Potassium Dichromate Induced Cytotoxicity, Genotoxicity and Oxidative Stress in Human Liver Carcinoma (HepG2) Cells




{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Cell Culture and Cytotoxicity
2.3. ROS Detection
2.4. Comet Assay
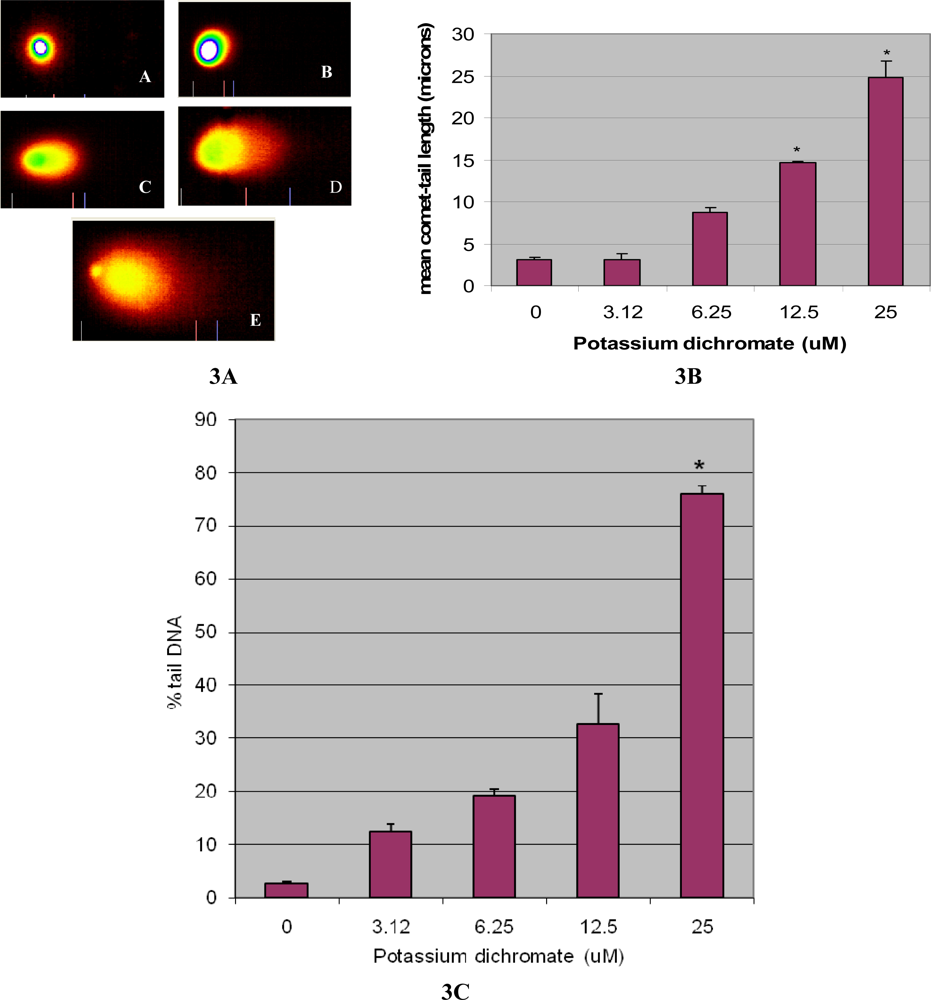
- DNA head (DNAH), sum of intensities of all points of the head.
- DNA tail (DNAT), sum of intensities of all points of the tail.
- Percent tail DNA (%DNAT) = 100DNAT/(DNAH+DNAT)
2.5. Malondialdehyde (MDA) Determination
3. Data Analysis and Statistics
4. Results
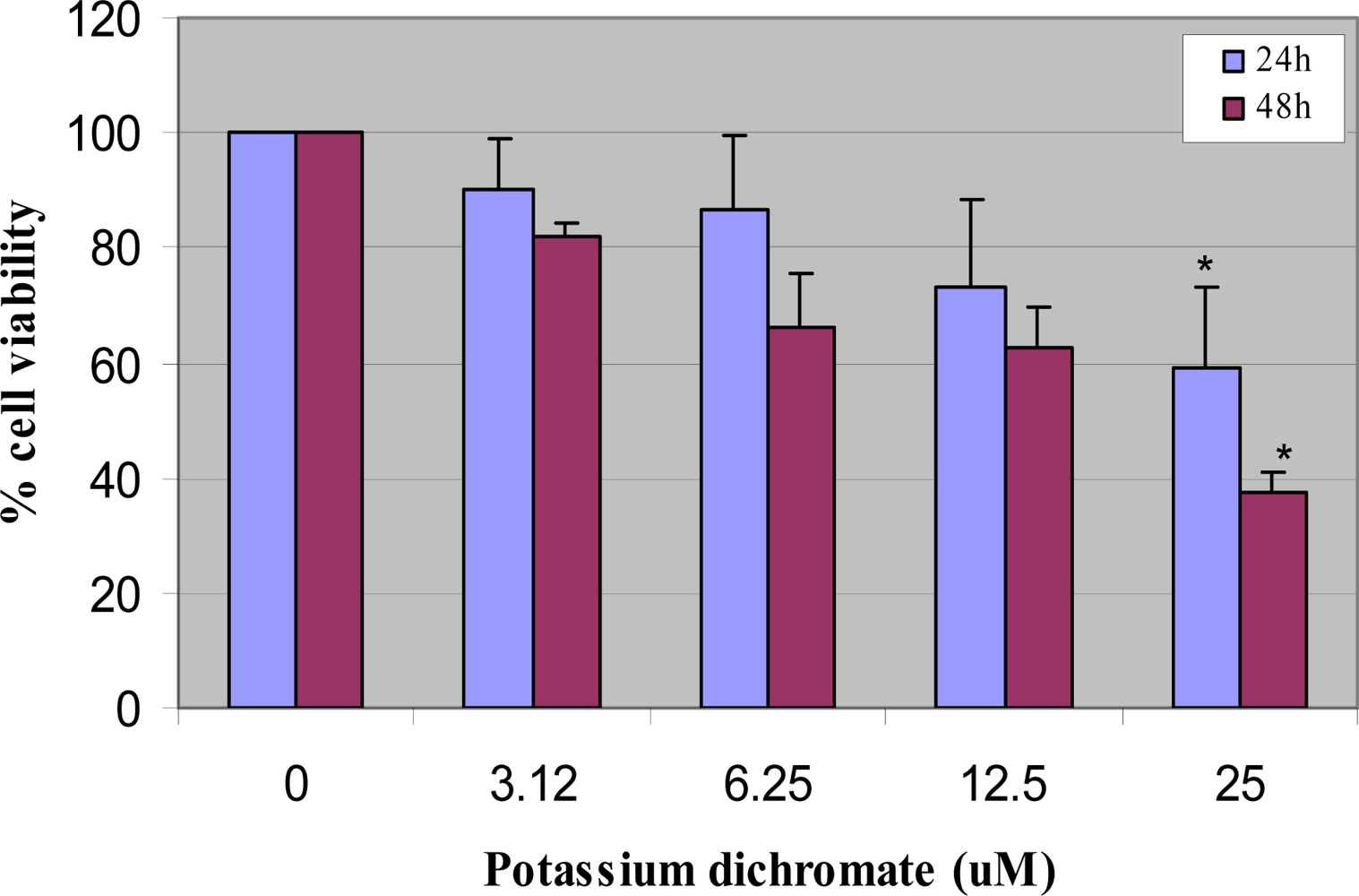
4.1. Cytotoxicity Assay
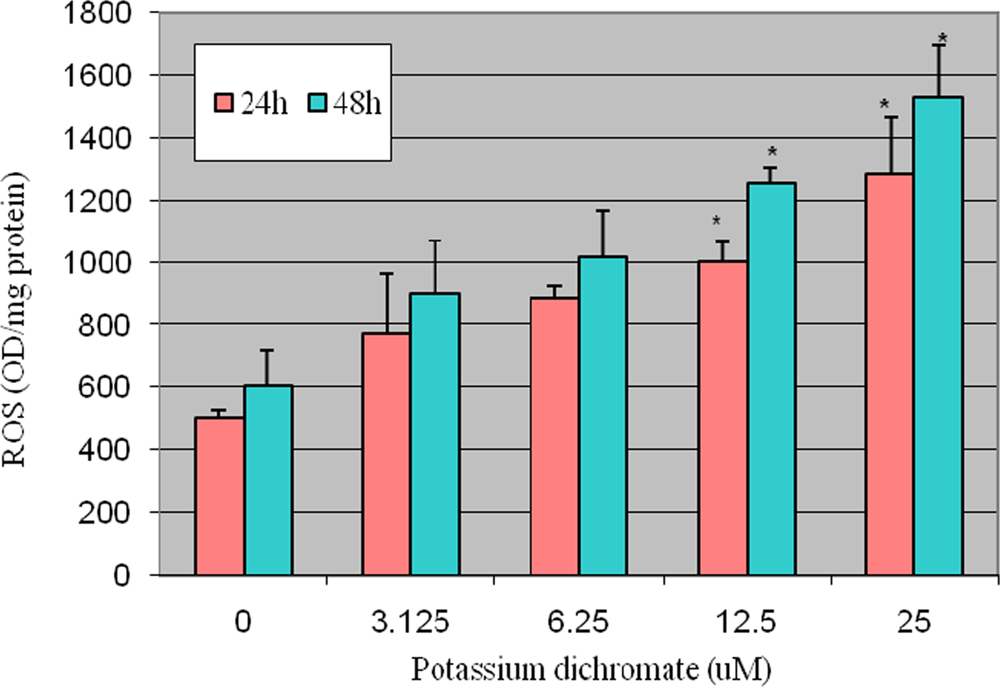
4.2. ROS Detection
4.3. DNA Damage [Comet assay]
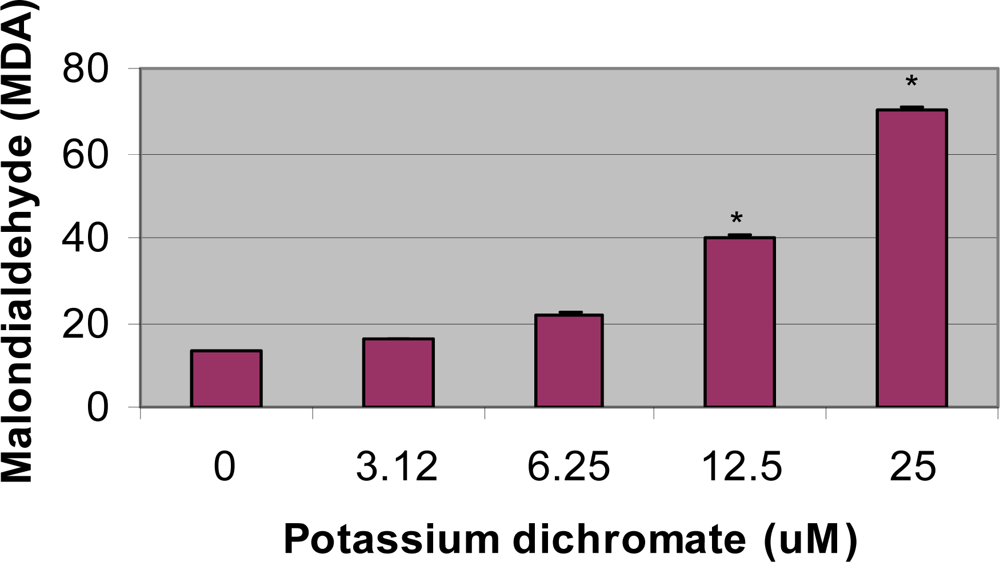
4.4. Lipid Peroxidation
5. Discussion
Acknowledgments
References
- Cohen, MD; Kargacin, B; Klein, CB; Costa, M. Mechanisms of chromium carcinogenicity and toxicity. Crit. Rev. Toxicol 1993, 23, 255–281. [Google Scholar]
- Norseth, T. The carcinogenicity of chromium - Review. Environ. Health. Perspect 1981, 40, 121–30. [Google Scholar]
- Wang, XF; Xing, ML; Shen, Y; Zhu, X; Xu, LH. Oral administration of Cr (VI) induced oxidative stress, DNA damage and apoptotic cell death in mice. Toxicology 2006, 228, 16–23. [Google Scholar]
- Kim, E; Na, KJ. Nephrotoxicity of sodium dichromate depending on the route of administration. Arch. Toxicol 1991, 65, 537–541. [Google Scholar]
- Gumbleton, M; Nicholls, PJ. Dose-response and time-response biochemical and histological study of potassium dichromate-induced nephrotoxicity in the rat. Food Chem. Toxicol 1988, 26, 37–44. [Google Scholar]
- Bagchi, D; Hassoun, EA; Bagchi, M; Muldoon, D; Stohs, SJ. Oxidative stress induced by chronic administration of sodium dichromate (Cr VI) to rats. Comp. Biochem. Physiol 1995a, 110C, 281–287. [Google Scholar]
- Bagchi, D; Vuchetich, PJ; Bagchi, M; Hassoun, EA; Tran, MX; Tang, L; Stohs, SJ. Induction of oxidative stress by chronic administration of sodium dichromate (chromium VI) and cadmium chloride (cadmium II) to rats. Free Rad. Biol. Med 1997b, 22, 471–478. [Google Scholar]
- Costa, M. Toxicity and carcinogenicity of Cr (VI) in animal models and humans. Crit Rev Toxico 1997, 27, 431–42. [Google Scholar]
- Dayan, AD; Paine, AJ. Mechanisms of chromium toxicity, carcinogenicity and allergenicity: review of the literature from 1985 to 2000. Hum Exp Toxicol 2001, 20, 439–51. [Google Scholar]
- Gambelunghe, A; Piccinini, R; Ambrogi, M; Villarini, M; Moretti, M; Marchetti, C; Abbritti, G; Muzi, G. Primary DNA damage in chrome-plating workers. Toxicology 2003, 188, 187–95. [Google Scholar]
- Goulart, M; Batoreu, MC; Rodrigues, AS; Laires, A; Rueff, J. Lipoperoxidation products and thiol antioxidants in chromium exposed workers. Mutagenesis 2005, 20, 311–15. [Google Scholar]
- Connett, PH; Wetterhahn, KE. Metabolism of carcinogenic chromate by cellular constituents. Struct. Bonding 1983, 54, 93–24. [Google Scholar]
- De Flora, S; Bagnasco, M; Serra, D; Zanacchi, P. Genotoxicity of chromium compounds: a review. Mutat. Res 1990, 238, 99–172. [Google Scholar]
- De Flora, S; Wetterhahn, KE. Mechanisms of chromium metabolism and genotoxicity. Life Chem. Rep 1989, 7, 169–244. [Google Scholar]
- Shi, X; Chiu, A; Chen, CT; Halliwell, B; Castranova, V; Vallyathan, V. Reduction of chromium (VI) and its relationship to carcinogenesis. J. Toxicol. Environ. Health 1999, 2, 87–104. [Google Scholar]
- O’Brien, TJ; Ceryak, S; Patierno, SR. Complexities of chromium carcinogenesis: role of cellular response, repair and recovery mechanisms. Mutat. Res 2003, 533, 3–36. [Google Scholar]
- Nordberg, J; Arner, ES. Reactive oxygen species, antioxidants, and the mammalian thioredoxin system. Free Radical Biol. Med 2001, 31, 1287–1312. [Google Scholar]
- Gutteridge, JMC; Quinlan, GJ. Malondialdehyde formation from lipid peroxides in thiobarbituric acid test. The role of lipid radicals, iron salts and metal chelator. J. Appl. Biochem 1983, 5, 293–299. [Google Scholar]
- Halliwell, B. Oxygen radicals: A common sense look at their nature and medical importance. Med. Biol 1984, 62, 71–77. [Google Scholar]
- Murray, RK; Granner, DK; Mayes, PA; Rodwell, VW. Harper’s Biochemistry, 21st Edition ed; Prentice Hall: NJ, USA, 1988; pp. 138–139. [Google Scholar]
- Holvoet, P; Collen, D. Oxidation of low density lipoproteins in the pathogenesis of atherosclerosis. Atherosclerosis 1998, 137(Suppl), S33–38. [Google Scholar]
- Spiteller, G. Lipid peroxidation in aging and age-dependent diseases. Exp. Gerontol 2001, 36, 1425–1457. [Google Scholar]
- Henrotin, Y; Deby-Dupont, G; Deby, C; Franchimont, P; Emerit, I. Active oxygen species, articular inflammation and cartilage damage. EXS 1992, 62, 308–322. [Google Scholar]
- Marnett, LJ. Oxyradicals and DNA damage. Carcinogenesis 2000, 21, 361–370. [Google Scholar]
- Bismuth, C; Garnier, R; Baud, FJ; Muszynski, J; Keyes, C. Paraquat poisoning; An overview of current status. Drug Saf 1990, 5, 243–251. [Google Scholar]
- Brattin, WJ; Glende, EA; Recknagel, RO. Pathological mechanisms in carbon tetrachloride hepatotoxicity. J. Free Radic. Biol. Med 1985, 1, 27–38. [Google Scholar]
- Kasprzak, KS. Possible role of oxidative damage in metal induced carcinogensis. Cancer Inv 1995, 13, 411–430. [Google Scholar]
- De Zwart, LL; Meerman, JH; Commandeur, JN; Vermeulen, NP. Biomarkers of free radical damage applications in experimental animals and in humans. Free Radic Biol Med 1999, 26, 202–226. [Google Scholar]
- Gutteridge, JMC. Lipid peroxidation and antioxidants as biomarkers of tissue damage. Clin. Chem 1995, 41, 1819–1828. [Google Scholar]
- Lawler, JM; Song, W; Demaree, SR. Hindlimb unloading increases oxidative stress and disrupts antioxidant capacity in skeletal muscle. Free Radical. Biol. Med 2003, 35, 9–16. [Google Scholar]
- Singh, NP; McCoy, MT; Tice, RR; Schneider, EL. A simple technique for quantitation of low levels of DNA damage in individual cells. Exp. Cell Res 1988, 175, 184–191. [Google Scholar]
- Bagchi, D; Bagchi, M; Stohl, SJ. Chromium (VI)-induced oxidative stress, apoptotic cell death and modulation of p53 tumor suppressor gene. Mol. Cell Biochem 2001, 222, 149–158. [Google Scholar]
- Sugiyama, M. Role of physiological antioxidants in Cr (VI) induced cellular injury. Free Rad. Biol. Med 1992, 12, 397–407. [Google Scholar]
- Miesel, R; Kroger, H; Kurpisz, M; Wesser, U. Induction of arthritis in mice and rats by potassium peroxochromate and assessment of disease activity by whole blood chemiluminescence and 99mpertechnetate-imaging. Free Radic. Res 1995, 23, 213–227. [Google Scholar]
- Kadiiska, MB; Xiang, QH; Mason, RP. In vivo free radical generation by chromium (VI): An electron-spin resonance spin-trapping investigation. Chem. Res. Toxicol 1994, 7, 800–805. [Google Scholar]
- Dana Devi, K; Rozati, R; Saleha Banu, B; Jamil, K; Grover, P. In vivo genotoxic effect of potassium dichromate in mice leukocytes using comet assay. Food Chem. Toxicol 2001, 39, 859–865. [Google Scholar]
- Wise, SS; Holmes, AL; Wise, JP, Sr. Hexavalent chromium-induced DNA damage and repair mechanisms. Rev Environ Health 2008, 23, 39–57. [Google Scholar]
- Blasiak, J; Kowalik, J. A comparison of the in vitro genotoxicity of tri and hexavalent chromium. Mutat Res 2000, 469, 135–145. [Google Scholar]




Share and Cite
Patlolla, A.K.; Barnes, C.; Hackett, D.; Tchounwou, P.B. Potassium Dichromate Induced Cytotoxicity, Genotoxicity and Oxidative Stress in Human Liver Carcinoma (HepG2) Cells. Int. J. Environ. Res. Public Health 2009, 6, 643-653. https://doi.org/10.3390/ijerph6020643
Patlolla AK, Barnes C, Hackett D, Tchounwou PB. Potassium Dichromate Induced Cytotoxicity, Genotoxicity and Oxidative Stress in Human Liver Carcinoma (HepG2) Cells. International Journal of Environmental Research and Public Health. 2009; 6(2):643-653. https://doi.org/10.3390/ijerph6020643
Chicago/Turabian StylePatlolla, Anita K., Constance Barnes, Diahanna Hackett, and Paul B. Tchounwou. 2009. "Potassium Dichromate Induced Cytotoxicity, Genotoxicity and Oxidative Stress in Human Liver Carcinoma (HepG2) Cells" International Journal of Environmental Research and Public Health 6, no. 2: 643-653. https://doi.org/10.3390/ijerph6020643
APA StylePatlolla, A. K., Barnes, C., Hackett, D., & Tchounwou, P. B. (2009). Potassium Dichromate Induced Cytotoxicity, Genotoxicity and Oxidative Stress in Human Liver Carcinoma (HepG2) Cells. International Journal of Environmental Research and Public Health, 6(2), 643-653. https://doi.org/10.3390/ijerph6020643