Cutaneous Involvement in Diseases with Plasma Cell Differentiation: Diagnostic Approach

 ,
,  , ,
, ,  , , ,
, , ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. PCMZL
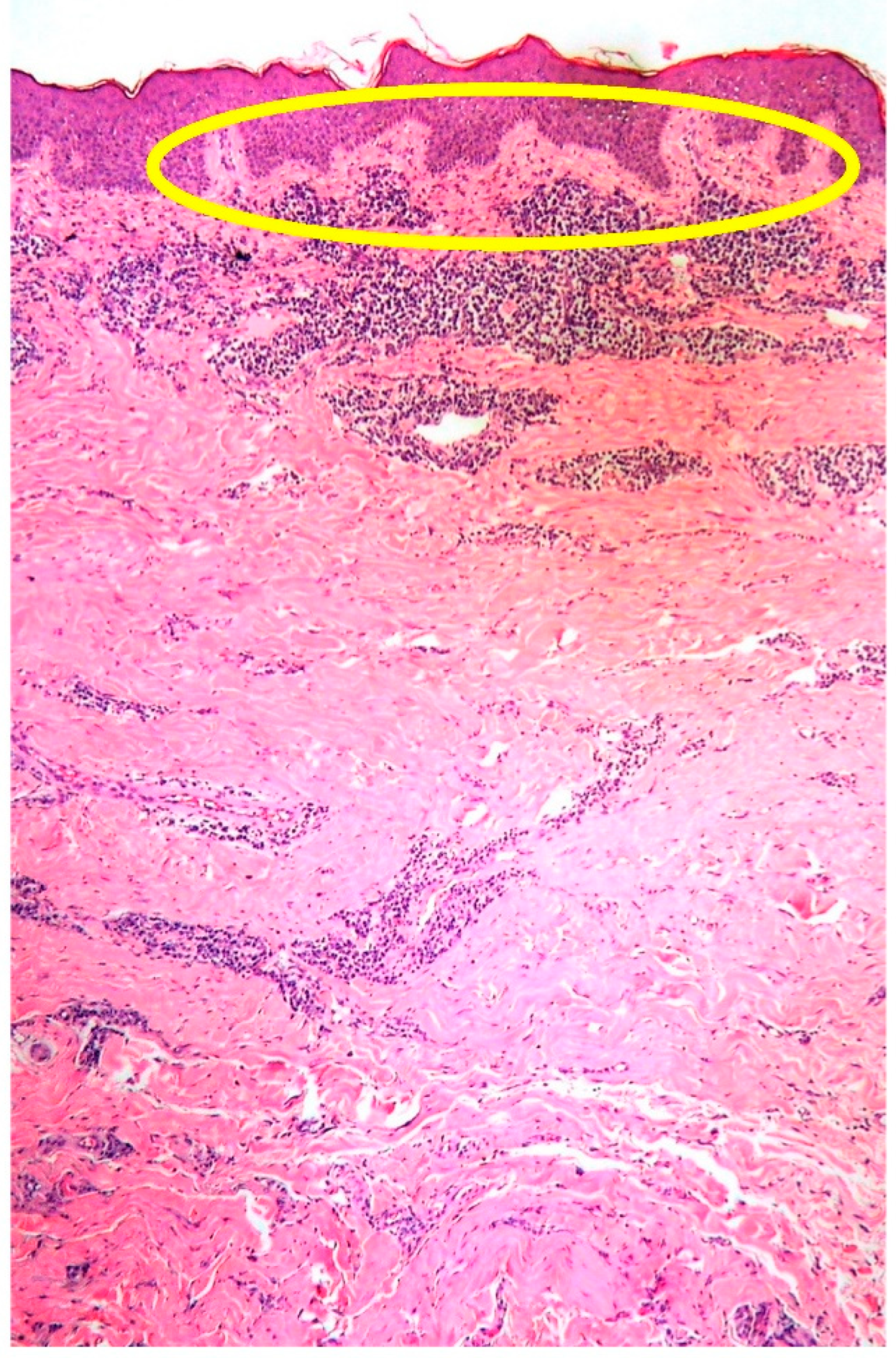
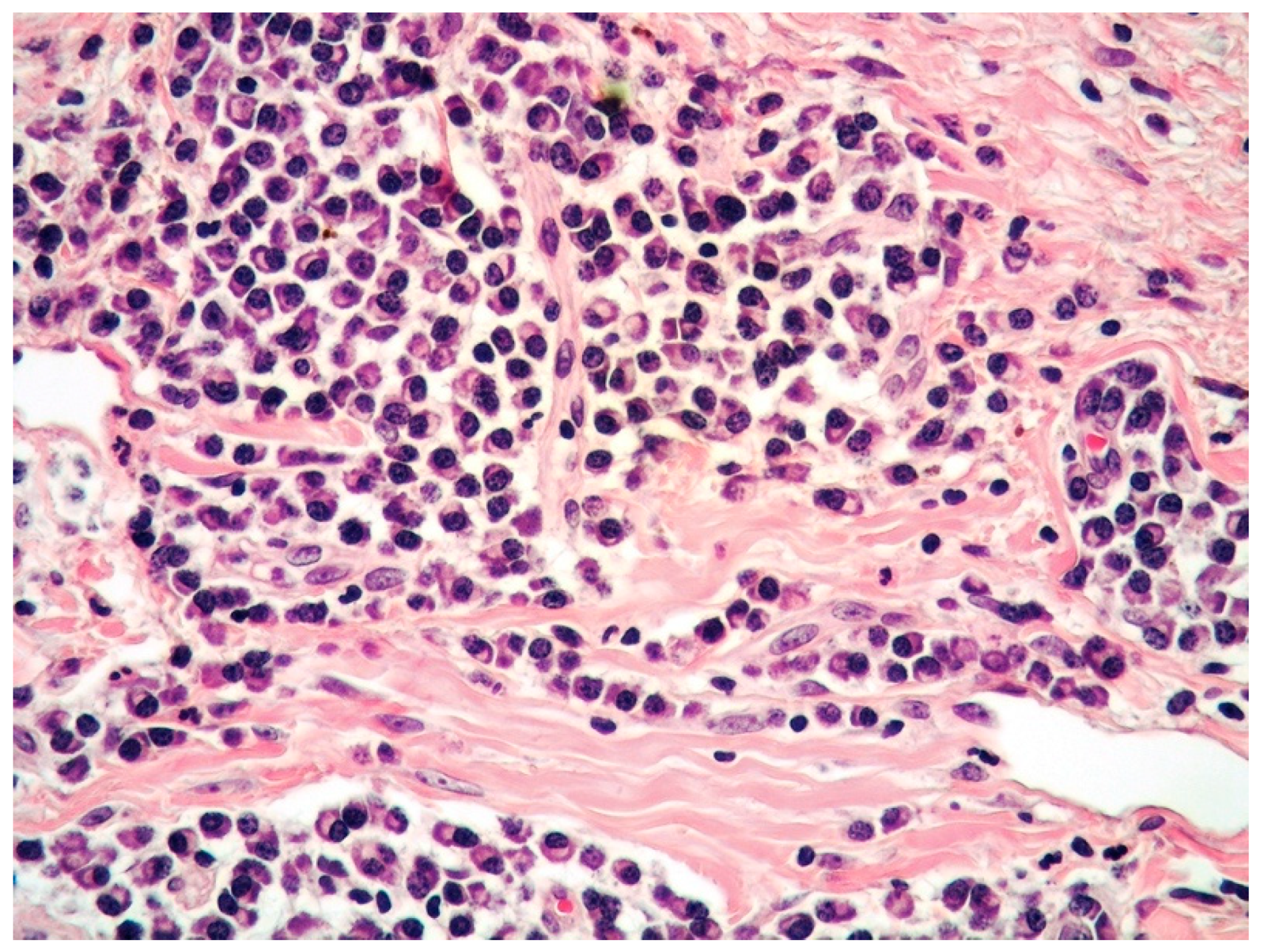
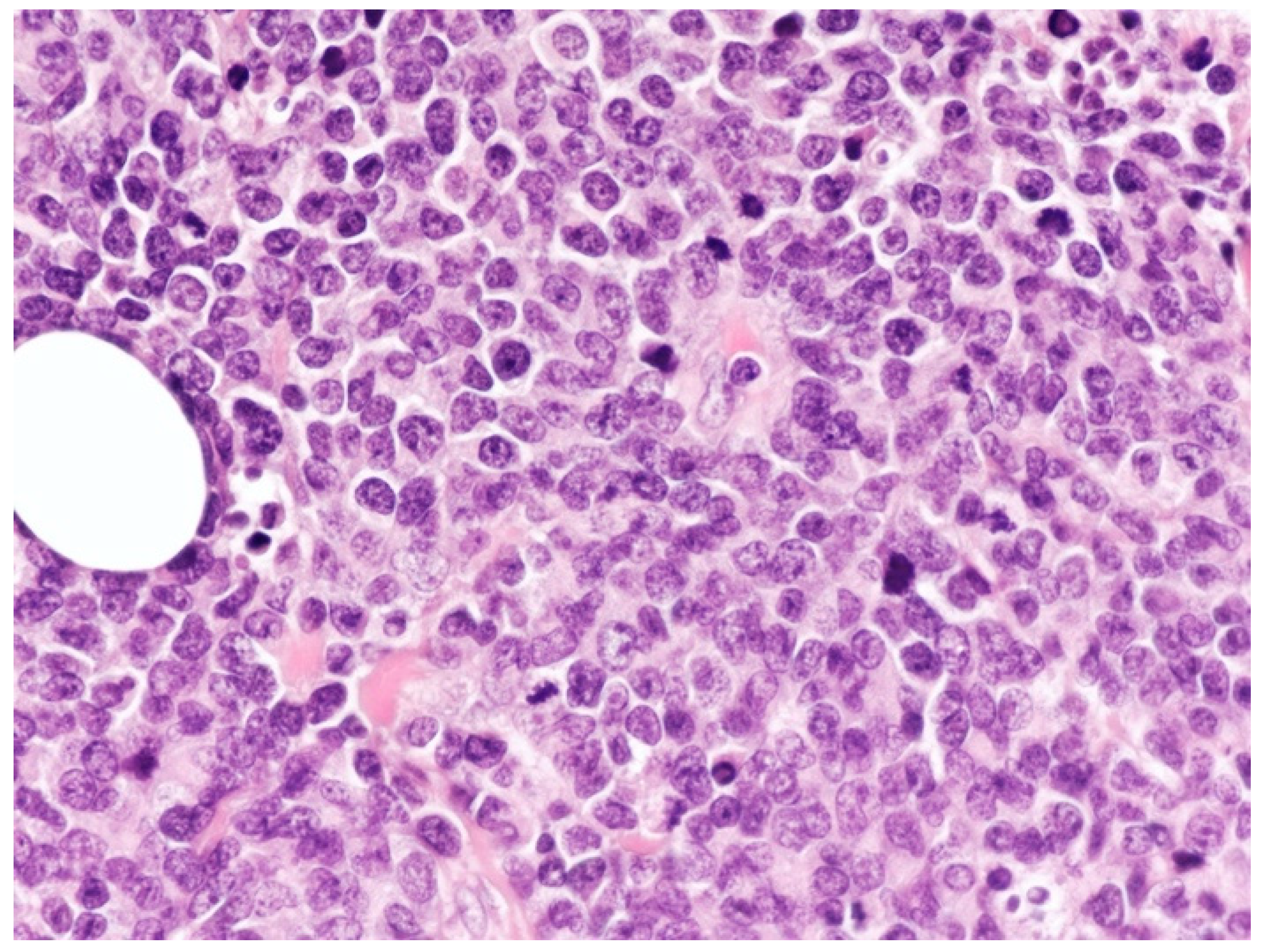
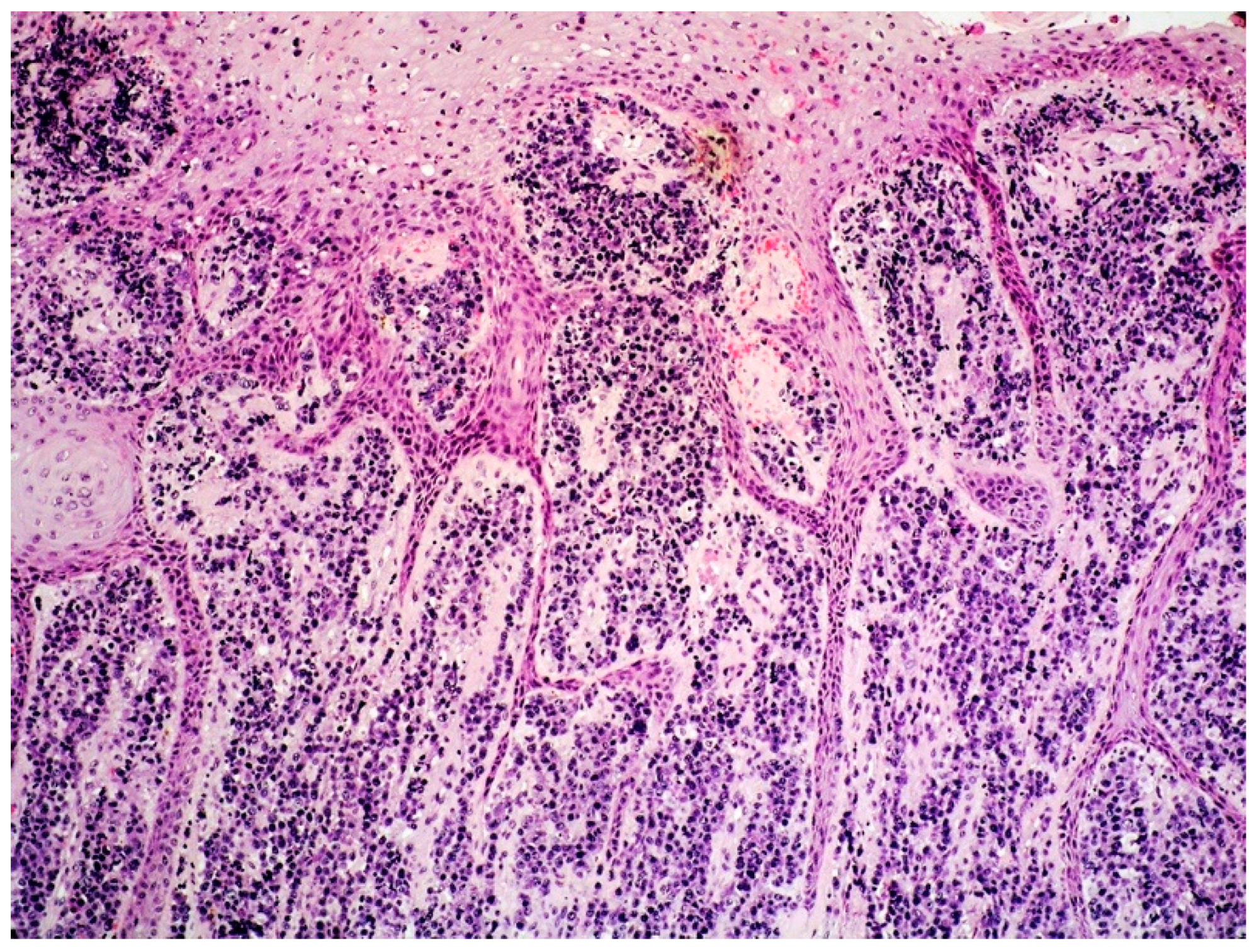
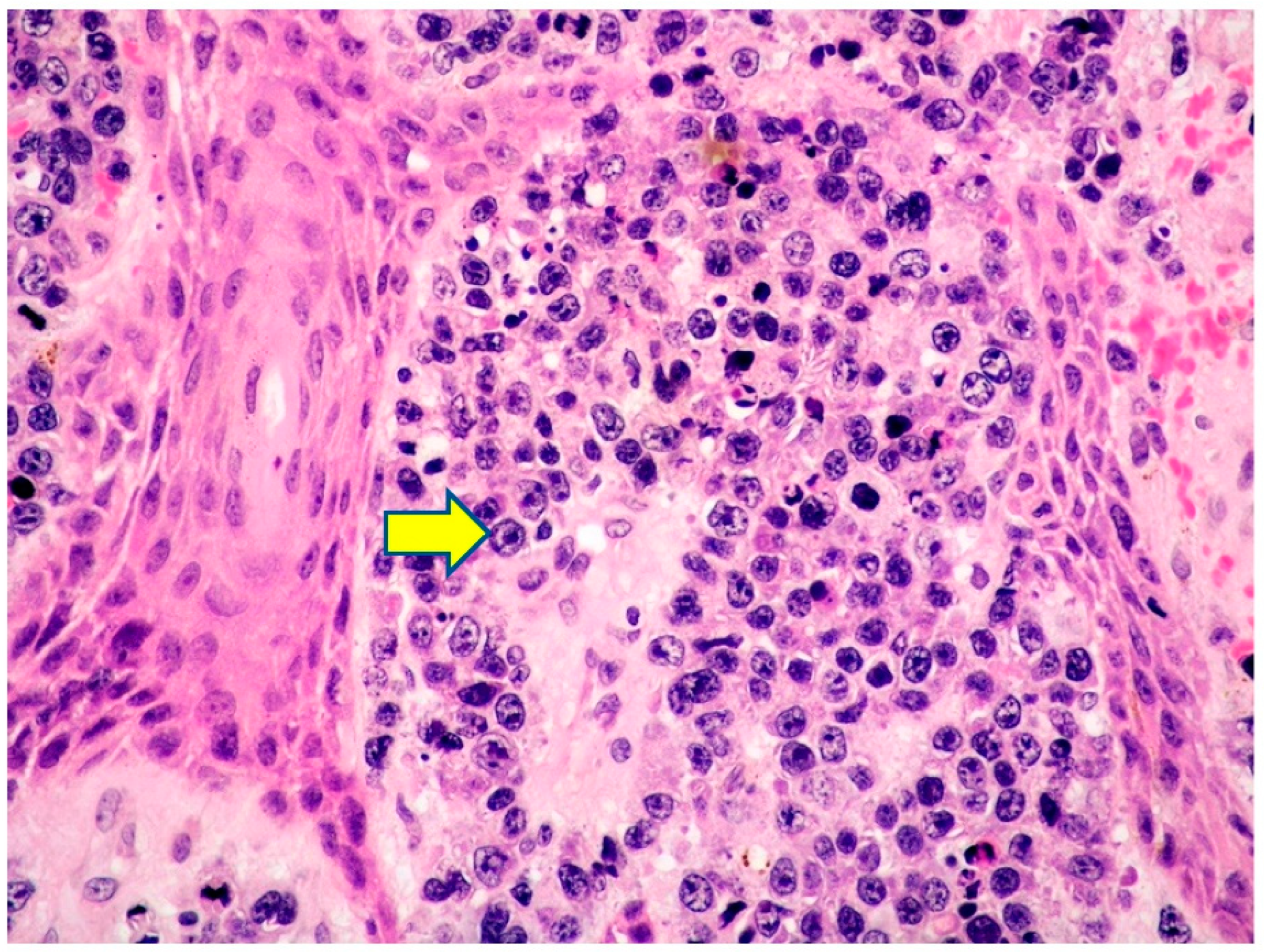
2.1. Clinicopathological Example of PCMZL, Plasmacytic Variant
2.2. Epidemiology, Clinical Features, Outcome and Treatment
2.3. PCMZL: Histopathological Variants
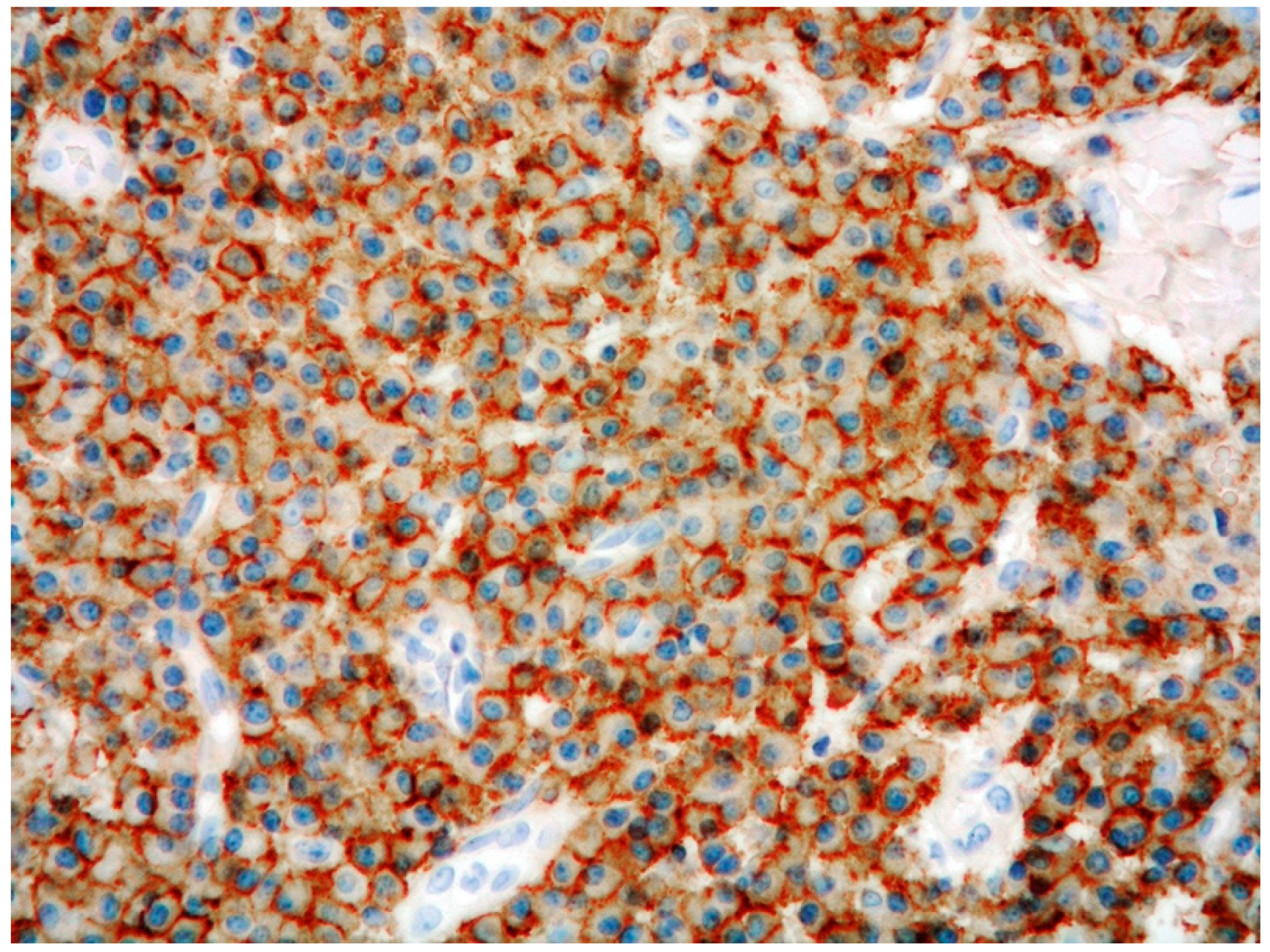
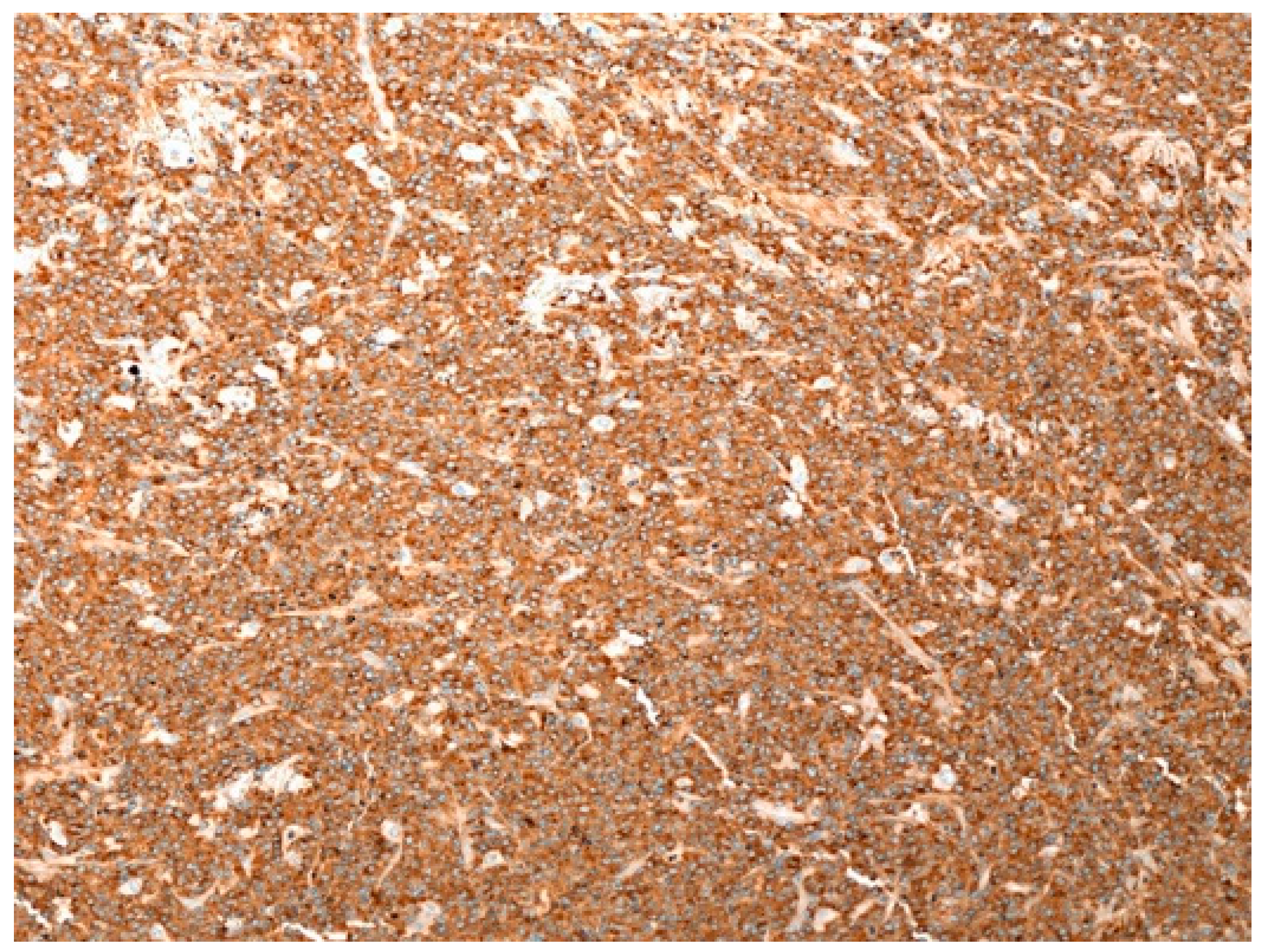
2.4. PCMZL: Immunohistochemical and Genetic Profile
3. Cutaneous Nodular Amyloidosis/Amyloidoma
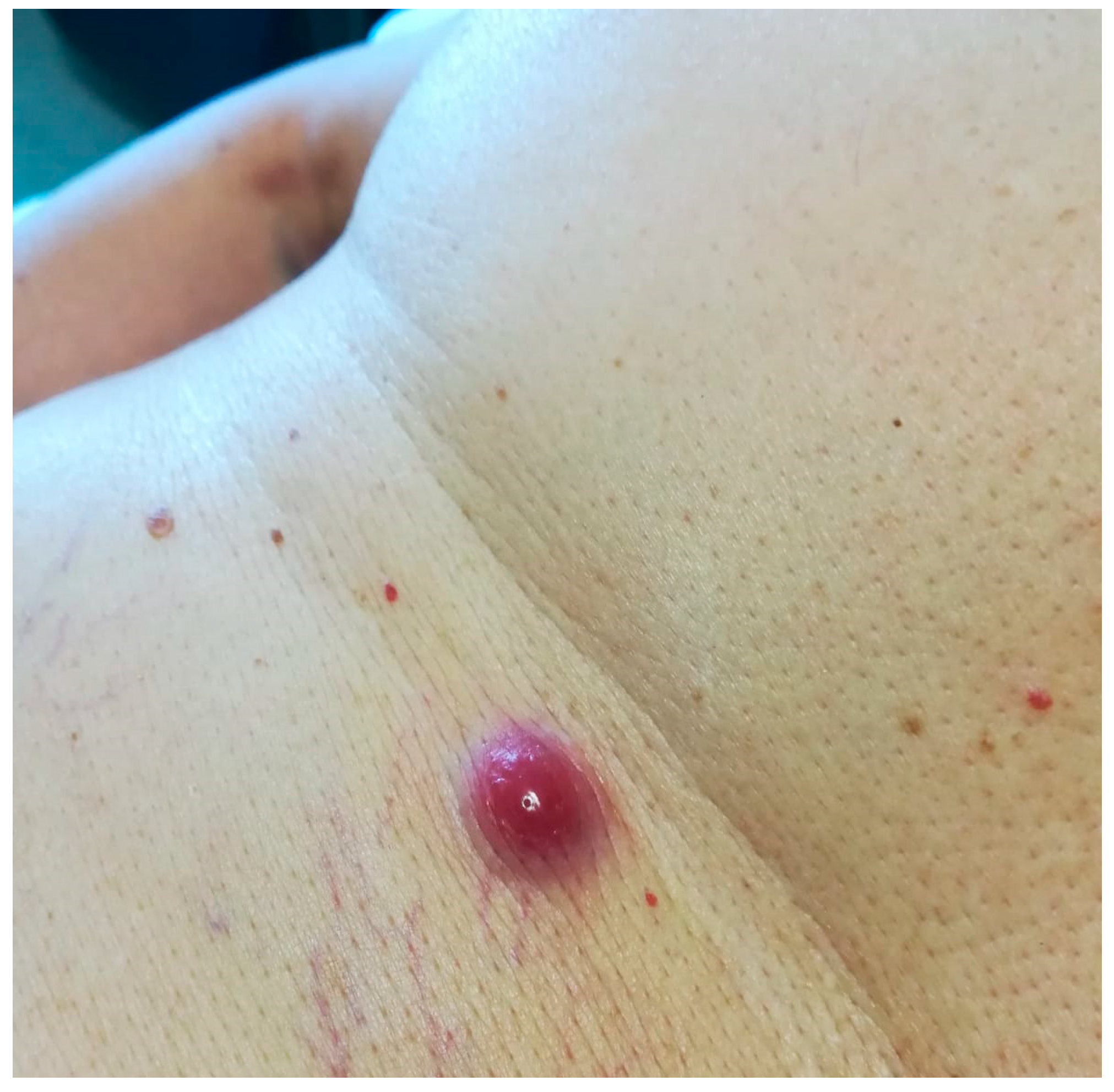
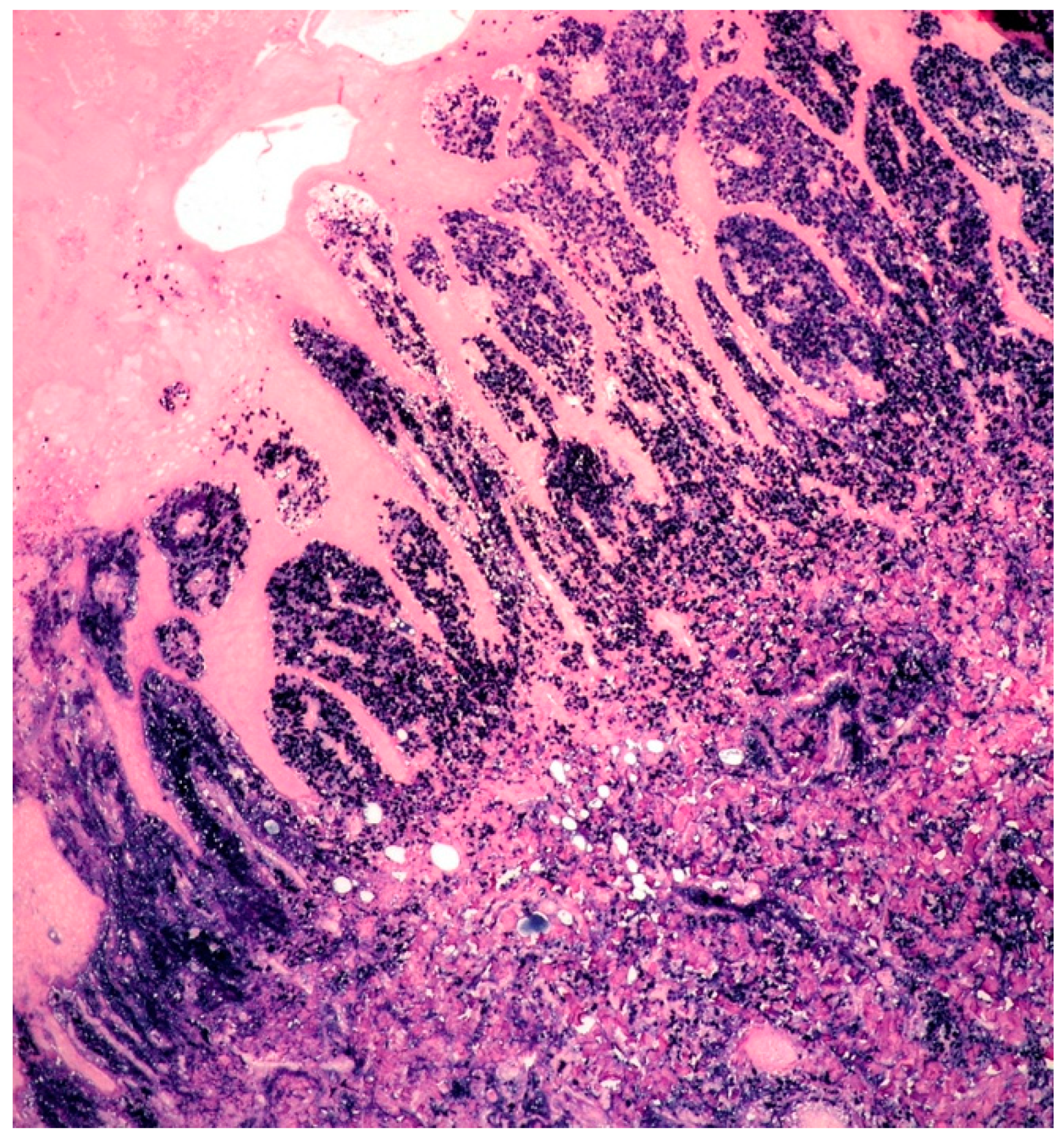
3.1. Clinicopathological Example of Cutaneous Amyloidoma
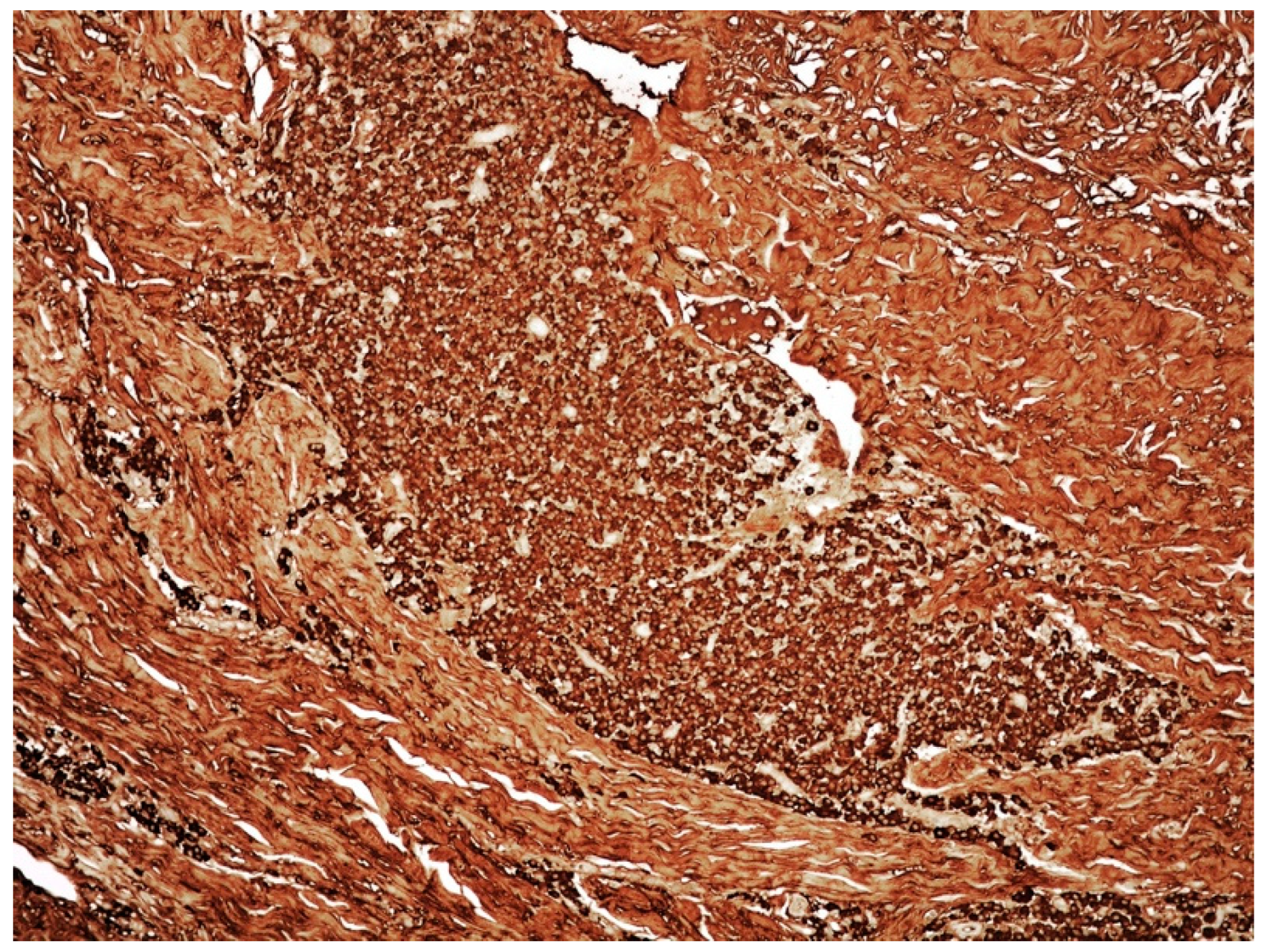
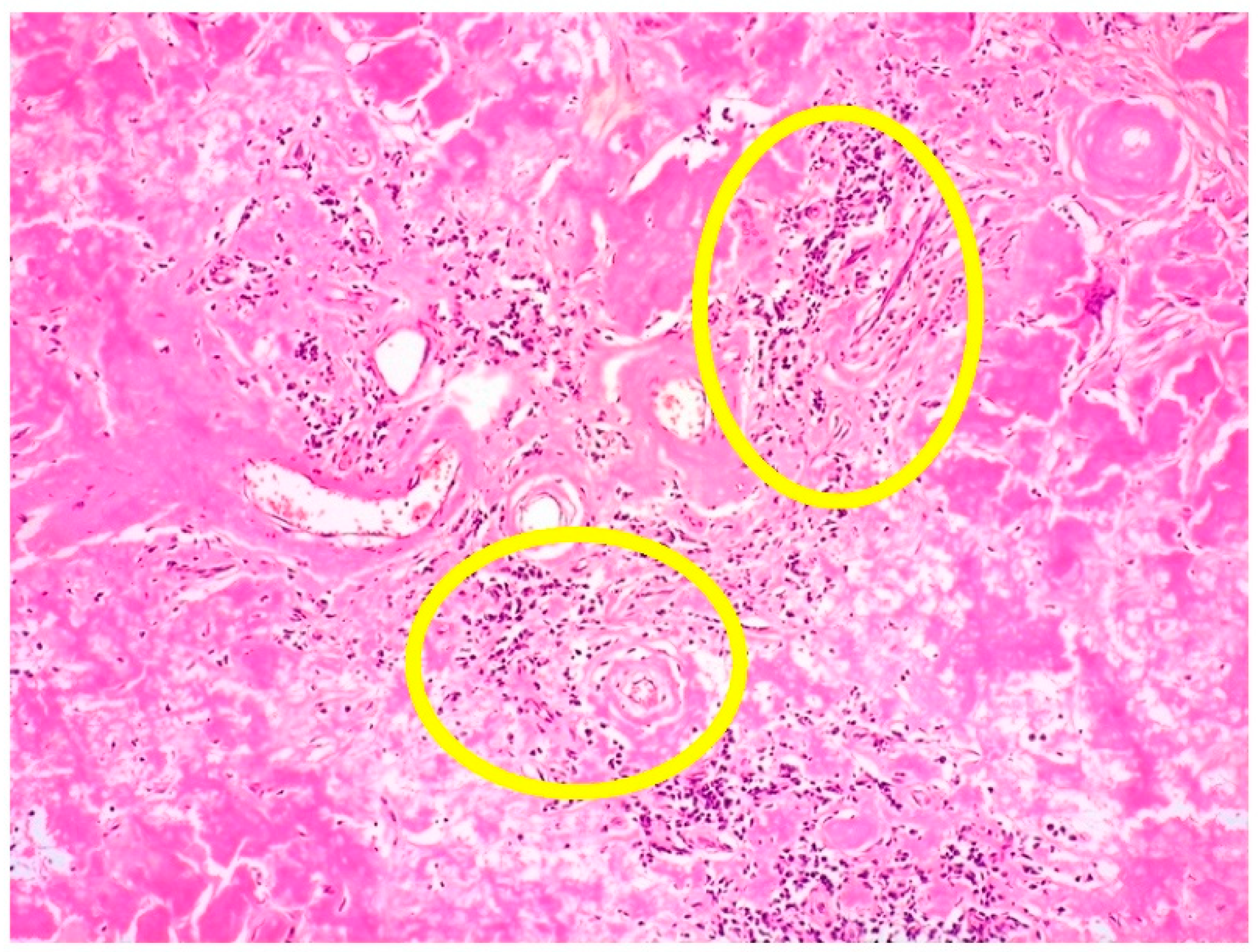
3.2. Clinicopathological Features of Amyloid Deposits and Cutaneous Amyloidoma
4. Teaching Points of Case 1 (PCMZL) and Case 2 (Cutaneous Amyloidoma)
- PCMZL shows a spectrum of histopathological variants. The identification of small lymphocytes, marginal zone cells, and neoplastic plasma cells are helpful histological features for the diagnosis of classic cases of PCMZL. However, uncommon and atypical variants of PCMZL may be diagnostically challenging for pathologists. Case 1 represents an example of PCMZL, plasmacytic variant, characterized by an almost exclusive proliferation of neoplastic plasma cells.
- The plasmacytic variant of PCMZL is exceedingly rare, and the possibility of secondary cutaneous involvement by MM needs to be excluded through accurate staging procedures, including BM biopsy.
- The plasmacytic variant of PCMZL also raises the diagnostic possibility of primary cutaneous plasmacytoma. Extraosseous plasmacytoma is a plasma cell malignancy arising in patients with no evidence of MM. The majority of extraosseous plasmacytomas occurs in the upper respiratory tract. Currently, according to the WHO-EORTC classification, cases regarded in the past as primary cutaneous plasmacytoma need to be re-classified as PCMZL, plasmacytic variant.
- Cutaneous amyloidoma represents a rare manifestation of amyloidosis. The identification of amyloid deposition in the skin, as well in other body sites, imposes to rule out systemic diseases such systemic amyloidosis, lymphomas, and plasma cell dyscrasias, including MM.
5. MM
5.1. Clinicopathological Example of MM on Cutaneous Surgical Scar
5.2. Extramedullary Cutaneous Localization of MM: Clinical Features
5.3. Extramedullary Cutaneous Localization of MM: Histological Features
6. PBL
6.1. Clinicopathological Example of Cutaneous Localization of PBL
6.2. PBL: General Features
6.3. Cutaneous Localization of PBL: Clinical Features
6.4. PBL: Histology, Immunophenotype and Genetic Profile
6.5. PBL: Differential Diagnosis with Malignancies Showing Plasmablastic Features and Cutaneous Involvement
7. Teaching Points of Case 3 (Cutaneous Localization of MM) and Case 4 (Cutaneous Localization of PBL)
- Once a clonal plasma cell proliferation is observed in the skin, it is mandatory to evaluate clinical, laboratory, and radiologic data, including BM biopsy, in order to exclude the presence of a systemic disease.
- Extramedullary involvement of the skin in MM is a rare event; it is associated with a poor prognosis and may occur at any stage of the disease.
- MM with skin localization may be poorly differentiated, often showing a polymorphic or plasmablastic morphology; hence, the plasma cell lineage may be difficult to recognize without appropriate immunohistochemical stains.
- PM needs to be differentiated from PBL, which usually occurs in patients with IS and is strongly associated with EBV, unlike PM.
8. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Willemze, R.; Cerroni, L.; Kempf, W.; Berti, E.; Facchetti, F.; Swerdlow, S.H.; Jaffe, E.S. The 2018 update of the WHO-EORTC classification for primary cutaneous lymphomas. Blood 2019, 133, 1703–1714. [Google Scholar] [CrossRef] [PubMed]
- Cerroni, L. Skin Lymphoma: The Illustrated Guide, 5th ed.; Wiley-Backwell: Hoboken, NJ, USA, 2020. [Google Scholar]
- WHO Classification of Skin Tumours; IARC: Lyon, France, 2018.
- Cerroni, L.; Signoretti, S.; Hofler, G.; Annessi, G.; Putz, B.; Lackinger, E.; Metze, D.; Giannetti, A.; Kerl, H. Primary cutaneous marginal zone B-cell lymphoma: A recently described entity of low-grade malignant cutaneous B-cell lymphoma. Am. J. Surg. Pathol. 1997, 21, 1307–1315. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, S.H. Cutaneous marginal zone lymphomas. Semin. Diagn. Pathol. 2017, 34, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Inagaki, H.; Kuo, T.T.; Hu, S.; Okabe, M.; Eimoto, T. Primary cutaneous marginal zone B-cell lymphoma. A molecular and clinicopathologic study of 24 Asian cases. Am. J. Surg. Pathol. 2003, 27, 1061–1069. [Google Scholar] [CrossRef] [PubMed]
- Goodlad, J.R.; Davidson, M.M.; Hollowood, K.; Ling, C.; MacKenzie, C.; Christie, I.; Batstone, P.J.; Ho-Yen, D.O. Primary cutaneous B-cell lymphoma and Borrelia burgdorferi infection in patients from the Highlands of Scotland. Am. J. Surg. Pathol. 2000, 24, 1279–1285. [Google Scholar] [CrossRef]
- De la Fouchardiere, A.; Vandenesch, F.; Berger, F. Borrelia-associated primary cutaneous MALT lymphoma in a non-endemic region. Am. J. Surg. Pathol. 2003, 27, 702–703. [Google Scholar] [CrossRef] [PubMed]
- Goteri, G.; Ranaldi, R.; Simonetti, O.; Capretti, R.; Menzo, S.; Stramazzotti, D.; Morichetti, D.; Offidani, A.M.; Rupoli, S.; Leoni, P. Clinicopathological features of primary cutaneous B-cell lymphomas from an academic regional hospital in central Italy: No evidence of Borrelia burgdorferi association. Leuk. Lymphoma 2007, 48, 2184–2188. [Google Scholar] [CrossRef]
- Zinzani, P.L.; Quaglino, P.; Pimpinelli, N.; Berti, E.; Baliva, G.; Rupoli, S.; Martelli, M.; Alaibac, M.; Borroni, G.; Chimenti, S.; et al. Prognostic factors in primary cutaneous B-cell lymphoma: The Italian Study Group for Cutaneous lymphomas. J. Clin. Oncol. 2006, 24, 1376–1382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fink-Puches, R.; Zenahlik, P.; Back, B.; Smolle, J.; Kerl, H.; Cerroni, L. Primary cutaneous lymphomas: Applicability of current classification schemes (European Organization for Research and Treatment of Cancer, World Health Organization) based on clinicopathologic features observed in a large group of patients. Blood 2002, 99, 800–805. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.A.; Shah, F.; Chaganti, S.; Stevens, A.; Amel-Kashipaz, R.; Vydianath, B.; Scarisbrick, J.J. Primary cutaneous B-cell lymphoma: Systemic spread is rare while cutaneous relapses and secondary malignancies are frequent. Br. J. Dermatol. 2017, 177, 287–289. [Google Scholar] [CrossRef]
- Servitje, O.; Muniesa, C.; Benavente, Y.; Monsalvez, V.; Garcia-Muret, M.P.; Gallardo, F.; Domingo-Domenech, E.; Lucas, A.; Climent, F.; Rodriguez-Peralto, J.L.; et al. Primary cutaneous marginal zone B-cell lymphoma: Response to treatment and disease-free survival in a series of 137 patients. J. Am. Acad. Dermatol. 2013, 69, 357–365. [Google Scholar] [CrossRef]
- Willemze, R.; Hodak, E.; Zinzani, P.L.; Specht, L.; Ladetto, M. on behalf of the ESMO Guidelines committee. Primary cutaneous lymphomas: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2018, 29, iv30–iv40. [Google Scholar] [CrossRef]
- Duncan, L.N.; LeBoit, P.E. Are primary cutaneous immunocytoma and marginal zone lymphoma the same disease? Am. J. Surg. Pathol. 1997, 21, 1368–1372. [Google Scholar] [CrossRef]
- Hussong, J.W.; Perkins, S.L.; Schnitzer, B.; Hargreaves, H.; Frizzera, G. Extramedullary plasmacytoma. A form of marginal zone lymphoma? Am. J. Clin. Pathol. 1999, 111, 111–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turbiner Geyer, J.; Ferry, J.A.; Longtine, J.A.; Flotte, T.J.; Harris, N.L.; Zukenberg, L.R. Characteristics of cutaneous marginal zone lymphomas with marked plasmacytic differentiation and a T-cell-rich background. Am. J. Clin. Pathol. 2010, 133, 59–69. [Google Scholar] [CrossRef]
- Brenner, I.; Roth, S.; Puppe, B.; Wobser, M.; Rosenwald, A.; Geissinger, E. Primary cutaneous marginal zone lymphomas with plasmacytic differentiation show frequent IgG4 expression. Mod. Pathol. 2013, 26, 1568–1576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Souza, A.; Ferry, J.A.; Burghart, D.R.; Tinguely, M.; Goyal, A.; Duncan, L.M.; Kutzner, H.; Kempf, W. IgG4 expression in primary cutaneous marginal zone lymphoma: A multicenter study. Appl. Immunohistochem. Mol. Morphol. 2018, 26, 462–467. [Google Scholar] [CrossRef]
- Magro, C.M.; Yang, A.; Fraga, G. Blastic marginal zone lymphoma: A clinical and pathological study of 8 cases and review of the literature. Am. J. Dermatopathol. 2013, 35, 319–326. [Google Scholar] [CrossRef]
- WHO Classification of Tumours Haematopoietic and Lymphoid Tissues, Revised 4th ed.; WHO Classification of Tumours Editorial Board (Ed.) IARC: Lyon, France, 2017. [Google Scholar]
- Edinger, J.T.; Kant, J.A.; Swerdlow, S.H. Cutaneous marginal zone lymphomas have distinctive features and include 2 subsets. Am. J. Surg Pathol 2010, 34, 1830–1841. [Google Scholar] [CrossRef]
- Rochen, C.; Knut, S. Amyloid in surgical pathology. Virchows Arch. 2003, 443, 3–16. [Google Scholar]
- Mahmood, S.; Bridoux, F.; Venner, C.P.; Sachchithanantham, S.; Gilbertson, J.A.; Rowczenio, D.; Wagner, T.; Sayed, R.; Patel, K.; Fontana, M.; et al. Natural history and outcomes in localised immunoglobulin light-chain amyloidosis: A long-term observational study. Lancet Haematol. 2015, 2, e241–e250. [Google Scholar] [CrossRef]
- Kaltoft, B.; Schmidt, G.; Lauritzen, A.F.; Gimsing, P. Primary localized cutaneous amyloidosis-a systematic review. Dan. Med. J. 2013, 60, A4727. [Google Scholar] [PubMed]
- Walsh, N.M.; Lano, I.M.; Green, P.; Gallant, C.; Pasternak, S.; Yen Ly, T.; Requena, L.; Kutzner, H.; Chott, A.; Cerroni, L. AL Amyloidoma of the skin/subcutis: Cutaneous amyloidosis, plasma cell dyscrasia or a manifestation of primary cutaneous marginal zone lymphoma? Am. J. Surg. Pathol. 2017, 41, 1069–1076. [Google Scholar] [CrossRef]
- Dangien, A.; Beylot-Barry, M.; Battistella, M.; Ram-Woff, C.; Talbot, A.; Rybojad, M.; Vergier, B.; Jachiet, M.; Bouaziz, J.D.; Arnulf, B.; et al. Clinical presentation, therapeutic approach and outcome of primary cutaneous marginal zone B-cell lymphoma presenting as AL amyloidoma of the skin. Br. J. Dermatol. 2019, 181, 607–609. [Google Scholar] [CrossRef] [PubMed]
- Ueberdiek, S.; Kempf, W.; Kretschmer, L.; Shon, M.P.; Mitteldorf, C. AL-amyloidoma of the skin-a are manifestation of primary cutaneous marginal zone lymphoma. Am. J. Dermatopathol. 2019, 41, 518–521. [Google Scholar] [CrossRef] [PubMed]
- Grogg, K.L.; Aubry, M.C.; Vrana, J.A.; Theis, J.D.; Dogan, A. Nodular pulmonary amyloidosis is characterized by localized immunoglobulin deposition and is frequently associated with an indolent B-cell lymphoproliferative disorder. Am. J. Surg. Pathol. 2013, 37, 406–412. [Google Scholar] [CrossRef]
- Bhutani, M.; Shaid, Z.; Schnebelen, A.; Alapat, D.; Usmani, S.Z. Cutaneous manifestations of multiple myeloma and other plasma cell proliferative disorders. Semin. Oncol. 2016, 43, 395–400. [Google Scholar] [CrossRef]
- Kois, J.M.; Mack Sexton, F.; Lookinghill, D.P. Cutaneous manifestations of multiple myeloma. Arch. Dermatol. 1991, 127, 69–74. [Google Scholar] [CrossRef]
- Requena, L.; Kutzer, H.; Palmedo, G.; Calonje, E.; Requena, C.; Perez, G.; Pastor, M.A.; Sangueza, O.P. Cutaneous involvement in multiple myeloma. A clinicopathologic, immunohistochemical ad cytogenetic study of 8 cases. Arch. Dermatol. 2003, 139, 475–486. [Google Scholar] [CrossRef]
- Woo, Y.R.; Kim, J.S.; Lim, J.H.; Hwang, S.; Kim, M.; Bae, J.M.; Park, Y.M.; Min, C.K.; Kim, D.W.; Park, H.J. Prevalence and clinicopathologic characteristics of multiple myeloma with cutaneous involvement: A case series from Korea. J. Am. Acad. Dermatol. 2018, 78, 471–478. [Google Scholar] [CrossRef]
- Jurczyszyn, A.; Olszewska-Szopa, M.; Hungria, V.; Crusoe, E.; Pika, T.; Delforge, M.; Leleu, X.; Rasche, L.; Nooka, A.K.; Druzd-Sitek, A.; et al. Cutaneous involvement in multiple myeloma: A multi-institutional retrospective study of 53 patients. Leuk. Lymphoma 2016, 57, 2071–2076. [Google Scholar] [CrossRef] [PubMed]
- Rosenblum, M.D.; Bredeson, C.N.; Chang, C.C.; Rizzo, J.D. Subcutaneous plasmacytomas with tropism to sites of previous trauma in a multiple myeloma patient treated with an autologous bone marrow transplant. Am. J. Hematol. 2003, 72, 274–277. [Google Scholar] [CrossRef] [PubMed]
- Boyle, S.; Hourigan, M. Cutaneous seeding of neoplastic plasma cells to a site of bruising in a patient with relapsed myeloma. Br. J. Haematol. 2018, 181, 723. [Google Scholar] [CrossRef] [Green Version]
- Malysz, J.; Talamo, G.; Zhu, J.; Clarke, L.E.; Bayerl, M.G.; Ali, L.; Helm, K.F.; Chung, C.G. Cutaneous involvement in multiple myeloma (MM): A case series with clinicopathologic correlation. J. Am. Acad. Dermatol. 2016, 74, 784–787. [Google Scholar] [CrossRef]
- Marchica, V.; Accardi, F.; Storti, P.; Mancini, C.; Martella, E.; Dalla Palma, B.; Bolzoni, M.; Todoerti, K.; Marcatti, M.; Schifano, C.; et al. Cutaneous localization in multiple myeloma in the context of bortezomib-based treatment: How do myeloma cells escape from the bone marrow to the skin? Int. J. Hematol. 2017, 105, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Varricchio, S.; Pagliuca, F.; Travaglino, A.; Gallo, L.; Villa, M.R.; Mascolo, M. Cutaneous localization of plasmablastic multiple myeloma with heterotopic expression of CD3 and CD4: Skin involvement revealing systemic disease. J. Cutan. Pathol. 2019, 46, 619–622. [Google Scholar] [CrossRef] [PubMed]
- Delecleuse, H.J.; Anagnostopoulos, I.; Dallenbach, F.; Hummel, M.; Marafioti, T.; Schneider, U.; Huhn, D.; Schmidt-Westhausen, A.; Reichart, P.A.; Gross, U.; et al. Plasmablastic lymphomas of the oral cavity: A new entity associated with the human immunodeficiency virus infection. Blood 1997, 89, 1413–1420. [Google Scholar] [CrossRef]
- Zizzo, M.; Zanelli, M.; Martiniani, R.; Sanguedolce, F.; De Marco, L.; Martino, G.; Parente, P.; Annessi, V.; Manzini, L.; Ascani, S. Oral plasmablastic lymphoma: A case report. Medicine 2020, 99, e22335. [Google Scholar] [CrossRef]
- Castillo, J.J.; Bibas, M.; Miranda, R.N. The biology and treatment of plasmablastic lymphoma. Blood 2015, 125, 2323–2330. [Google Scholar] [CrossRef] [Green Version]
- Morscio, J.; Dierickx, D.; Nijs, J.; Verhoef, G.; Bittoun, E.; Vanoeteren, X.; Wlodarska, I.; Sagaert, X.; Tousseyn, T. Clinicopathologic comparison of plasmablastic lymphoma in HIV-positive, immunocompetent and posttransplant patients single-center series of 25 cases and meta-analysis of 277 reported cases. Am. J. Surg. Pathol. 2014, 38, 875–886. [Google Scholar] [CrossRef]
- Harmon, C.M.; Smith, L.B. Plasmablastic lymphoma: A review of clinicopathologic features and differential diagnosis. Arch Pathol. Lab. Med. 2016, 140, 1074–1078. [Google Scholar] [CrossRef] [Green Version]
- Zanelli, M.; Sanguedolce, F.; Palicelli, A.; Zizzo, M.; Martino, G.; Caprera, C.; Fragliasso, V.; Soriano, A.; Valle, L.; Ricci, S.; et al. EBV-driven lymphoproliferative disorders and lymphomas of the gastrointestinal tract: A spectrum of entities with a common denominator (Part 2). Cancers 2021, 13, 4527. [Google Scholar] [CrossRef]
- Sanguedolce, F.; Zanelli, M.; Zizzo, M.; Martino, G.; Rossi, C.; Parente, P.; Ascani, S. Clinical, pathological and molecular features of plasmablastic lymphoma arising in the gastrointestinal tract: A review and reappraisal. Pathol. Res. Pract. 2020, 216, 152973. [Google Scholar] [CrossRef]
- Lopez, A.; Abrisqueta, P. Plasmablastic lymphoma: Current perspectives. Blood Lymph. Cancer Targets Ther. 2018, 8, 63–70. [Google Scholar]
- Jordan, L.B.; Lessells, A.M.; Goodlad, J.R. Plasmablastic lymphoma arising at a cutaneous site. Histopathology 2005, 46, 108–117. [Google Scholar] [CrossRef]
- Heiser, D.; Muller, H.; Kempf, W.; Eisendle, K.; Zelger, B. Primary cutaneous plasmablastic lymphoma of the lower leg in an HIV-negative patient. J. Am. Acad. Dermatol. 2012, 67, e202–e205. [Google Scholar] [CrossRef]
- Jambusaria, A.; Shafer, D.; Wu, H.; Al-Saleem, T.; Perlis, C. Cutaneous plasmablastic lymphoma. J. Am. Acad. Dermatol. 2008, 58, 676–678. [Google Scholar] [CrossRef]
- Tiong, I.S.; Strauss, M.; Lau, M.B.Y.; Chiruka, S. Cutaneous plasmablastic lymphoma in an immunocompetent patient with long-term pyrimethamine use for essential thrombocytemia: A case report and literature review. Case Rep. Hematol. 2013, 2013, 541783. [Google Scholar]
- Castillo, J.J. Plasmablastic lymphoma: Are more intensive regimens needed? Leuk. Res. 2011, 35, 1547–1548. [Google Scholar] [CrossRef]
- Pretscher, D.; Kalish, A.; Wilhelm, M.; Birkmann, J. Refractory plasmablastic lymphoma. A review of treatment options beyond standard therapy. Ann. Hematol. 2017, 96, 967–970. [Google Scholar] [CrossRef]
- Castillo, J.J.; Reagan, J.L.; Sikov, W.M.; Winer, E.S. Bortezomib in combination with infusional dose-adjusted EPOCH for the treatment of plasmablastic lymphoma. Br. J. Haematol. 2015, 169, 352–355. [Google Scholar] [CrossRef]
- Al-Malki, M.M.; Castillo, J.J.; Sloan, J.M.; Re, A. Hematopoietic cell transplantation for plasmablastic lymphoma: A review. Biol. Blood Marrow Transplant. 2014, 20, 1877–1884. [Google Scholar] [CrossRef] [Green Version]
- Laurent, C.; Fabiani, B.; Do, C.; Tchernonog, E.; Cartron, G.; Gravelle, P.; Amara, N.; Malot, S.; Palisoc, M.M.; Copie-Bergman, C.; et al. Immune-checkpoint expression in Epstein-Barr virus positive and negative plasmablastic lymphoma: A clinical and pathological study in 82 patients. Haematologica 2016, 101, 976–984. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.J.; Chuang, S.S. Lymphoid neoplasms with plasmablastic differentiation: A comprehensive review and diagnostic approaches. Adv. Anat. Pathol. 2020, 27, 61–74. [Google Scholar] [CrossRef]
- Valera, A.; Balague, O.; Colomo, L.; Martinez, A.; Delabie, J.; Taddesse-Heath, L.; Jaffe, E.S.; Campo, E. IG/MYC rearrangements are the main cytogenetic alteration in plasmablastic lymphomas. Am. J. Surg. Pathol. 2010, 34, 1686–1694. [Google Scholar] [CrossRef] [Green Version]
- Zanelli, M.; Valli, R.; Capodanno, I.; Ragazzi, M.; Ascani, S. Anaplastic lymphoma kinase-positive large B-cell lymphoma: Description of a case with an unexpected clinical outcome. Int. J. Surg. Pathol. 2015, 23, 78–83. [Google Scholar] [CrossRef]
- Chadburn, A.; Hyjek, E.; Mathew, S.; Cesarman, E.; Said, J.; Knowles, D.M. KSHV-positive solid lymphomas represent an extra-cavitary variant of primary effusion lymphoma. Am. J. Surg. Pathol. 2004, 28, 1401–1416. [Google Scholar] [CrossRef]
- Kim, Y.; Leventaki, V.; Bhaijee, F.; Jackson, C.C.; Medeiros, L.J.; Vega, F. Extracavitary/solid variant of primary effusion lymphoma. Ann. Diagn. Pathol. 2012, 16, 441–446. [Google Scholar] [CrossRef]
- Zanelli, M.; Sanguedolce, F.; Zizzo, M.; Palicelli, A.; Bassi, M.C.; Santandrea, G.; Martino, G.; Soriano, A.; Caprera, C.; Corsi, M.; et al. Primary effusion lymphoma occurring in the setting of transplanted patients: A systematic review of a rare, life-threatening post-transplantation occurrence. BMC Cancer 2021, 21, 468. [Google Scholar] [CrossRef]
- Nicolae, A.; Pittaluga, S.; Abdullah, S.; Steinberg, S.M.; Pham, T.A.; Davies-Hill, T.; Xi, L.-Q.; Raffeld, M.; Jaffe, E.S. EBV-positive large B-cell lymphomas in young patients: A nodal lymphoma with evidence for a tolerogenic immune environment. Blood 2015, 126, 863–872. [Google Scholar] [CrossRef] [Green Version]
- Uccini, S.; Al-Jadiry, M.F.; Scarpino, S.; Ferraro, D.; Alsaadawi, A.R.; Al-Darraji, A.F.; Moletti, M.L.; Testi, A.M.; Al-Hadad, S.A.; Ruco, L. Epstein-Barr virus-positive diffuse large B-cell lymphoma in children: A disease reminiscent of Epstein Barr virus-positive diffuse large B-cell lymphoma of the elderly. Hum. Pathol. 2015, 46, 716–724. [Google Scholar] [CrossRef]
- Zanelli, M.; Sanguedolce, F.; Palicelli, A.; Zizzo, M.; Martino, G.; Caprera, C.; Fragliasso, V.; Soriano, A.; Valle, L.; Ricci, S.; et al. EBV-driven lymphoproliferative disorders and lymphomas of the gastrointestinal tract: A spectrum of entities with a common denominator (Part 1). Cancers 2021, 13, 4578. [Google Scholar] [CrossRef]












Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zanelli, M.; Palicelli, A.; Sanguedolce, F.; Zizzo, M.; Filosa, A.; Ricci, L.; Cresta, C.; Martino, G.; Bisagni, A.; Zanetti, E.; et al. Cutaneous Involvement in Diseases with Plasma Cell Differentiation: Diagnostic Approach. Curr. Oncol. 2022, 29, 3026-3043. https://doi.org/10.3390/curroncol29050246
Zanelli M, Palicelli A, Sanguedolce F, Zizzo M, Filosa A, Ricci L, Cresta C, Martino G, Bisagni A, Zanetti E, et al. Cutaneous Involvement in Diseases with Plasma Cell Differentiation: Diagnostic Approach. Current Oncology. 2022; 29(5):3026-3043. https://doi.org/10.3390/curroncol29050246
Chicago/Turabian StyleZanelli, Magda, Andrea Palicelli, Francesca Sanguedolce, Maurizio Zizzo, Alessandra Filosa, Linda Ricci, Camilla Cresta, Giovanni Martino, Alessandra Bisagni, Eleonora Zanetti, and et al. 2022. "Cutaneous Involvement in Diseases with Plasma Cell Differentiation: Diagnostic Approach" Current Oncology 29, no. 5: 3026-3043. https://doi.org/10.3390/curroncol29050246
APA StyleZanelli, M., Palicelli, A., Sanguedolce, F., Zizzo, M., Filosa, A., Ricci, L., Cresta, C., Martino, G., Bisagni, A., Zanetti, E., di Donato, F., Melli, B., Soriano, A., Cimino, L., Cavazza, A., Vivian, L. F., & Ascani, S. (2022). Cutaneous Involvement in Diseases with Plasma Cell Differentiation: Diagnostic Approach. Current Oncology, 29(5), 3026-3043. https://doi.org/10.3390/curroncol29050246