1. Introduction
Lots of bioinformatics techniques have been developed for the disease detection and diagnosis of patients with incurable diseases such as cancer for several decades [
1]. However, it still remains challenging to deal with cancer patients. Furthermore, the development of appropriate classification models using the gene which is extracted from patients is useful for early diagnosis of both patients and normal people. Cancer is a major disease which causes death, involving abnormal cell differentiation [
2]. It is caused by many reasons, but the majority of cancers (90~95%) are due to genetic mutations from lifestyle factors such as smoking, obesity, alcohol and so on. The remaining 5~10% are caused due to inherited genes [
3].
A hematopoietic malignancy is a neoplasm from hematopoietic cells in the bone marrow, lymph nodes, peripheral blood and lymphatic system which is related to organs of the hematopoietic system. Furthermore, it can be found in other organs such as the gastrointestinal system and the central nervous system [
4]. Given that hematopoietic malignancies occur in the hematopoietic system, this malignancy is called a liquid tumor. While uncommon in other cancers, chromosomal translocations are a common cause of these diseases. Hematopoietic cancer accounts for 8~10% of all cancer diagnoses, and its mortality rate is also similar to this [
5].
Historically, hematopoietic cancer is divided by whether the malignant location is in the blood or the lymph nodes. However, in 2001, the World Health Organization (WHO) introduced the WHO classification of tumors of hematopoietic and lymphoid tissue as a standard and updated it in 2008 and 2016 [
6]. This WHO classification criterion focused on cell linkage rather than the location of the occurrence. According to the WHO classification, hematopoietic malignancies are mainly divided into leukemia, lymphoma and myeloma [
7].
Leukemia is one type of hematopoietic cancer that results from genetic changes in hematopoietic cells in the blood or bone marrow. If an abnormality occurs in the bone marrow, the abnormally generated blood cells mix with blood in the body and spread widely into the body through the blood stream [
8]. Most leukemia cases are diagnosed in adults aged over 65 years, but it is also commonly observed in children under the age of 15 [
7]. The American Cancer Society (ACS) reported, in 2020, that the United States will see about 60,530 new cases and 23,100 deaths from leukemia [
9]. Lymphoma is usually found in distinct stationary masses of lymphocytes, such as the lymph node, thymus or spleen. Like leukemia, lymphoma can also travel through the whole body by the blood stream. Commonly, lymphoma cases are divided into Hodgkin lymphoma, non-Hodgkin lymphoma, acquired immune deficiency syndrome (AIDS)-related lymphoma and primary central nervous system (CNS) lymphoma [
8]. In 2020, the ACS reported that there will be approximately 85,720 new cases and 20,910 deaths from lymphoma [
9]. Myeloma is a tumor that occurs in plasma cells which are differentiated from bone marrow, blood or other tissue. Plasma cells generate antibodies that protect against disease or infection, but when they develop abnormally, it interferes with antibody generation and causes confusion in the human immune system [
8]. According to the estimation by the ACS, there would be 32,270 new cases and 12,830 deaths in 2020 [
9].
There have been several studies conducted using genetic data-based cancer classification by machine learning and data mining approaches [
10,
11,
12,
13,
14,
15,
16,
17,
18]. For instance, one study [
10] utilized several machine learning approaches with naïve Bayes and k-nearest neighbor classification algorithm for breast cancer classification. Another study [
11] applied classification algorithms for binary subtype cancer classification on acute myeloblastic leukemia microarray data. Other kinds of research also typically use machine learning algorithms such as support vector machine, decision tree, random forest, lasso and neural network [
12,
13,
14,
15,
16,
17,
18].
Over the years, various approaches for data mining have been applied on many cancer research studies. Specifically, a deep learning method was applied in this area [
19,
20,
21,
22,
23]. Ahmed M et al. [
19] developed a breast cancer classification model using deep belief networks in an unsupervised part for learning input feature statistics. Additionally, in the supervised part, they adopted a conjugate gradient and Levenberg–Marquardt algorithm.
Furthermore, there are several studies on cancer subtype classification. A study [
20] used a deep learning approach for kidney cancer subtype classification using miRNA data from The Cancer Genome Atlas (TCGA), which contained five different subtypes. They employed neighborhood component analysis for feature extraction and a long short-term memory (LSTM)-based classifier. DeepCC [
22] architecture using the gene set enrichment analysis (GSEA) method and artificial neural network generated deep cancer subtype classification frameworks that made comparisons between machine learning algorithms such as support vector machine, logistic regression and gradient boost using a colon cancer dataset from TCGA which contains 14 subtype cancer labels, and a Deeptype [
23] framework has been made for cancer subtype classification based on PAM50 [
24]. They used a multi-layer neural network structure for adopting representation power to project on representation space.
Traditional approaches [
10,
11,
12,
13,
14,
15,
16,
17,
18] have used statistical techniques for cancer classification. Sun et al. [
17] used an entropy-based approach for feature extraction in a cancer classification model. However, this method has a disadvantage; that is, multiple classes cannot be applied at once because each cancer is classified one by one through binary classification for cancer classification. In the case of Deeptype [
23], clustering is established using a specific gene set called PAM50, previously known for breast cancer.
However, since this PAM50 indicator is used as an already known indicator, subtypes can be classified through some valid information about breast cancer. Above all, for other cancers, including hematopoietic cancer, there is no gene set for subtype classification. In view of this, our work has a difference in feature extraction and subtype classification only from the gene expression data of hematologic cancer. In order to overcome the demerit of the multi-class classification task and the limitations due to the absence of a gene set, we applied a method of feature extraction using an autoencoder-based method among deep learning methods. In addition, we propose a subtype classification method in which reconstruction error is generated by the autoencoder. The classification error generated by the classification model and merged error are used as the loss function by referring to the loss function application methods of Deeptype [
23].
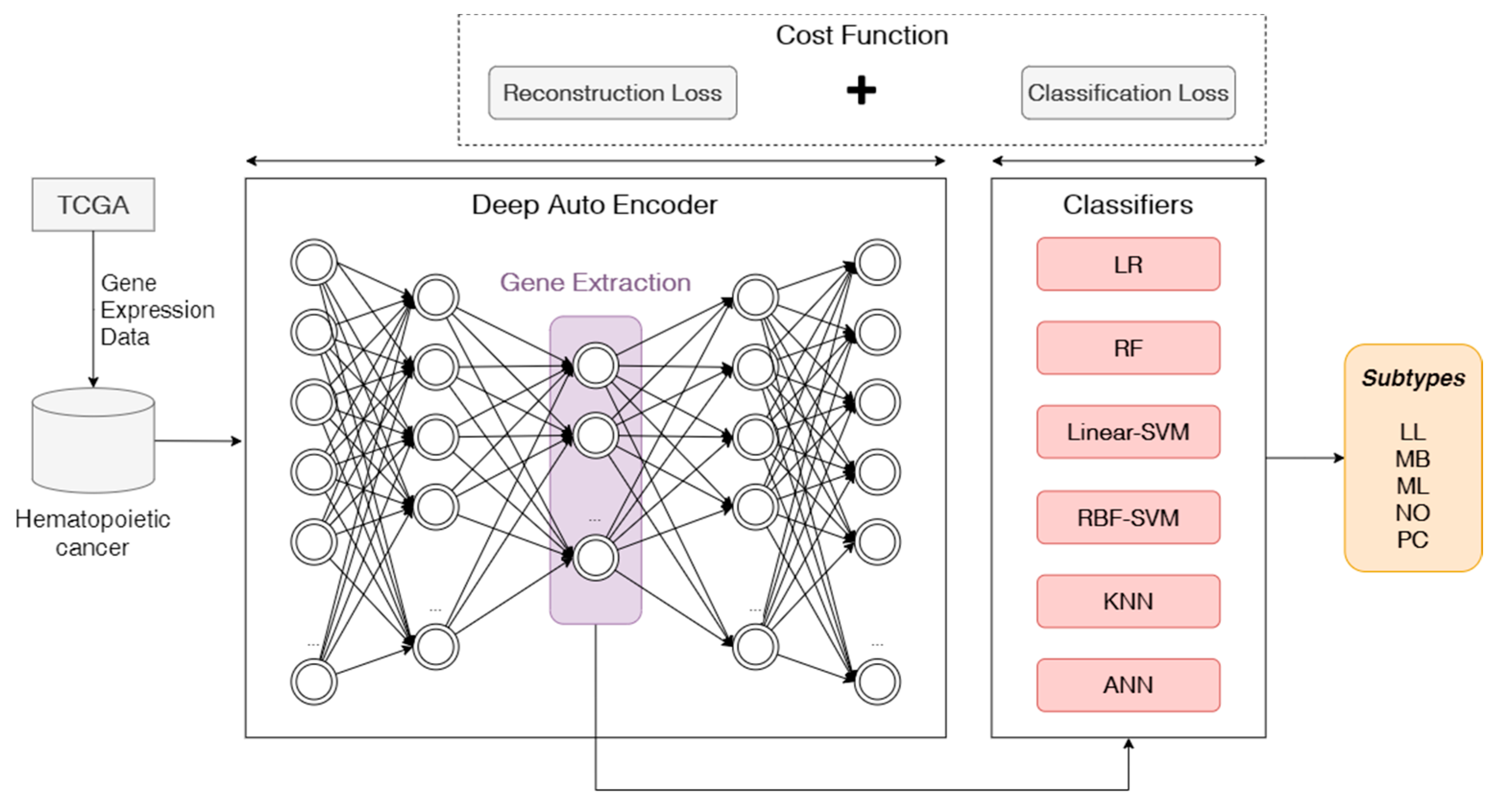
The goal of this work is to develop an autoencoder-based feature extraction approach for hematopoietic cancer subtype classification. We not only focus on the five subtypes of hematopoietic cancer and conduct a study on classifying by applying deep learning techniques, but we also perform a feature extraction and calculation of two kinds of errors, which are reconstruction error and classification error. In the process, first a deep autoencoder approach is used for extracting suited features for building a classification model, and then the reconstruction error and classification error (cross-entropy loss and focal loss) are calculated for considering the data imbalance problem when building the classification model using the extracted features. To validate the deep autoencoder-based classification model, we compared other traditional feature selection algorithms and classification algorithms with an oversampling approach. We compared five widely used classification algorithms including logistic regression (LR), random forest (RF), k-nearest neighbor (KNN), artificial neural network (ANN) and support vector machine (SVM).
We compared our proposed method with traditional cancer classification and cancer subtype classification methods such as data mining and machine learning approaches which are not able to be used in the previous end-to-end approaches. Our end-to-end approach has multiple steps including feature engineering, data imbalance handling and a classification task. The objectives of this study are to extract features from a deep learning-based approach on the gene expression data for predicting hematopoietic cancer subtypes and develop an end-to-end deep learning-based classification model. The major contributions of this study are, briefly, as follows:
We propose an end-to-end approach without any manual engineering, which classifies hematopoietic cancer subtypes;
We adopt a non-linear transformation step by using a deep autoencoder to select deep features from gene expression data in hematopoietic cancer by adopting a deep learning architecture-based feature engineering task;
We implement a mixed loss function for the proposed deep learning model, considering both the compression of knowledge representation and the data imbalance problem.
The remainder of this paper is organized as follows:
Section 2 introduces the hematopoietic cancer gene expression dataset from TCGA. Furthermore, the proposed deep autoencoder-based approach is explained in detail. In
Section 3, the experimental results are provided. Finally,
Section 4 discusses the experimental results with our conclusion.
4. Discussion
In this paper, we suggested an autoencoder-based feature extraction approach for hematopoietic cancer subtype classification. The five major hematopoietic cancer subtypes were selected to create experimental data based on gene expression level. We also compared our approach with traditional feature extraction algorithms, PCA and NMF, which are widely used in cancer classification based on gene expression data. In addition, in consideration of the class imbalance problem occurring in multi-label classification, we applied the SMOTE oversampling algorithm.
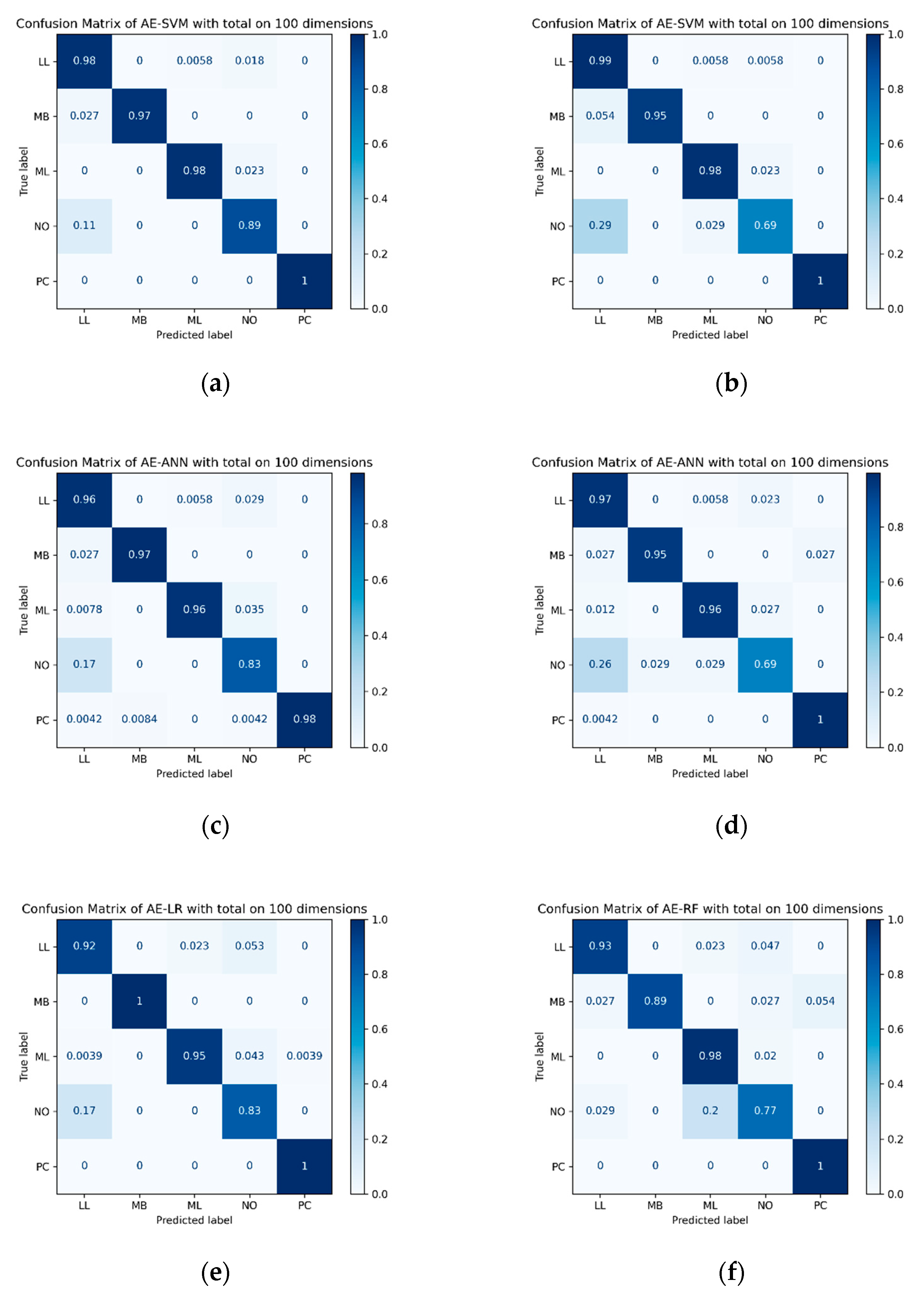
In the experimental results, the traditional feature selection approaches, NMF and PCA, showed good performance, but our proposed DAE-based approach for subtype classification showed a better performance. For example, in the results of the SVM classifier using the SMOTE oversampling method, the PCA and NMF feature extraction approaches showed 90.63% and 90.22% accuracy, respectively, and the AE-based feature extraction approaches with cross-entropy error (CE), reconstruction error (RE) and merged error (CE + RE) showed 93.34%, 96.06% and 96.88% classification accuracy, respectively. This result was also the same when focal loss was applied instead of cross-entropy loss. The accuracy for each focal loss case that applied the AE-based feature extraction approach with focal loss (FL), reconstruction error (RE) and merged error (FL + RE) was 90.08%, 95.65% and 97.01%, respectively. Although SVM showed the best result with merged error (FL + RE) using focal loss, in other cases, we found that the merged error (CE + RE) using cross-entropy error showed the best performance in the other feature extraction approaches. Using those extracted results on classification algorithm, the result of subtype classification using the DAE-based feature selection approach showed better performance than traditional statistics and machine learning feature extraction approaches.
Furthermore, as shown in
Table 8, when all of the results were summarized, we found that the AE-based feature extraction approach shows better performance than other feature extraction methods. In addition, when comparing the loss function, the results of applying both the classification error (FL/CE) and the reconstruction error (RE) together showed better performance rather than the single loss function, and the sampling method also showed better results when applying the SMOTE oversampling technique.
5. Conclusions
In this paper, we focused on the autoencoder-based feature extraction method to extract biological information from complex cancer data such as gene expression, clinical data and methylation data. We evaluated the proposed method on TCGA data samples from 2457 patients with hematopoietic cancer: lymphoid leukemia, myeloid leukemia, leukemia nos (not otherwise specified), mature B-cell leukemia and plasma cell neoplasm. To the best of our knowledge, there is no other research work on hematopoietic cancer using deep learning-based feature extraction techniques. We compared the proposed autoencoder-based method to the traditional state-of-the-art algorithms PCA and NMF, as well as another generative deep learning technique, VAE. We provided comprehensive experimental results that show the efficiency of our proposed method.
As shown in the experimental results, the proposed method shows higher performance than the other compared techniques in terms of various evaluation metrics. The proposed method with TOF loss achieved the highest accuracy (97.01%), precision (92.68%), recall (94.60%), specificity (99.52%), F1-measure (93.53%), G-mean (97.87%) and index imbalanced accuracy (95.46%) followed by the SVM classifier, which was trained on the sampled data by SMOTE. The learned representations contain rich, valuable information related to hematopoietic cancer which also can be used for other downstream tasks such as regression, classification, survival analysis, etc. We also applied the SHAP feature interpretation technique to our pre-trained model to explain the black box and show the importance of each bio-marker. By extracting bio-markers using deep learning structures, this study is expected to be helpful in enabling gene-specific treatment of patients. Furthermore, it is expected that this model will be helpful in the development of public healthcare through extensibility that can be applied not only to cancer but also to various diseases.
In conclusion, we found that our autoencoder-based feature extraction approach for hematopoietic cancer subtype classification algorithm showed good classification performance in multiclass classification, and our suggested approach showed better performance than PCA and NMF, which are widely used feature extraction methods for cancer classification. Furthermore, the problem of unbalanced data can be solved by applying the SMOTE method.


 ,
, 















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}