Heterogeneous Catalysis in (Bio)Ethanol Conversion to Chemicals and Fuels: Thermodynamics, Catalysis, Reaction Paths, Mechanisms and Product Selectivities

 , and
, and 
Abstract
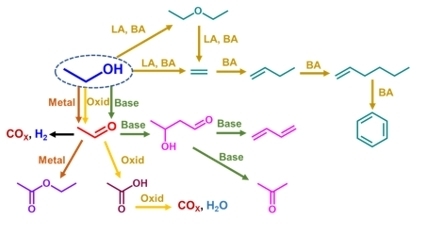
:
1. Introduction
2. Methods
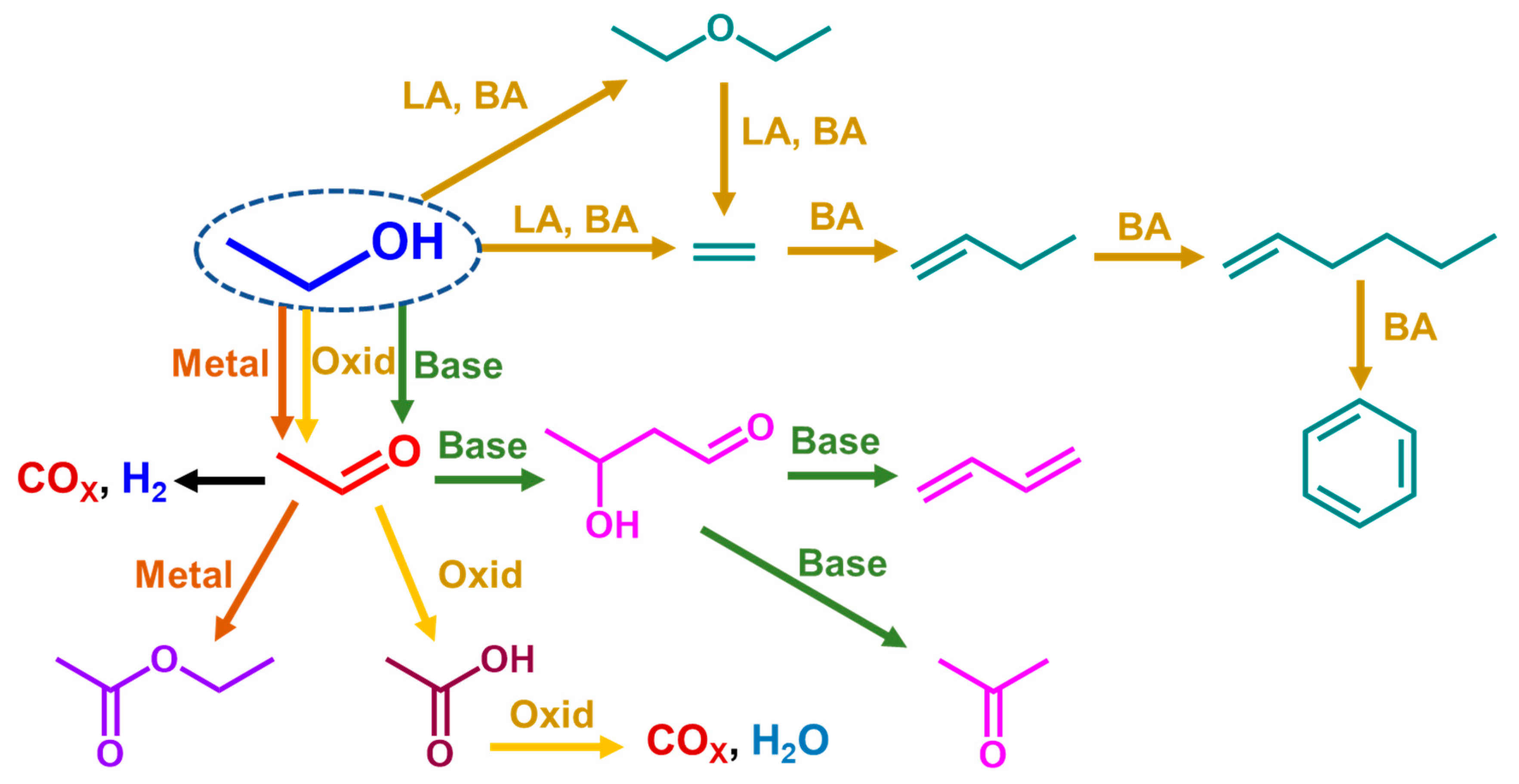
3. Results
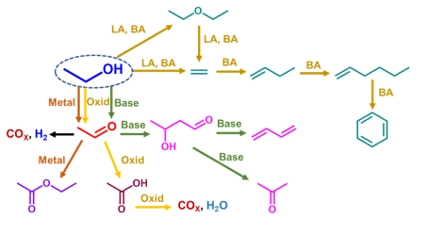
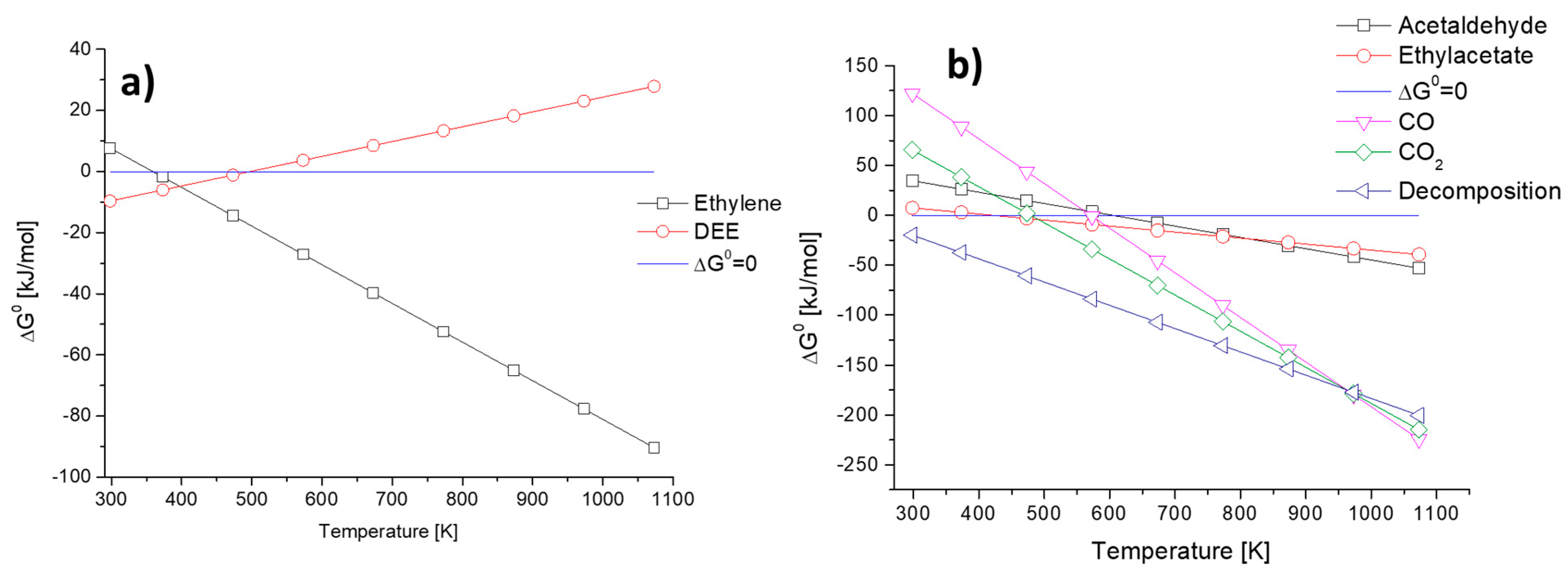
3.1. Evaluation of the Thermodynamic Equilibria in Ethanol Conversion to Chemicals
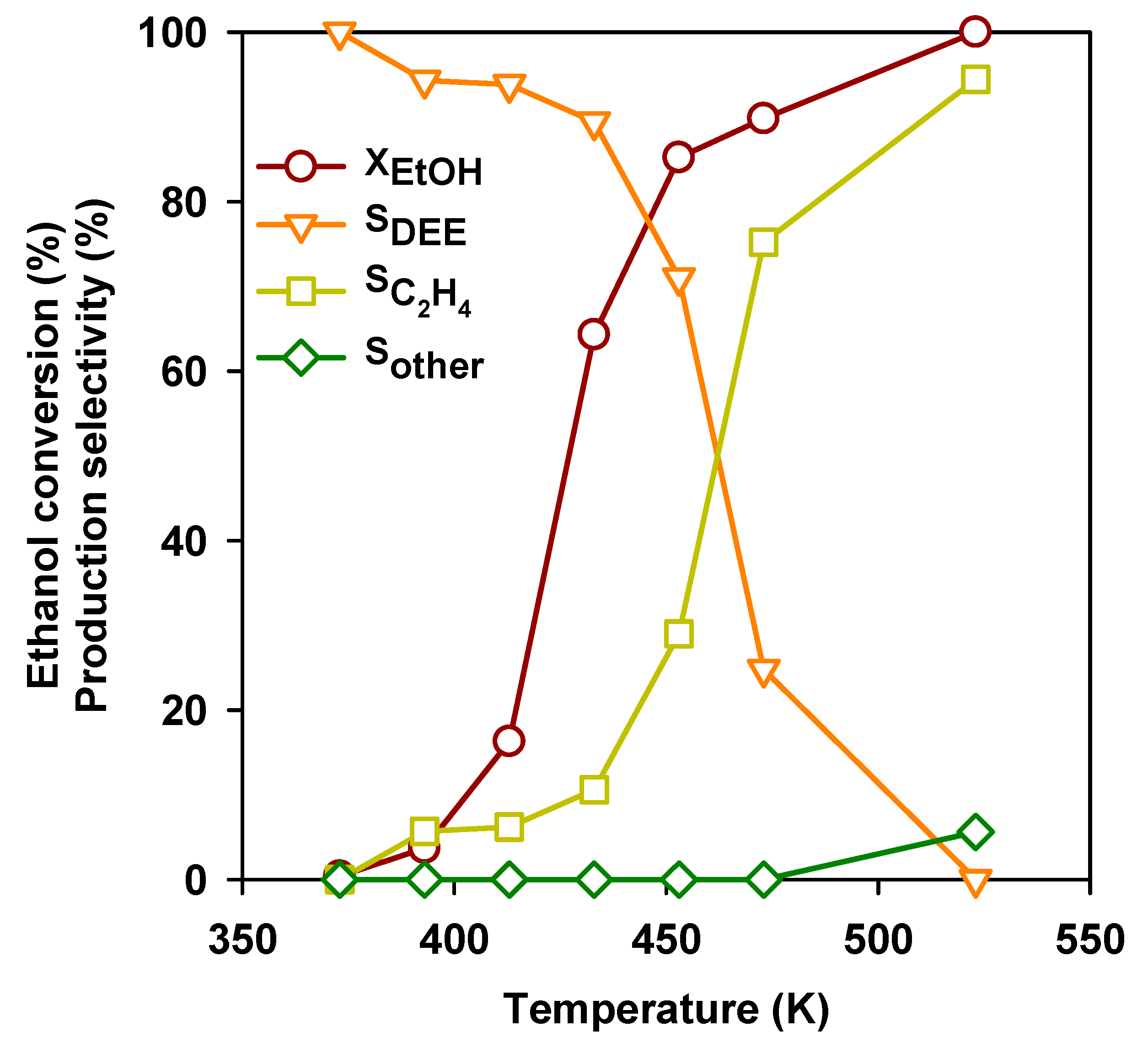
3.2. Ethanol to Ethyl Ether
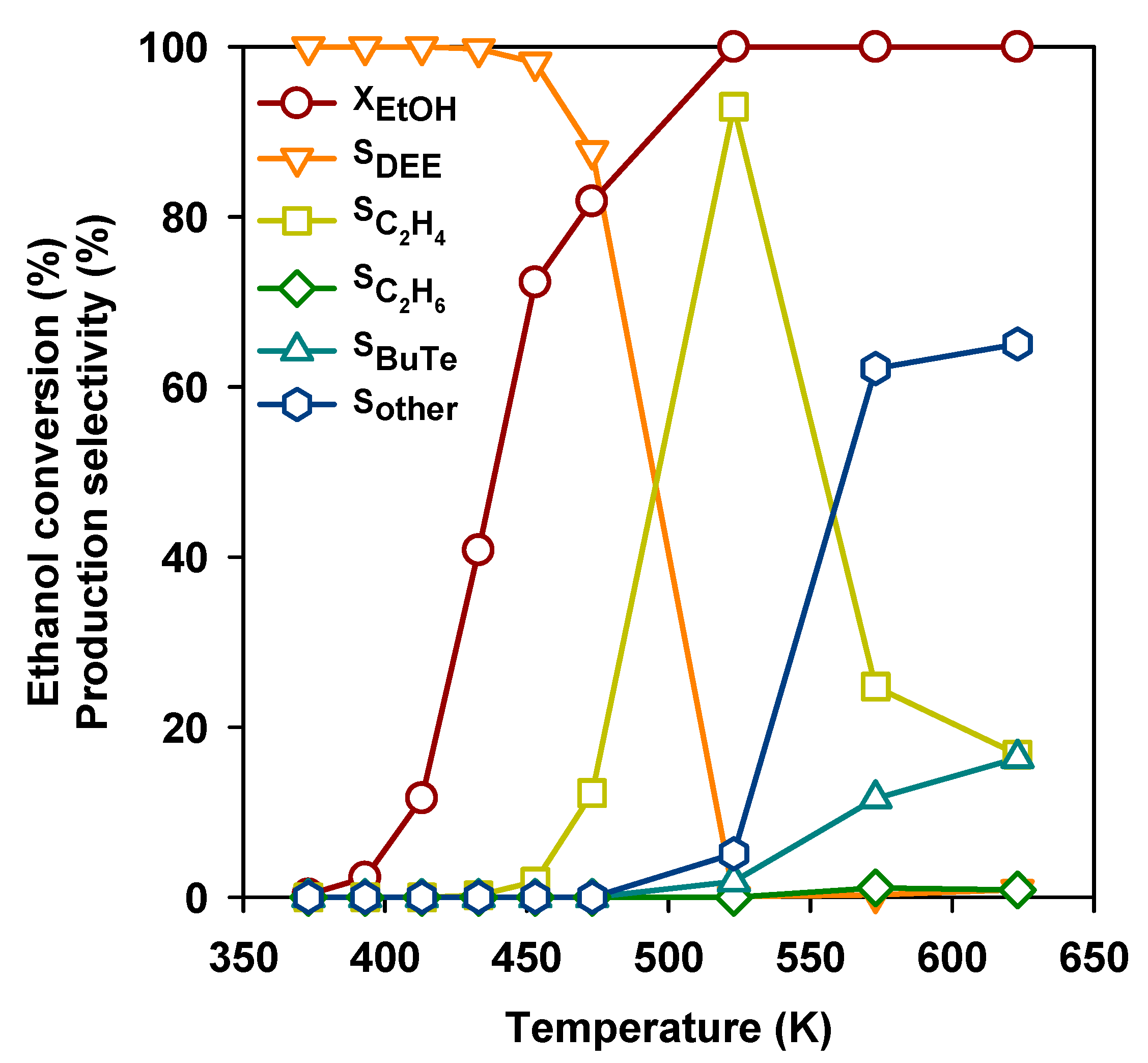
3.3. Ethanol to Ethylene
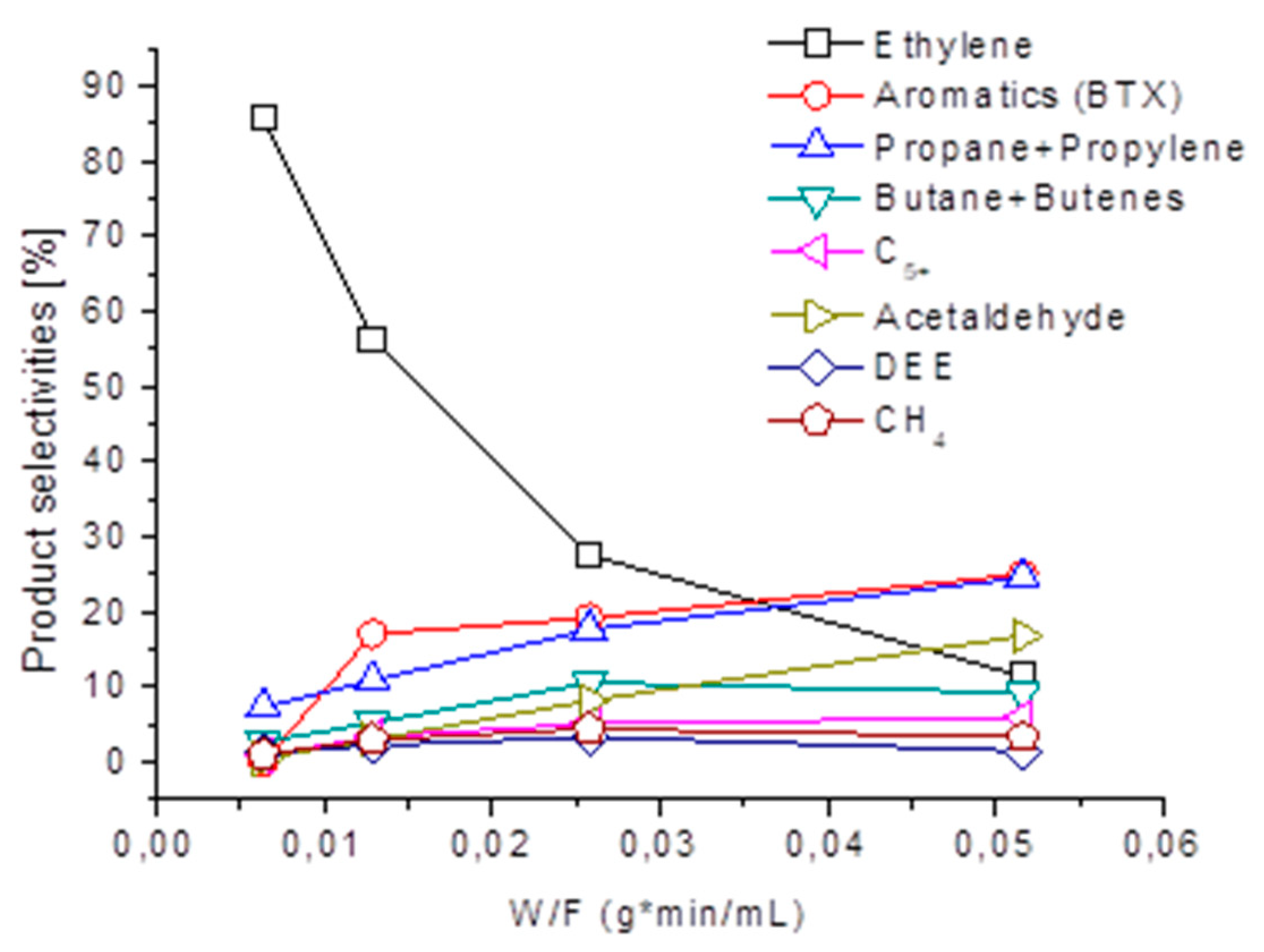
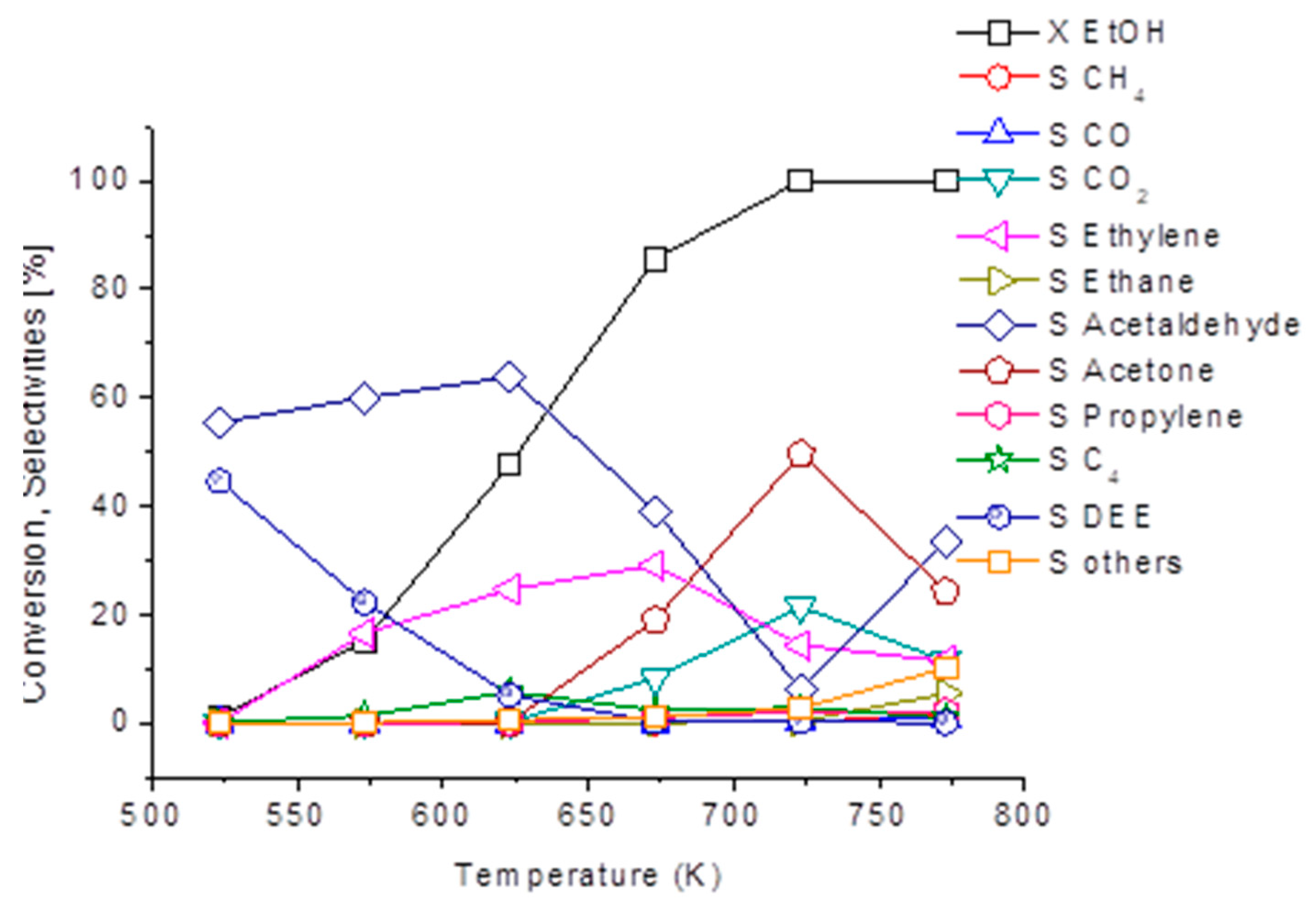
3.4. Ethanol to Hydrocarbons
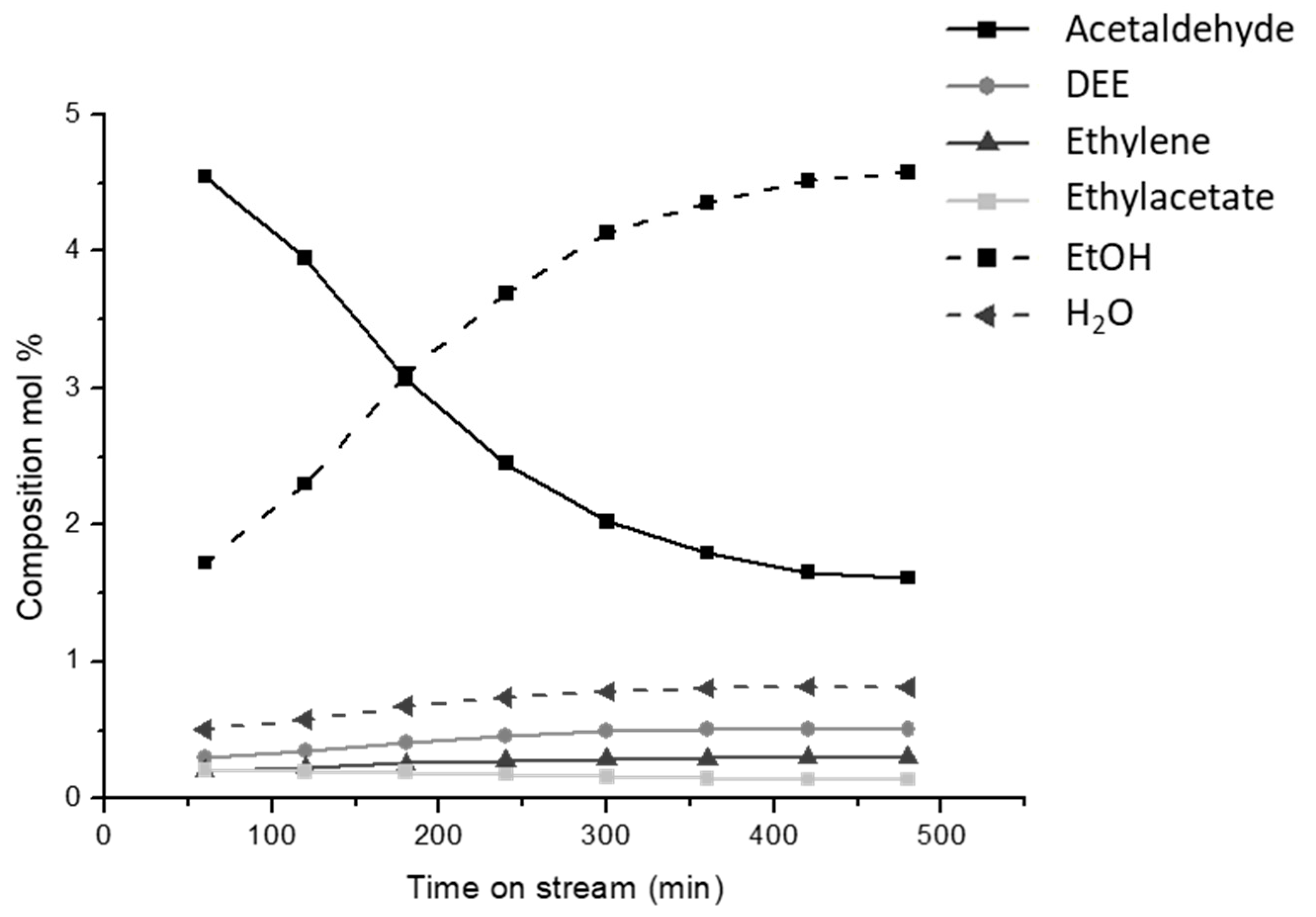
3.5. Ethanol to Acetaldehyde by Dehydrogenation
3.6. Acetaldehyde Over-Conversion: Ethanol to Butadiene and Acetone
3.7. Ethanol to Acetaldehyde by Oxidative Dehydrogenation
3.8. Ethanol Total Oxidation
3.9. Ethanol to Syngas by Partial Oxidation
3.10. Ethanol to Syngas by Steam Reforming
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ASA | Amorphous Silica–Alumina |
| BA | Brønsted Acid |
| BTX | Benzene-Toluene-Xylenes |
| C2 | Carbon Atom in Position 2 |
| DEE | Diethylether |
| ESR | Ethanol Steam Reforming |
| EtOH | Ethanol |
| FID | Flame Ionization Detector |
| GHSV | Gas Hourly Space Velocity |
| H-BEA | Protonic Zeolite Beta |
| H-FER | Protonic Zeolite Ferrierite |
| H-MFI | Protonic Zeolite Mobil Five |
| H-MOR | Protonic Zeolite Mordenite |
| HY | Protonic zeolite Faujasite |
| LA | Lewis Acid |
| MSR | Methanol Steam Reforming |
| NIST | National Institute of Standards and Technology |
| Oxid | Oxide Catalyst |
| SS | Steady State |
| TCD | Thermo-Conductivity Detector |
| TPSR | Temperature Programmed Surface Reaction |
| VOC | Volatile Organic Compounds |
| WHSV | Weight Hourly Space Velocity |
| USY | Ultra Stable Y zeolite |
| Symbols used | |
| G0 | standard reaction free energy (kJ/mol) |
| ΔH0298 | standard reaction enthalpy at 298 K (kJ/mol) |
| ΔS0298 | standard reaction entropy at 298 K (J/mol K) |
| F | molar flow (mol/time) |
| F | volumetric flow (mL/min) |
| n | stoichiometric coefficients ratio (-) |
| S | selectivity (-) |
| T | temperature (K) |
| X | conversion (-) |
| Y | yield (-) |
| W | catalyst weight (g) |
| Subscripts | |
| in | reactor entrance |
| out | reactor exit |
| P | product |
| R | reactant |
References
- Jambo, S.A.; Abdulla, R.; Mohd Azhar, S.H.; Marbawi, H.; Gansau, J.A.; Ravindra, P. A review on third generation bioethanol feedstock. Renew. Sustain. Energy Rev. 2016, 65, 756–769. [Google Scholar] [CrossRef]
- Soccol, C.R.; Faraco, V.; Karp, S.G.; Vandenberghe, L.P.S.; Thomaz-Soccol, V.; Woiciechowski, A.L.; Pandey, A. Lignocellulosic Bioethanol: Current Status and Future Perspectives, 2nd ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2019; ISBN 9780128168561. [Google Scholar]
- Cavani, F.; Albonetti, S.; Basile, F.; Gandini, A. Chemicals and Fuels from Biobased Bulding Blocks, 1st ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2016; ISBN 9789811518034. [Google Scholar]
- Mohsenzadeh, A.; Zamani, A.; Taherzadeh, M.J. Bioethylene Production from Ethanol: A Review and Techno-economical Evaluation. ChemBiolEng Rev. 2017, 4, 75–91. [Google Scholar] [CrossRef] [Green Version]
- Makshina, E.V.; Dusselier, M.; Janssens, W.; Degrève, J.; Jacobs, P.A.; Sels, B.F. Review of old chemistry and new catalytic advances in the on-purpose synthesis of butadiene. Chem. Soc. Rev. 2014, 43, 7917–7953. [Google Scholar] [CrossRef] [Green Version]
- Eckert, M.; Fleischmann, G.; Jira, R.; Bolt, H.M.; Golka, K. Acetaldehyde. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2006. [Google Scholar]
- Sun, J.; Wang, Y. Recent Advances in Catalytic Conversion of Ethanol to Chemicals. ACS Catal. 2014, 4, 1078–1090. [Google Scholar] [CrossRef]
- Phung, T.K.; Busca, G. Selective Bioethanol Conversion to Chemicals and Fuels via Advanced Catalytic Approaches. In Biorefinery of Alternative Resources: Targeting Green Fuels and Platform Chemicals; Springer: Singapore, 2020; pp. 75–103. [Google Scholar]
- Sawatmongkhon, B.; Theinnoi, K.; Wongchang, T.; Haoharn, C.; Wongkhorsub, C.; Tsolakis, A. Hydrogen production via the catalytic partial oxidation of ethanol on a platinum-rhodium catalyst: Effect of the oxygen-to-ethanol molar ratio and the addition of steam. Energy Fuels 2019, 33, 6742–6753. [Google Scholar] [CrossRef]
- Poling, B.E.; Prausnitz, J.M.; O’Connell, J.P. Properties of Gases and Liquids, Fifth Edition; McGraw-Hill Education: New York, NY, USA, 2001; ISBN 9780070116825. [Google Scholar]
- Subramani, V.; Song, C. Catalysis; Spivey, J.J., Ed.; Royal Society of Chemistry: Cambridge, UK, 2007; Volume 20, ISBN 978-0-85404-244-9. [Google Scholar]
- Subramani, V.; Sharma, P.; Zhang, L.; Liu, K. Catalytic Steam Reforming Technology for the Production of Hydrogen and Syngas. In Hydrogen and Syngas Production and Purification Technologies, 1st ed.; Liu, K., Song, C., Subramani, V., Eds.; John Wiley & Sons Ltd, Inc.: Hoboken, NJ, USA, 2009; pp. 14–126. ISBN 9780470561256. [Google Scholar]
- Garbarino, G.; Pugliese, F.; Cavattoni, T.; Busca, G.; Costamagna, P. A Study on CO2 Methanation and Steam Methane Reforming over Commercial Ni/Calcium Aluminate Catalysts. Energies 2020, 13, 2792. [Google Scholar] [CrossRef]
- Sakuth, M.; Mensing, T.; Schuler, J.; Heitmann, W.; Strehlke, G.; Mayer, D. Ethers, Aliphatic. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2010. [Google Scholar]
- Phung, T.K.; Proietti Hernández, L.; Lagazzo, A.; Busca, G. Dehydration of ethanol over zeolites, silica alumina and alumina: Lewis acidity, Brønsted acidity and confinement effects. Appl. Catal. A Gen. 2015, 493, 77–89. [Google Scholar] [CrossRef]
- Phung, T.K.; Hernández, L.P.; Busca, G. Conversion of Ethanol over transition metal oxide catalysts: Effect of tungsta addition on catalytic behaviour of titania and zirconia. Appl. Catal. A Gen. 2015, 489, 180–187. [Google Scholar] [CrossRef]
- Phung, T.K.; Busca, G. Diethyl ether cracking and ethanol dehydration: Acid catalysis and reaction paths. Chem. Eng. J. 2015, 272, 92–101. [Google Scholar] [CrossRef]
- Phung, T.K.; Lagazzo, A.; Rivero Crespo, M.Á.; Sánchez Escribano, V.; Busca, G. A study of commercial transition aluminas and of their catalytic activity in the dehydration of ethanol. J. Catal. 2014, 311, 102–113. [Google Scholar] [CrossRef]
- Garbarino, G.; Prasath Parameswari Vijayakumar, R.; Riani, P.; Finocchio, E.; Busca, G. Ethanol and diethyl ether catalytic conversion over commercial alumina and lanthanum-doped alumina: Reaction paths, catalyst structure and coking. Appl. Catal. B Environ. 2018, 236, 490–500. [Google Scholar] [CrossRef]
- Garbarino, G.; Travi, I.; Pani, M.; Carnasciali, M.M.; Busca, G. Pure vs. ultra-pure γ-alumina: A spectroscopic study and catalysis of ethanol conversion. Catal. Commun. 2015, 70, 77–81. [Google Scholar] [CrossRef]
- Phung, T.K.; Busca, G. Ethanol dehydration on silica-aluminas: Active sites and ethylene/diethyl ether selectivities. Catal. Commun. 2015, 68, 110–115. [Google Scholar] [CrossRef]
- Phung, T.K.; Herrera, C.; Larrubia, M.Á.; García-Diéguez, M.; Finocchio, E.; Alemany, L.J.; Busca, G. Surface and catalytic properties of some γ-Al2O3 powders. Appl. Catal. A Gen. 2014, 483, 255–264. [Google Scholar] [CrossRef]
- Garbarino, G.; Wang, C.; Valsamakis, I.; Chitsazan, S.; Riani, P.; Finocchio, E.; Flytzani-Stephanopoulos, M.; Busca, G. Acido-basicity of lanthana/alumina catalysts and their activity in ethanol conversion. Appl. Catal. B Environ. 2017, 200, 458–468. [Google Scholar] [CrossRef] [Green Version]
- Costa, E.; Uguina, A.; Aguado, J.; Hernandez, P.J. Ethanol to gasoline process: Effect of variables, mechanism, and kinetics. Ind. Eng. Chem. Process Des. Dev. 1985, 24, 239–244. [Google Scholar] [CrossRef]
- Eagan, N.M.; Kumbhalkar, M.D.; Buchanan, J.S.; Dumesic, J.A.; Huber, G.W. Chemistries and processes for the conversion of ethanol into middle-distillate fuels. Nat. Rev. Chem. 2019, 3, 223–249. [Google Scholar] [CrossRef]
- Phung, T.K.; Radikapratama, R.; Garbarino, G.; Lagazzo, A.; Riani, P.; Busca, G. Tuning of product selectivity in the conversion of ethanol to hydrocarbons over H-ZSM-5 based zeolite catalysts. Fuel Process. Technol. 2015, 137, 290–297. [Google Scholar] [CrossRef]
- Li, Z.; Lepore, A.W.; Salazar, M.F.; Foo, G.S.; Davison, B.H.; Wu, Z.; Narula, C.K. Selective conversion of bio-derived ethanol to renewable BTX over Ga-ZSM-5. Green Chem. 2017, 19, 4344–4352. [Google Scholar] [CrossRef]
- Cimino, S.; Lisi, L.; Romanucci, S. Catalysts for conversion of ethanol to butanol: Effect of acid-base and redox properties. Catal. Today 2018, 304, 58–63. [Google Scholar] [CrossRef]
- Garbarino, G.; Riani, P.; Villa García, M.; Finocchio, E.; Sánchez Escribano, V.; Busca, G. A study of ethanol conversion over zinc aluminate catalyst. React. Kinet. Mech. Catal. 2018, 124, 503–522. [Google Scholar] [CrossRef]
- Lari, G.M.; Desai, K.; Mondelli, C.; Pérez-Ramírez, J. Selective dehydrogenation of bioethanol to acetaldehyde over basic USY zeolites. Catal. Sci. Technol. 2016, 6, 2706–2714. [Google Scholar] [CrossRef]
- Wang, C.; Garbarino, G.; Allard, L.F.; Wilson, F.; Busca, G.; Flytzani-Stephanopoulos, M. Low-Temperature Dehydrogenation of Ethanol on Atomically Dispersed Gold Supported on ZnZrOX. ACS Catal. 2016, 6, 210–218. [Google Scholar] [CrossRef]
- Finger, P.H.; Osmari, T.A.; Costa, M.S.; Bueno, J.M.C.; Gallo, J.M.R. The role of the interface between Cu and metal oxides in the ethanol dehydrogenation. Appl. Catal. A Gen. 2020, 589, 117236. [Google Scholar] [CrossRef]
- Garbarino, G.; Riani, P.; Villa García, M.; Finocchio, E.; Sanchez Escribano, V.; Busca, G. A study of ethanol dehydrogenation to acetaldehyde over copper/zinc aluminate catalysts. Catal. Today 2020, 354, 167–175. [Google Scholar] [CrossRef]
- Pampararo, G.; Garbarino, G.; Riani, P.; Garcia, M.V.; Escribano, V.S.; Busca, G. A study of ethanol dehydrogenation to acetaldehyde over supported copper catalysts: Catalytic activity, deactivation and regeneration. Appl. Catal. A Gen. 2020, 117710. [Google Scholar] [CrossRef]
- Carotenuto, G.; Tesser, R.; Di Serio, M.; Santacesaria, E. Kinetic study of ethanol dehydrogenation to ethyl acetate promoted by a copper/copper-chromite based catalyst. Catal. Today 2013, 203, 202–210. [Google Scholar] [CrossRef]
- Morales, M.V.; Asedegbega-Nieto, E.; Castillejos-López, E.; Bachiller-Baeza, B.; Guerrero-Ruiz, A. Difference in the deactivation of Au catalysts during ethanol transformation when supported on ZnO and on TiO2. RSC Adv. 2018, 8, 7473–7485. [Google Scholar] [CrossRef] [Green Version]
- Ochoa, J.V.; Bandinelli, C.; Vozniuk, O.; Chieregato, A.; Malmusi, A.; Recchi, C.; Cavani, F. An analysis of the chemical, physical and reactivity features of MgO–SiO2 catalysts for butadiene synthesis with the Lebedev process. Green Chem. 2016, 18, 1653–1663. [Google Scholar] [CrossRef]
- Subramani, V.; Song, C. Advances in Catalysis and Processes for Hydrogen Production from Ethanol Reforming. In Catalysis; Royal Society of Chemistry: Cambridge, UK, 2007; pp. 65–106. [Google Scholar]
- Liu, P.; Hensen, E.J.M. Highly Efficient and Robust Au/MgCuCr2O4 Catalyst for Gas-Phase Oxidation of Ethanol to Acetaldehyde. J. Am. Chem. Soc. 2013, 135, 14032–14035. [Google Scholar] [CrossRef]
- Mostrou, S.; Nagl, A.; Ranocchiari, M.; Föttinger, K.; van Bokhoven, J.A. The catalytic and radical mechanism for ethanol oxidation to acetic acid. Chem. Commun. 2019, 55, 11833–11836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Čičmanec, P.; Ganjkhanlou, Y.; Kotera, J.; Hidalgo, J.M.; Tišler, Z.; Bulánek, R. The effect of vanadium content and speciation on the activity of VOx/ZrO2 catalysts in the conversion of ethanol to acetaldehyde. Appl. Catal. A Gen. 2018, 564, 208–217. [Google Scholar] [CrossRef]
- Chumbhale, V.R.; Sonsale, A.Y.; Awasarkar, P.A. Oxidative Dehydrogenation of Ethanol over Binary Oxides of Sn and Mo. In Studies in Surface Science and Catalysis; Elsevier Inc.: Amsterdams, The Netherlands, 1998; Volume 113, pp. 479–485. [Google Scholar]
- Li, X.; Iglesia, E. Selective Catalytic Oxidation of Ethanol to Acetic Acid on Dispersed Mo-V-Nb Mixed Oxides. Chem. A Eur. J. 2007, 13, 9324–9330. [Google Scholar] [CrossRef] [PubMed]
- Folco, F.; Velasquez Ochoa, J.; Cavani, F.; Ott, L.; Janssen, M. Ethanol gas-phase ammoxidation to acetonitrile: The reactivity of supported vanadium oxide catalysts. Catal. Sci. Technol. 2017, 7, 200–212. [Google Scholar] [CrossRef]
- Liotta, L.F. Catalytic oxidation of volatile organic compounds on supported noble metals. Appl. Catal. B Environ. 2010, 100, 403–412. [Google Scholar] [CrossRef]
- Peluso, M.A.; Pronsato, E.; Sambeth, J.E.; Thomas, H.J.; Busca, G. Catalytic combustion of ethanol on pure and alumina supported K-Mn oxides: An IR and flow reactor study. Appl. Catal. B Environ. 2008, 78. [Google Scholar] [CrossRef]
- Gallegos, M.V.; Peluso, M.A.; Finocchio, E.; Thomas, H.J.; Busca, G.; Sambeth, J.E. Removal of VOCs by catalytic process. A study of MnZnO composites synthesized from waste alkaline and Zn/C batteries. Chem. Eng. J. 2017, 313. [Google Scholar] [CrossRef]
- Trevisanut, C.; Mari, M.; Millet, J.-M.M.; Cavani, F. Chemical-loop reforming of ethanol over metal ferrites: An analysis of structural features affecting reactivity. Int. J. Hydrogen Energy 2015, 40, 5264–5271. [Google Scholar] [CrossRef]
- Ochoa, J.V.; Trevisanut, C.; Millet, J.-M.M.; Busca, G.; Cavani, F. In situ DRIFTS-MS study of the anaerobic oxidation of ethanol over spinel mixed oxides. J. Phys. Chem. C 2013, 117, 23908–23918. [Google Scholar] [CrossRef]
- Livio, D.; Diehm, C.; Donazzi, A.; Beretta, A.; Deutschmann, O. Catalytic partial oxidation of ethanol over Rh/Al2O3: Spatially resolved temperature and concentration profiles. Appl. Catal. A Gen. 2013, 467, 530–541. [Google Scholar] [CrossRef]
- Nahar, G.; Dupont, V. Hydrogen via steam reforming of liquid biofeedstock. Biofuels 2012, 3, 167–191. [Google Scholar] [CrossRef]
- Bion, N.; Duprez, D.; Epron, F. Design of Nanocatalysts for Green Hydrogen Production from Bioethanol. ChemSusChem 2012, 5, 76–84. [Google Scholar] [CrossRef]
- Busca, G.; Costantino, U.; Montanari, T.; Ramis, G.; Resini, C.; Sisani, M. Nickel versus cobalt catalysts for hydrogen production by ethanol steam reforming: Ni-Co-Zn-Al catalysts from hydrotalcite-like precursors. Int. J. Hydrog. Energy 2010, 35, 5356–5366. [Google Scholar] [CrossRef]
- Resini, C.; Concepción Herrera Delgado, M.; Presto, S.; Alemany, L.J.; Riani, P.; Marazza, R.; Ramis, G.; Busca, G. Yttria-stabilized zirconia (YSZ) supported Ni-Co alloys (precursor of SOFC anodes) as catalysts for the steam reforming of ethanol. Int. J. Hydrog. Energy 2008, 33, 3728–3735. [Google Scholar] [CrossRef]
- Moretti, E.; Storaro, L.; Talon, A.; Chitsazan, S.; Garbarino, G.; Busca, G.; Finocchio, E. Ceria-zirconia based catalysts for ethanol steam reforming. Fuel 2015, 153, 166–175. [Google Scholar] [CrossRef]
- Riani, P.; Garbarino, G.; Cavattoni, T.; Busca, G. CO2 hydrogenation and ethanol steam reforming over Co/SiO2 catalysts: Deactivation and selectivity switches. Catal. Today 2020. [Google Scholar] [CrossRef]
- Garbarino, G.; Sanchez Escribano, V.; Finocchio, E.; Busca, G. Steam reforming of phenol-ethanol mixture over 5% Ni/Al2O3. Appl. Catal. B Environ. 2012, 147, 113–114. [Google Scholar] [CrossRef]
- Garbarino, G.; Romero Perez, A.; Finocchio, E.; Busca, G. A study of the deactivation of low loading Ni/Al2O3 steam reforming catalyst by tetrahydrothiophene. Catal. Commun. 2013, 38, 67–73. [Google Scholar] [CrossRef]
- Garbarino, G.; Riani, P.; Lucchini, M.A.; Canepa, F.; Kawale, S.; Busca, G. Cobalt-based nanoparticles as catalysts for low temperature hydrogen production by ethanol steam reforming. Int. J. Hydrogen Energy 2013, 38, 82–91. [Google Scholar] [CrossRef]
- Garbarino, G.; Lagazzo, A.; Riani, P.; Busca, G. Steam reforming of ethanol-phenol mixture on Ni/Al2O3: Effect of Ni loading and sulphur deactivation. Appl. Catal. B Environ. 2013, 129. [Google Scholar] [CrossRef]
- Garbarino, G.; Campodonico, S.; Perez, A.R.; Carnasciali, M.M.; Riani, P.; Finocchio, E.; Busca, G. Spectroscopic characterization of Ni/Al2O3 catalytic materials for the steam reforming of renewables. Appl. Catal. A Gen. 2013, 452, 163–173. [Google Scholar] [CrossRef]
- Chitsazan, S.; Sepehri, S.; Garbarino, G.; Carnasciali, M.M.; Busca, G. Steam reforming of biomass-derived organics: Interactions of different mixture components on Ni/Al2O3 based catalysts. Appl. Catal. B Eniron. 2016, 187, 386–398. [Google Scholar] [CrossRef]
- Riani, P.; Garbarino, G.; Lucchini, M.A.; Canepa, F.; Busca, G. Unsupported versus alumina-supported Ni nanoparticles as catalysts for steam/ethanol conversion and CO2 methanation. J. Mol. Catal. A Chem. 2014, 383, 10–16. [Google Scholar] [CrossRef]
- Garbarino, G.; Chitsazan, S.; Phung, T.K.; Riani, P.; Busca, G. Preparation of supported catalysts: A study of the effect of small amounts of silica on Ni/Al2O3 catalysts. Appl. Catal. A Gen. 2015, 505, 86–97. [Google Scholar] [CrossRef]
- Garbarino, G.; Wang, C.; Valsamakis, I.; Chitsazan, S.; Riani, P.; Finocchio, E.; Flytzani-Stephanopoulos, M.; Busca, G. A study of Ni/Al2O3 and Ni-La/Al2O3 catalysts for the steam reforming of ethanol and phenol. Appl. Catal. B Environ. 2015, 174, 21–34. [Google Scholar] [CrossRef] [Green Version]
- Riani, P.; Garbarino, G.; Canepa, F.; Busca, G. Cobalt nanoparticles mechanically deposited on α-Al2O3: A competitive catalyst for the production of hydrogen through ethanol steam reforming. J. Chem. Technol. Biotechnol. 2019, 94, 538–546. [Google Scholar] [CrossRef]
- Garbarino, G.; Cavattoni, T.; Riani, P.; Brescia, R.; Canepa, F.; Busca, G. On the Role of Support in Metallic Heterogeneous Catalysis: A Study of Unsupported Nickel–Cobalt Alloy Nanoparticles in Ethanol Steam Reforming. Catal. Lett. 2019, 149, 929–941. [Google Scholar] [CrossRef]
- Riani, P.; Garbarino, G.; Cavattoni, T.; Canepa, F.; Busca, G. Unsupported cobalt nanoparticles as catalysts: Effect of preparation method on catalytic activity in CO2 methanation and ethanol steam reforming. Int. J. Hydrogen Energy 2019, 44, 27319–27328. [Google Scholar] [CrossRef]
- Costamagna, P.; Pugliese, F.; Cavattoni, T.; Busca, G.; Garbarino, G. Modeling of Laboratory Steam Methane Reforming and CO2 Methanation Reactors. Energies 2020, 13, 2624. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reaction | GHSV (h−1) | T Range (K) | Gas Mixture | Mode |
|---|---|---|---|---|
| Dehydration to ethylene and DEE | 10,000–240,000 | 423–773 | N2/Ethanol | SS + TPSR |
| Dehydration to higher hydrocarbons | 10,000 | 423–773 | N2/Ethanol | SS |
| Dehydrogenation | 10,000–240,000 | 423–773 | N2/Ethanol | SS + TPSR |
| Oxidative dehydrogenation | 10,000 | 423–773 | N2/O2/Ethanol | SS |
| 240,000 | 423–773 | N2/O2/Ethanol | TPSR | |
| Steam reforming | 55,000 | 473–1073 | He/H2O/Ethanol | SS |
| Reactions | ΔH0298 (kJ/mol) | No. |
|---|---|---|
| CH3CH2OH(g) ⇌ ½ (CH3CH2)2O(g) + ½ H2O(g) | −11.5 | (1) |
| CH3CH2OH(g) ⇌ CH2=CH2(g)+ H2O(g) | +44.7 | (2) |
| CH3CH2OH(g) ⇌ CH3CHO(g) + H2(g) | +68.6 | (3) |
| CH3CH2OH(g) ⇌ ½ CH3COOCH2CH3(g) + H2(g) | +25.4 | (4) |
| CH3CH2OH(g) + H2O(g) ⇌ 2 CO(g) + 4 H2(g) | +255.6 | (5) |
| CH3CH2OH(g) + 3 H2O(g) ⇌ 2 CO2(g) + 6 H2(g) | +173.2 | (6) |
| CH3CH2OH(g) ⇌ CH4(g) + CO(g) + H2(g) | +49.7 | (7) |
| (CH3CH2)2O(g) ⇌ CH2=CH2(g) + CH3CH2OH(g) | +67.7 | (8) |
| CH3CH2OH → 1/3 CH2=CH-CH2-CH2-CH2-CH3 + H2O | −22.0 | (9) |
| CH3CH2OH → 1/3 C6H6 + H2 + H2O | +19.3 | (10) |
| CH3CHO ⇌ ½ CH3CHOH-CH2-CHO → ½ CH3COCH3 + ½ H2 + ½ CO | +6.4 | (11) |
| CH3CH2OH → ½ CH3COCH3 + 3/2 H2 + ½ CO | +71 | (12) |
| CH3CH2OH + ½ O2 → CH3CHO + H2O | −173.2 | (13) |
| CH3CH2OH + 3 O2 → 2 CO2 + 3 H2O | −1277.6 | (14) |
| CH3CH2OH + ½ O2 → 2 CO + 3 H2 | +13.8 | (15) |
| CO + H2O ⇌ CO2 + H2 | −41.2 | (16) |
| CO + 3 H2⇌ CH4+ H2O | −205.9 | (17) |
| CO2 + 4 H2⇌ CH4+ 2 H2O | −164.7 | (18) |
| CH3CHO ⇌ CH4 + CO | −18.9 | (19) |
| Catalyst | Producer | T (K) | XEtOH % | YDEE % | YBy-Products % |
|---|---|---|---|---|---|
| H-BEA | Zeolyst | 473 | 84.5 | 74.6 | 9.4 ethylene, 0.5 butene |
| WO3/TiO2 | Bayer | 473 | 87.3 | 73.2 | 13.9 ethylene, 0.3 acetaldehyde |
| H-MFI | Zeolyst | 473 | 82.0 | 71.9 | 10.1 ethylene |
| H-FER | Zeolyst | 473 | 70.0 | 66.1 | 3.9 ethylene |
| γ-Al2O3 | Sasol | 523 | 77.8 | 61.8 | 15.6 ethylene, 0.1 butene |
| H-MOR | Zeolyst | 453 | 85.2 | 60.5 | 24.7 ethylene |
| Catalyst | Producer | T (K) | Maximum YEtylene % | By-Products at Maximum Ethylene Yield |
|---|---|---|---|---|
| USY | Zeolyst | 573 | >99.5 | DEE |
| SO42−/ZrO2 | Home-made | 573 | >99.5 | – |
| ASA | Strem | 623 | >99.5 | – |
| H-Y | Zeolyst | 573 | 99.4 | DEE (hydrocarbons) |
| WO3/ZrO2 | Home-made | 623 | 99.0 | hydrocarbons |
| H-MFI (280) | Zeolyst | 573 | 98.9 | hydrocarbons |
| H-FER | Zeolyst | 523 | 98.7 | DEE (hydrocarbons) |
| γ-Al2O3 | Sasol | 623 | 98.4 | hydrocarbons |
| WO3/TiO2 | Home-made | 623 | 98.3 | hydrocarbons |
| H-BEA | Zeolyst | 623 | 95.6 | hydrocarbons |
| H-MOR | Zeolyst | 523 | 94.4 | hydrocarbons |
| WO3/TiO2 | Bayer | 523 | 92.7 | hydrocarbons |
| H-MFI (50) | Zeolist | 523 | 92.1 | hydrocarbons |
| WO3/SiO2 | Home-made | 723 | 92.0 | hydrocarbons (DEE) |
| T (K) | ESR | MSR | |||
|---|---|---|---|---|---|
| XEtOH % | YH2 % | YCH4 % | XCH4 % | YH2 % | |
| 773 | 100 | 82 | 14 | 60 | 60 |
| 853 | 100 | 93 | 0 | 91 | 87 |
| 893 | 100 | 93 | 0 | 97 | 91 |
| 973 | 100 | 90 | 0 | 100 | 92 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garbarino, G.; Pampararo, G.; Phung, T.K.; Riani, P.; Busca, G. Heterogeneous Catalysis in (Bio)Ethanol Conversion to Chemicals and Fuels: Thermodynamics, Catalysis, Reaction Paths, Mechanisms and Product Selectivities. Energies 2020, 13, 3587. https://doi.org/10.3390/en13143587
Garbarino G, Pampararo G, Phung TK, Riani P, Busca G. Heterogeneous Catalysis in (Bio)Ethanol Conversion to Chemicals and Fuels: Thermodynamics, Catalysis, Reaction Paths, Mechanisms and Product Selectivities. Energies. 2020; 13(14):3587. https://doi.org/10.3390/en13143587
Chicago/Turabian StyleGarbarino, Gabriella, Giovanni Pampararo, Thanh Khoa Phung, Paola Riani, and Guido Busca. 2020. "Heterogeneous Catalysis in (Bio)Ethanol Conversion to Chemicals and Fuels: Thermodynamics, Catalysis, Reaction Paths, Mechanisms and Product Selectivities" Energies 13, no. 14: 3587. https://doi.org/10.3390/en13143587
APA StyleGarbarino, G., Pampararo, G., Phung, T. K., Riani, P., & Busca, G. (2020). Heterogeneous Catalysis in (Bio)Ethanol Conversion to Chemicals and Fuels: Thermodynamics, Catalysis, Reaction Paths, Mechanisms and Product Selectivities. Energies, 13(14), 3587. https://doi.org/10.3390/en13143587