Detailing the Self-Discharge of a Cathode Based on a Prussian Blue Analogue

 , ,
, ,  , and
, and 
Abstract
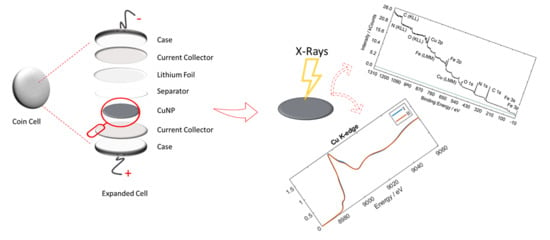
:
1. Introduction
2. Materials and Methods
2.1. Material Synthesis and Electrode Preparation
2.2. Material Characterization
3. Results and Discussion
3.1. Powder Characterization
3.2. Formulated Pellet and Pellet in Contact with Electrolyte
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Leonard, M.D.; Michaelides, E.E.; Michaelides, D.N. Energy storage needs for the substitution of fossil fuel power plants with renewables. Renew. Energy 2020, 145, 951–962. [Google Scholar] [CrossRef]
- Goodenough, J.B.; Kim, Y. Challenges for rechargeable Li batteries. Chem. Mater. 2010, 22, 587–603. [Google Scholar] [CrossRef]
- Elia, G.A.; Marquardt, K.; Hoeppner, K.; Fantini, S.; Lin, R.; Knipping, E.; Peters, W.; Drillet, J.F.; Passerini, S.; Hahn, R. An overview and future perspectives of aluminum batteries. Adv. Mater. 2016, 28, 7564–7579. [Google Scholar] [CrossRef] [PubMed]
- Kiani, M.A.; Mousavi, M.F.; Rahmanifar, M.S. Synthesis of nano- and micro-particles of LiMn2O4: Electrochemical investigation and assessment as a cathode in li battery. Int. J. Electrochem. Sci. 2011, 6, 2581–2595. [Google Scholar]
- Tarascon, J.M.; Armand, M. Issues and challenges facing rechargeable lithium batteries. Nature 2001, 414, 359–367. [Google Scholar] [CrossRef]
- Edström, K.; Gustafsson, T.; Thomas, J.O. The cathode-electrolyte interface in the Li-ion battery. Electrochim. Acta 2004, 50, 397–403. [Google Scholar] [CrossRef]
- Duncan, H.; Abu-Lebdeh, Y.; Davidson, I.J. Study of the cathode–electrolyte interface of LiMn1.5Ni0.5O4 synthesized by a Sol–Gel method for Li-Ion batteries. J. Electrochem. Soc. 2010, 157, A528. [Google Scholar] [CrossRef]
- Malmgren, S.; Ciosek, K.; Lindblad, R.; Plogmaker, S.; Kühn, J.; Rensmo, H.; Edström, K.; Hahlin, M. Consequences of air exposure on the lithiated graphite SEI. Electrochim. Acta 2013, 105, 83–91. [Google Scholar] [CrossRef] [Green Version]
- Andersson, A. Surface Phenomena in Li-Ion Batteries. Ph.D. Thesis, Uppsala University, Uppsala, Sweden, 2001. [Google Scholar]
- Leanza, D.; Vaz, C.A.F.; Melinte, G.; Mu, X.; Novák, P.; El Kazzi, M. Revealing. The dual surface reactions on a HE-NCM Li-Ion battery cathode and their impact on the surface chemistry of the counter electrode. ACS Appl. Mater. Interfaces 2019, 11, 6054–6065. [Google Scholar] [CrossRef]
- Lin, R.; Hu, E.; Liu, M.; Wang, Y.; Cheng, H.; Wu, J.; Zheng, J.C.; Wu, Q.; Bak, S.; Tong, X.; et al. Anomalous metal segregation in lithium-rich material provides design rules for stable cathode in lithium-ion battery. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Malmgren, S.; Ciosek, K.; Hahlin, M.; Gustafsson, T.; Gorgoi, M.; Rensmo, H.; Edström, K. Comparing anode and cathode electrode/electrolyte interface composition and morphology using soft and hard X-ray photoelectron spectroscopy. Electrochim. Acta 2013, 97, 23–32. [Google Scholar] [CrossRef]
- Yazami, R.; Reynier, Y.F. Mechanism of self-discharge in graphite-lithium anode. Electrochim. Acta 2002, 47, 1217–1223. [Google Scholar] [CrossRef]
- Wang, L.; Menakath, A.; Han, F.; Wang, Y.; Zavalij, P.Y.; Gaskell, K.J.; Borodin, O.; Iuga, D.; Brown, S.P.; Wang, C.; et al. Identifying the components of the solid–electrolyte interphase in Li-ion batteries. Nat. Chem. 2019, 11, 789–796. [Google Scholar] [CrossRef] [PubMed]
- Xu, K. Electrolytes and interphases in Li-ion batteries and beyond. Chem. Rev. 2014, 114, 11503–11618. [Google Scholar] [CrossRef]
- Persson, K.; Sethuraman, V.A.; Hardwick, L.J.; Hinuma, Y.; Meng, Y.S.; Van Der Ven, A.; Srinivasan, V.; Kostecki, R.; Ceder, G. Lithium diffusion in graphitic carbon. J. Phys. Chem. Lett. 2010, 1, 1176–1180. [Google Scholar] [CrossRef] [Green Version]
- Nie, M.; Chalasani, D.; Abraham, D.P.; Chen, Y.; Bose, A.; Lucht, B.L. Lithium ion battery graphite solid electrolyte interphase revealed by microscopy and spectroscopy. J. Phys. Chem. C 2013, 117, 1257–1267. [Google Scholar] [CrossRef]
- Aurbach, D.; Markovsky, B.; Shechter, A.; Ein-Eli, Y.; Cohen, H. A comparative study of synthetic graphite and Li electrodes in electrolyte solutions based on ethylene carbonate-Dimethyl carbonate mixtures. J. Electrochem. Soc. 1996, 143, 3809–3820. [Google Scholar] [CrossRef]
- Verma, P.; Maire, P.; Novak, P. A review of the features and analyses of the solid electrolyte interphase in Li-ion batteries. Electrochim. Acta 2010, 55, 6332–6341. [Google Scholar] [CrossRef]
- Ventosa, E.; Madej, E.; Zampardi, G.; Mei, B.; Weide, P.; Antoni, H.; La Mantia, F.; Muhler, M.; Schuhmann, W. Solid electrolyte interphase (SEI) at TiO2 electrodes in Li-Ion batteries: Defining Apparent and Effective SEI based on evidence from X-ray photoemission spectroscopy and scanning electrochemical microscopy. ACS Appl. Mater. Interfaces 2017, 9, 3123–3130. [Google Scholar] [CrossRef]
- Hekmatfar, M.; Kazzazi, A.; Eshetu, G.G.; Hasa, I.; Passerini, S. Understanding the electrode/electrolyte interface layer on the Li- Rich nickel manganese cobalt layered oxide cathode by XPS. ACS Appl. Mater. Interfaces 2019, 11, 43166–43179. [Google Scholar] [CrossRef]
- Erickson, E.M.; Li, W.; Dolocan, A.; Manthiram, A.; Erickson, E.M.; Li, W.; Dolocan, A.; Manthiram, A. Insights into the cathode-electrolyte interphases of high-energy-density cathodes in lithium-ion batteries. ACS Appl. Mater. Interfaces 2020, 12, 16451–16461. [Google Scholar] [CrossRef]
- Kang, S.H.; Abraham, D.P.; Xiao, A.; Lucht, B.L. Investigating the solid electrolyte interphase using binder-free graphite electrodes. J. Power Sources 2008, 175, 526–532. [Google Scholar] [CrossRef]
- Tsubouchi, S.; Domi, Y.; Doi, T.; Ochida, M.; Nakagawa, H.; Yamanaka, T.; Abe, T.; Ogumi, Z. Spectroscopic characterization of surface films formed on edge plane graphite in ethylene carbonate-based electrolytes containing film-forming additives. J. Electrochem. Soc. 2012, 159, A1786–A1790. [Google Scholar] [CrossRef]
- Michan, A.L.; Leskes, M.; Grey, C.P. Voltage dependent solid electrolyte interphase formation in silicon electrodes: Monitoring the formation of organic decomposition products. Chem. Mater. 2016, 28, 385–398. [Google Scholar] [CrossRef]
- Li, J.; Xing, L.; Zhang, L.; Yu, L.; Fa, W.; Xu, M.; Li, W. Insight into self-discharge of layered lithium-rich oxide cathode in carbonate-based electrolytes with and without additive. J. Power Sources 2016, 324, 17–25. [Google Scholar] [CrossRef]
- Ware, M. Prussian blue: Artists’ pigment and chemists’ sponge. J. Chem. Educ. 2008, 85, 612. [Google Scholar] [CrossRef]
- Mullaliu, A.; Aquilanti, G.; Conti, P.; Plaisier, J.R.; Fehse, M.; Stievano, L.; Giorgetti, M. Copper electroactivity in prussian blue-based cathode disclosed by operando XAS. J. Phys. Chem. C 2018, 122, 15868–15877. [Google Scholar] [CrossRef]
- Mullaliu, A.; Sougrati, M.-T.; Louvain, N.; Aquilanti, G.; Doublet, M.-L.; Stievano, L.; Giorgetti, M. The electrochemical activity of the nitrosyl ligand in copper nitroprusside: A new possible redox mechanism for lithium battery electrode materials? Electrochim. Acta 2017, 257, 364–371. [Google Scholar] [CrossRef]
- Hesse, R.; Chassé, T.; Szargan, R. Unifit 2002-universal analysis software for photoelectron spectra. Anal. Bioanal. Chem. 2003, 375, 856–863. [Google Scholar] [CrossRef]
- Hesse, R.; Streubel, P.; Szargan, R. Product or sum: Comparative tests of Voigt, and product or sum of Gaussian and Lorentzian functions in the fitting of synthetic Voigt-based X-ray photoelectron spectra. Surf. Interface Anal. 2007, 39, 381–391. [Google Scholar] [CrossRef]
- Yatsimirskii, K.B.; Nemoshkalenko, V.V.; Nazarenko, Y.P.; Aleshin, V.G.; Zhilinskaya, V.V.; Tomashevsky, N.A. Use of X-ray photoelectron and Mössbauer spectroscopies in the study of iron pentacyanide complexes. J. Electron Spectros. Relat. Phenomena 1977, 10, 239–245. [Google Scholar] [CrossRef]
- Aquilanti, G.; Giorgetti, M.; Dominko, R.; Stievano, L.; Arčon, I.; Novello, N.; Olivi, L. Operando characterization of batteries using x-ray absorption spectroscopy: Advances at the beamline XAFS at synchrotron Elettra. J. Phys. D Appl. Phys. 2017, 50, 1–12. [Google Scholar] [CrossRef]
- Cano, A.; Rodríguez-Hernández, J.; Shchukarev, A.; Reguera, E. Intercalation of pyrazine in layered copper nitroprusside: Synthesis, crystal structure and XPS study. J. Solid State Chem. 2019, 273, 1–10. [Google Scholar] [CrossRef]
- Biesinger, M.C.; Lau, L.W.M.; Gerson, A.R.; Smart, R.S.C. Resolving surface chemical states in XPS analysis of first row transition metals, oxides and hydroxides: Sc, Ti, V., Cu and Zn. Appl. Surf. Sci. 2010, 257, 887–898. [Google Scholar] [CrossRef]
- Gomez, A.; Rodriguez-Hernandez, J.; Reguera, E. Unique coordination in metal nitroprussides: The structure of Cu[Fe(CN)5NO] 2H2O and Cu[Fe(CN)5NO]. J. Chem. Crystallogr. 2004, 34, 893–903. [Google Scholar] [CrossRef]
- Mullaliu, A.; Aquilanti, G.; Stievano, L.; Conti, P.; Plaisier, J.R.; Cristol, S.; Giorgetti, M. Beyond the oxygen redox strategy in designing cathode material for batteries: Dynamics of a prussian blue-like cathode revealed by operando X-ray diffraction and X-ray absorption fine structure and by a theoretical approach. J. Phys. Chem. C 2019, 123, 8588–8598. [Google Scholar] [CrossRef]
- Greczynski, G.; Hultman, L. Compromising science by ignorant instrument calibration-need to revisit half a century of published XPS data. Angew. Chemie 2020, 132, 5034–5038. [Google Scholar] [CrossRef]
- Terborg, L.; Nowak, S.; Passerini, S.; Winter, M.; Karst, U.; Haddad, P.R.; Nesterenko, P.N. Ion chromatographic determination of hydrolysis products of hexafluorophosphate salts in aqueous solution. Anal. Chim. Acta 2012, 714, 121–126. [Google Scholar] [CrossRef]
- Siow, K.S.; Britcher, L.; Kumar, S.; Griesser, H.J. XPS study of sulfur and phosphorus compounds with different oxidation states. Sains Malays. 2018, 47, 1913–1922. [Google Scholar]
- Tisato, F.; Marzano, C.; Peruzzo, V.; Tegoni, M.; Giorgetti, M.; Damjanovic, M.; Trapananti, A.; Bagno, A.; Santini, C.; Pellei, M.; et al. Insights into the cytotoxic activity of the phosphane copper(I) complex [Cu(thp)4][PF6]. J. Inorg. Biochem. 2016, 165, 80–91. [Google Scholar] [CrossRef]
- Chaboy, J.; Muñoz-Páez, A.; Carrera, F.; Merkling, P.; Marcos, E.S. Ab initio x-ray absorption study of copper K -edge XANES spectra in Cu(II) compounds. Phys. Rev. B 2005, 71, 134208. [Google Scholar] [CrossRef]
- Tomson, N.C.; Williams, K.D.; Dai, X.; Sproules, S.; Debeer, S.; Warren, T.H.; Wieghardt, K. Re-evaluating the Cu K pre-edge XAS transition in complexes with covalent metal-ligand interactions. Chem. Sci. 2015, 6, 2474–2487. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
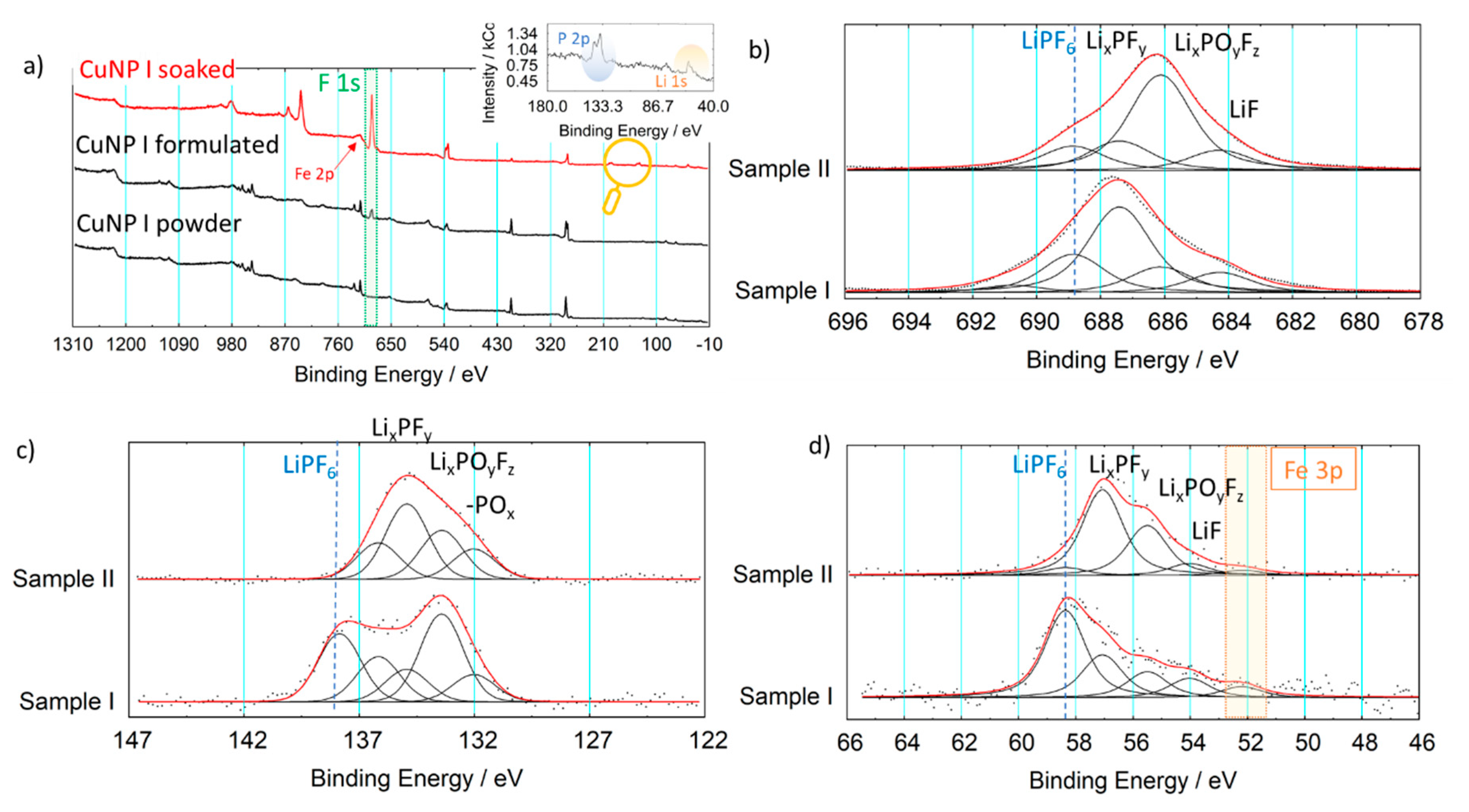
| Binding Energies (eV) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C 1s | N 1s | O 1s | Cu 2p | Fe 2p | ||||||||
| CN | Cadv | CN | NO | NO | H2O | Cu (+) | Cu (2+) | Cu Sat | Fe (II) High Spin | Fe (II) Low Spin | Fe Sat | |
| CuNP I | 283.7 | 284.6 | 397.2 | 402.2 | 530.3 | 531.6 | 932.0 | 935.4 | 942.0 | 708.0 | 709.8 | - |
| CuNP II | 283.6 | 284.9 | 397.2 | 402.2 | 529.7 | 531.5 | 932.1 | 935.4 | 942.1 | 708.2 | 710.2 | - |
| Ref [33] | 283.4/284.3 | 284.8 | 397.2/397.8 | 402.6 | 530.8 | 532.1 | 932.4 | 936 | 941.8 | 708.8 | 710.1 | 714.8 |
| C 1s (CN) | N 1s (CN) | N 1s (NO) | O 1s (NO) | Cu 2p | Fe 2p | Formula | Expected Formula | |
|---|---|---|---|---|---|---|---|---|
| I | 6.1 | 6.4 | 1 | 1.1 | 0.9 | 1.0 | Cu0.9Fe1(C6.1N6.7) (N1.0O1.1) | Cu0.8Fe1.2(CN)5(NO) |
| II | 6.5 | 6.7 | 1 | 1.0 | 1.7 | 1.4 | Cu1.7Fe 1.4(C6.5N6.7) (NO)1 | CuFe(CN)5(NO) |
| C 1s (CN) | N 1s (CN) | N 1s (NO) | O 1s (NO) | Cu 2p | Fe 2p | Formula | |
|---|---|---|---|---|---|---|---|
| I_formulated | 6.1 | 6.8 | 1.0 | 1.1 | 1.1 | 1.3 | Cu1.1Fe1.3(C6.1N6.8) (N1O1.1) |
| II_soaked | 6.4 | 6.1 | 1.0 | 1.0 | 1.4 | x | Cu1.4Fex(C6.4N6.1) (NO)1.0 |
| II_formulated | 6.7 | 6.8 | 1.0 | 1.0 | 1.5 | 1.7 | Cu1.5Fe1.7(C6.7N6.8) (NO)1.0 |
| II_soaked | 6.3 | 6.0 | 1.0 | 1.0 | 1.4 | x | Cu1.4 Fex(C6.3N6.0) (NO)1.0 |
| Assignment | Measured Binding Energy/eV | ||
|---|---|---|---|
| F 1s | Li 1s | P 2p | |
| LiF | 685.1 | 54.1 | - |
| LiPF6 | 688.8 | 58.4 | 137.8 |
| LixPFy | 687.6 | 57.1 | 136.5 |
| LixPOyFz | 686.2 | 55.5 | 134.5–135 |
| -POx | - | - | 133.0 |
| Teflon Area | 684–693 | - | - |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Musella, E.; Mullaliu, A.; Ruf, T.; Huth, P.; Tonelli, D.; Aquilanti, G.; Denecke, R.; Giorgetti, M. Detailing the Self-Discharge of a Cathode Based on a Prussian Blue Analogue. Energies 2020, 13, 4027. https://doi.org/10.3390/en13154027
Musella E, Mullaliu A, Ruf T, Huth P, Tonelli D, Aquilanti G, Denecke R, Giorgetti M. Detailing the Self-Discharge of a Cathode Based on a Prussian Blue Analogue. Energies. 2020; 13(15):4027. https://doi.org/10.3390/en13154027
Chicago/Turabian StyleMusella, Elisa, Angelo Mullaliu, Thomas Ruf, Paula Huth, Domenica Tonelli, Giuliana Aquilanti, Reinhard Denecke, and Marco Giorgetti. 2020. "Detailing the Self-Discharge of a Cathode Based on a Prussian Blue Analogue" Energies 13, no. 15: 4027. https://doi.org/10.3390/en13154027
APA StyleMusella, E., Mullaliu, A., Ruf, T., Huth, P., Tonelli, D., Aquilanti, G., Denecke, R., & Giorgetti, M. (2020). Detailing the Self-Discharge of a Cathode Based on a Prussian Blue Analogue. Energies, 13(15), 4027. https://doi.org/10.3390/en13154027