2 g of residue, and then the n-BuOH portion was subjected to column chromatography on silica, using CHCl3/MeOH (from 10:0 to 0:10) as eluent. By combining the fractions with TLC (GF254) monitoring, 8 fractions were obtained. Fraction 2 was chromatographed over Sephadex LH-20 eluting with CHCl3/MeOH (1:1) to obtain three sub-fractions (A–C). Sub-fraction B was purified over semi-preparative HPLC with MeOH/water (50:50) to yield 1 (10 mg) and 3 (4.0 mg). Sub-fraction C were purified over semi-preparative HPLC eluted with MeOH/H2O (60:40) to yield 2 (10.1 mg), 4 (13.0 mg), and 5 (2.3 mg). The EtOAc extracts were concentrated in vacuo to afford 33.5 g of residue. The EtOAc portion was subjected to column chromatography (CC) on silica, using petroleum ether-EtOAc (from 10:1 to 0:10) as eluent. By combining the fractions with TLC (GF254) monitoring, 16 fractions were obtained. Fraction 7 was purified by silica gel column, eluted with petroleum ether-EtOAc (2:1) to yield 8 (17.0 mg). Fraction 8 was subjected to CC on silica gel, eluted with CHCl3-Me2CO (from 100:5 to 0:10), and then purified with semi-preparative HPLC, using MeOH-water as eluent to afford 6 (10.0 mg) and 9 (6.4 mg). Fraction 10 was chromatographed over Sephadex LH-20 eluting with CHCl3/MeOH(1:1), then repeatedly subjected to CC on Si gel, eluted with CHCl3/MeOH (from 10:0 to 6:4) to yield 7 (10.3 mg).
Pseudozoanthoxanthin III (
1): Yellow oil; UV (MeOH) λ
max 221, 257, 304, 362 nm; IR (KBr) ν
max 3400, 3300, 1750, 1690, 1620 cm
−1;
1H NMR and
13C NMR spectral data, see
Table 1; ESI-MS (+)
m/z 371 [M + H]
+; HRESIMS
m/z 371.2159 [M + H]
+ (calcd for C
19H
27N
6O
2 371.2195).
Pseudozoanthoxanthin IV (
2): Yellow oil; UV (MeOH) λ
max 221, 257, 304, 362 nm; IR (KBr) ν
max 3407, 3313, 1752, 1694, 1623 cm
−1;
1H NMR and
13C NMR spectral data see
Table 1; ESI-MS(−)
m/z 381 [M − H]
−; HRESIMS
m/z 381.2075 [M − H]
− (calcd for C
20H
25N
6O
2, 381.2039).
8a-hydroxy-6β-methoxy-14-oxooplop-6,12-olide (6): Colorless oil; [α]25 D +0.3 (c 0.10, MeOH); UV (MeOH): 225 nm; IR (KBr) νmax 3276, 1723, 1625 cm−1; 1H NMR(500 MHz, CDCl3) δH: 1.94, 1.63 (each 1H, m, H-1), 2.28, 1.77 (each 1H, m, H-2), 3.31 (1H, ddd, J = 8.5, 11.0, 16.8 Hz, H-3), 2.65 (1H, J = 11.0, 12.5 Hz, H-4), 2.53, 1.77 (each 1H, d, J = 13.5 Hz, H-7), 1.84 (1H, m, H-9), 1.36 (3H, s, Me-10), 1.80 (3H, s, Me-13), 2.26 (3H, s, Me-15), 3.14 (3H, s, OMe-16); 13C NMR (125 MHz, CDCl3) δC: 23.4 (C-1), 27.0 (C-2), 52.1 (C-3), 42.1 (C-4), 157.5 (C-5), 106.9 (C-6), 51.2 (C-7), 71.5 (C-8), 56.7 (C-9), 21.9 (C-10), 121.4 (C-11), 171.9 (C-12), 8.7 (C-13), 208.2 (C-14), 28.7 (C-15), 50.0 (C-16); HR-EI-MS m/z 294.1472 [M]+ (calcd for C16H22O5, 294.1467).
3β-methoxyguaian-10(14)-en-2β-ol (7): Colorless oil; [α]25 D +0.8 (c 0.10, MeOH); IR (KBr) νmax 3446, 1648, 1456 cm−1; 1H NMR(500 MHz, CDCl3) δH: 2.97 (1H, t, J = 7.0 Hz, H-1), 4.15 (1H, dd, J = 7.0, 10.8 Hz, H-2), 3.67 (1H, dd, J = 7.0, 11.0 Hz, H-3), 1.73 (1H, m, H-4), 2.14 (1H, m, H-5), 1.97, 1.53 (each 1H, m, H-6), 1.28 (1H, m, H-7), 1.67 (2H, m, H-8), 2.04, 2.24 (2H, m, H-9), 1.73 (1H, m, H-11), 0.78 (3H, d, J = 6.9 Hz, Me-12), 0.95 (3H, d, J = 6.9 Hz, Me-13), 4.64, 4.62 (each 1H, s, H-14), 1.15 (3H, d, J = 6.5 Hz, Me-15), 3.37 (3H, s, OMe-16); 13C NMR (125 MHz, CDCl3) δC: 53.3 (C-1), 78.3 (C-2), 90.6 (C-3), 44.5 (C-4), 45.3 (C-5), 25.9 (C-6), 43.7 (C-7), 24.8 (C-8), 31.5 (C-9), 147.8 (C-10), 29.1 (C-11), 15.8 (C-12), 22.0 (C-13), 109.2 (C-14), 30.3 (C-15), 57.1 (C-16); HR-EI-MS m/z 252.2082 [M]+ (calcd for C16H28O2, 252.2089).
3.4. Viruses and Cells
HSV-1 (15577) strain and Vero cells were obtained from American Type Culture Collection. Cytotoxicity assay and cytopathic effect reduction assay were undertaken with the reported methods [
14]. ACV was used as the positive control.
3.5. Larval Settlement Bioassays
Antilarval activity of the compounds was evaluated in settlement inhibition assays with laboratory-reared
Balanus amphitrite and
Bugula neritina larvae. The procedures were the same as previously reported [
15].





{kind=link}
{kind=link}
{kind=link}