Modeling Adsorption in Silica Pores via Minkowski Functionals and Molecular Electrostatic Moments


Abstract
:1. Introduction
2. Methods
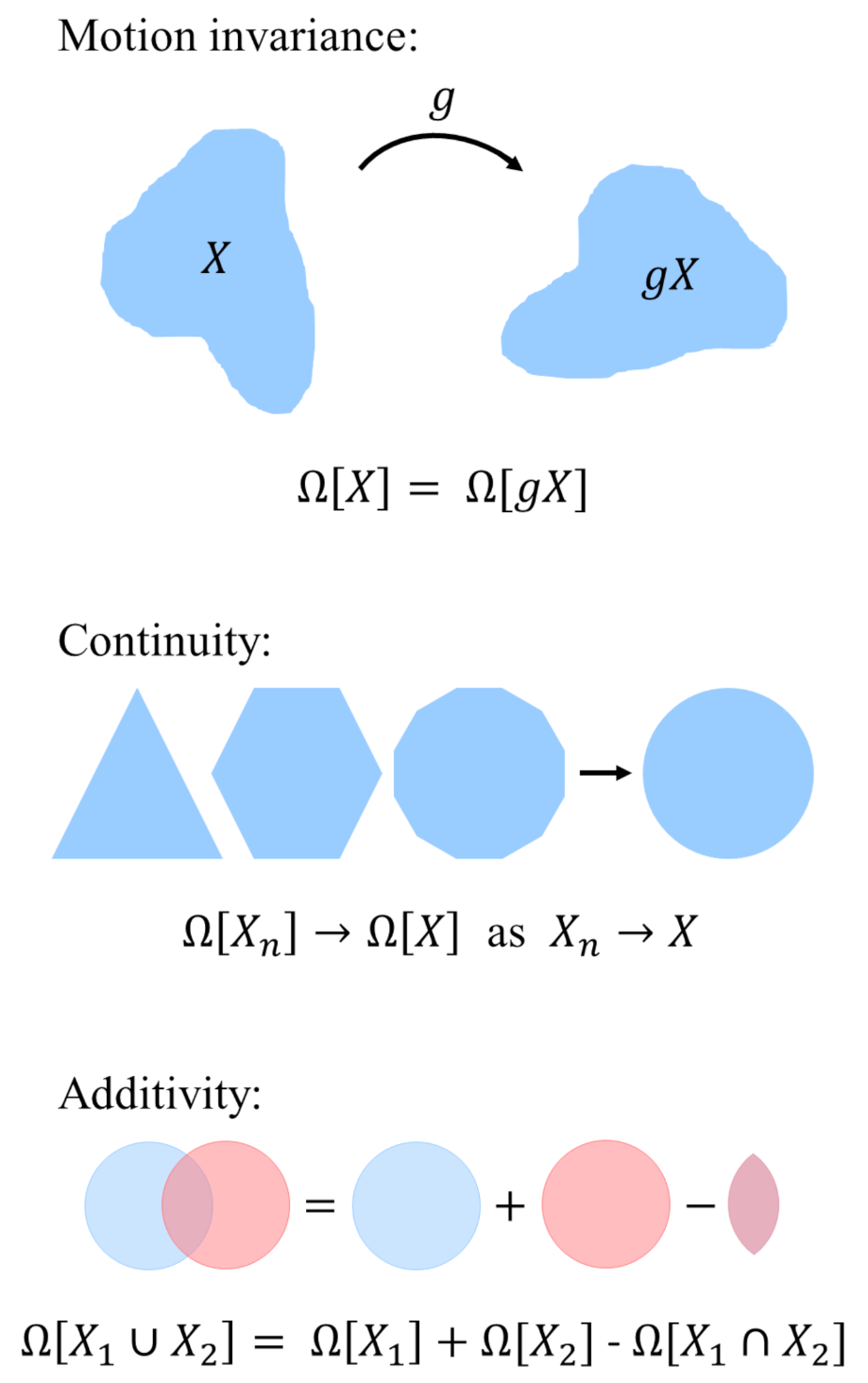
2.1. Mathematical Framework
- Motion invariance: The thermodynamic potential is independent of the system’s location and orientation in space. For any translation or rotation g:
- Continuity: If a sequence of convex sets converges to X for , then . This property states that an approximation of the convex domain also yields an approximation of the grand potential.
- Additivity: The functional union of two domains and is the sum of the functional of the individual domains subtracted by their intersection:
2.2. Numerical Simulations
3. Results and Discussion
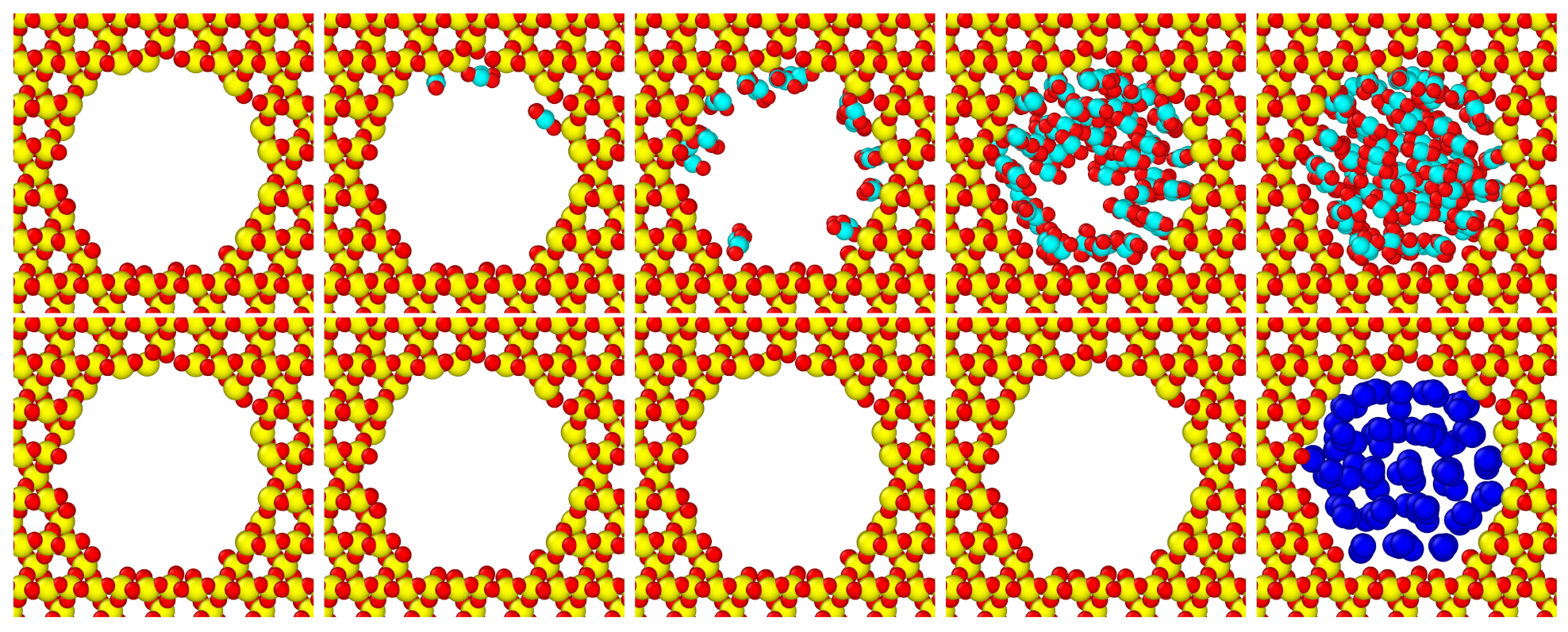
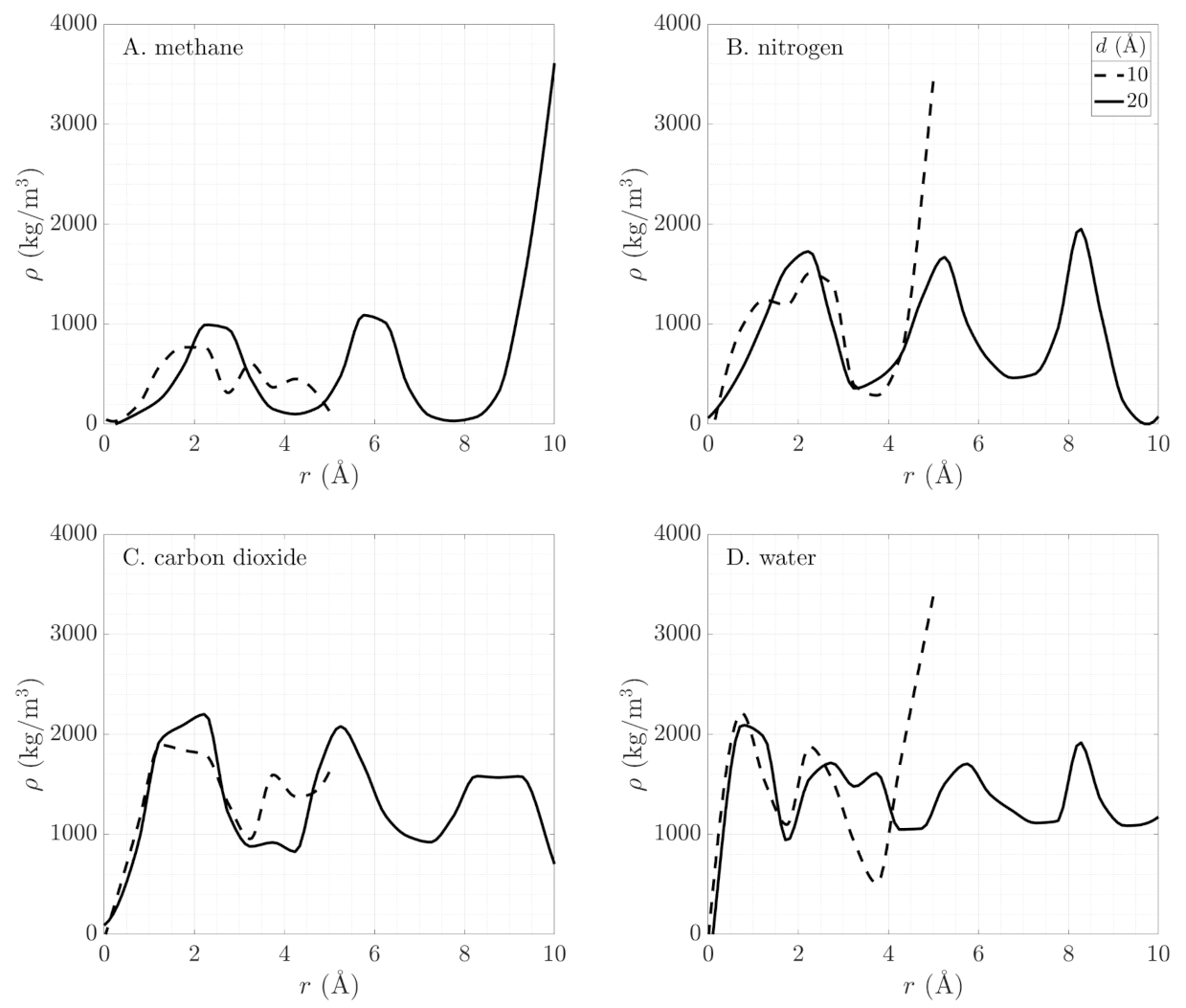
3.1. Influence of Molecular Polarity on Adsorption
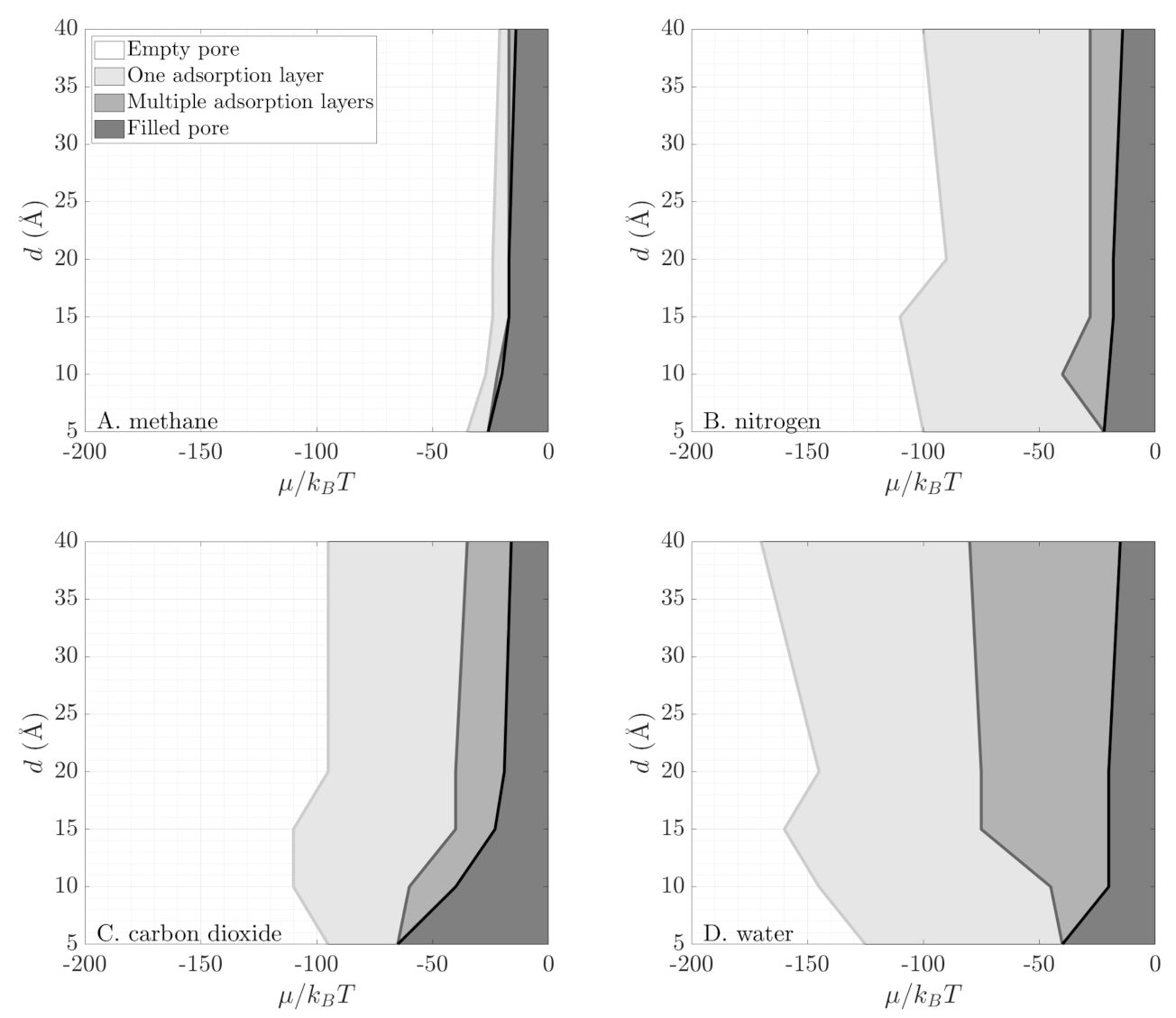
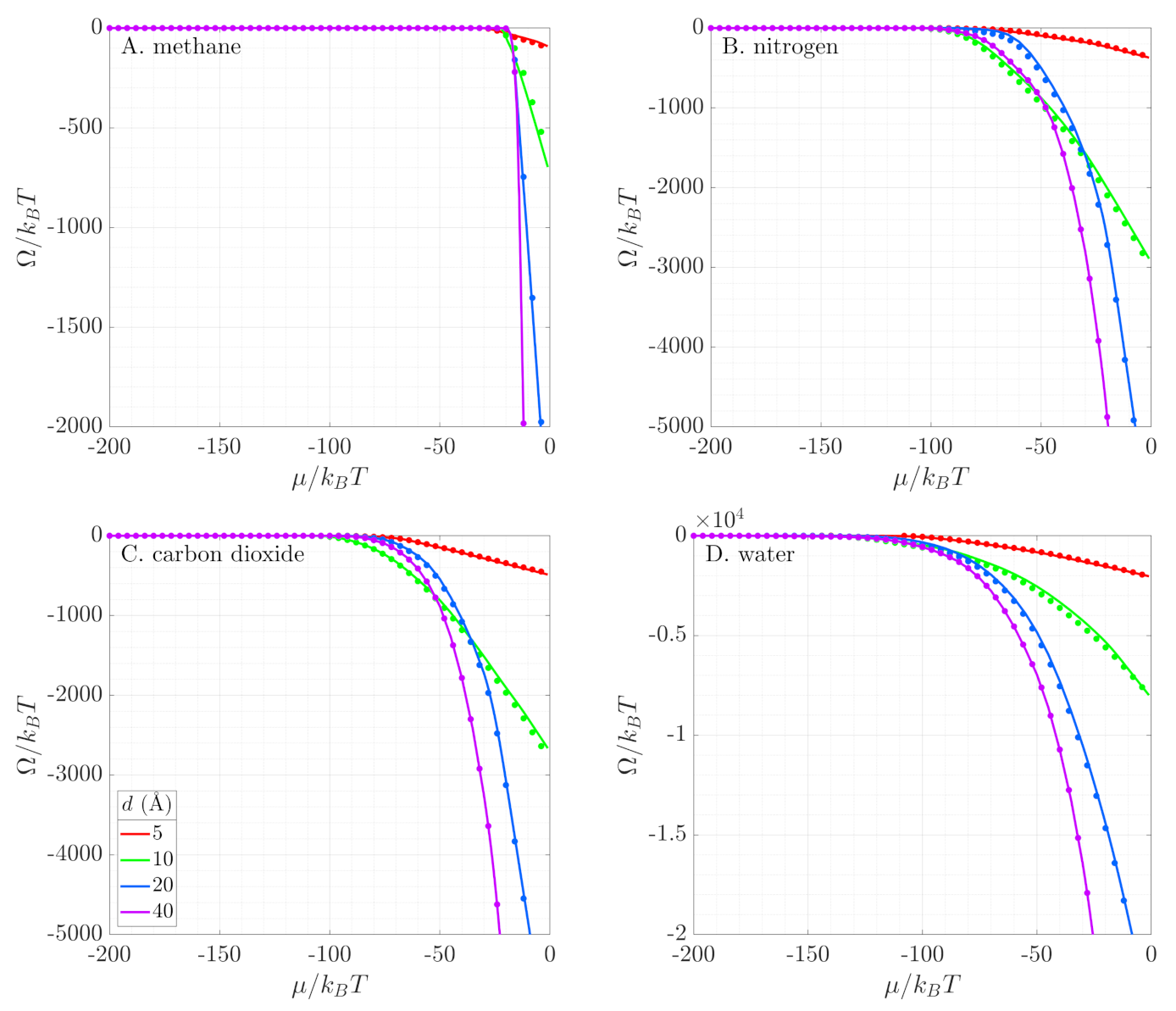
3.2. Grand Potential
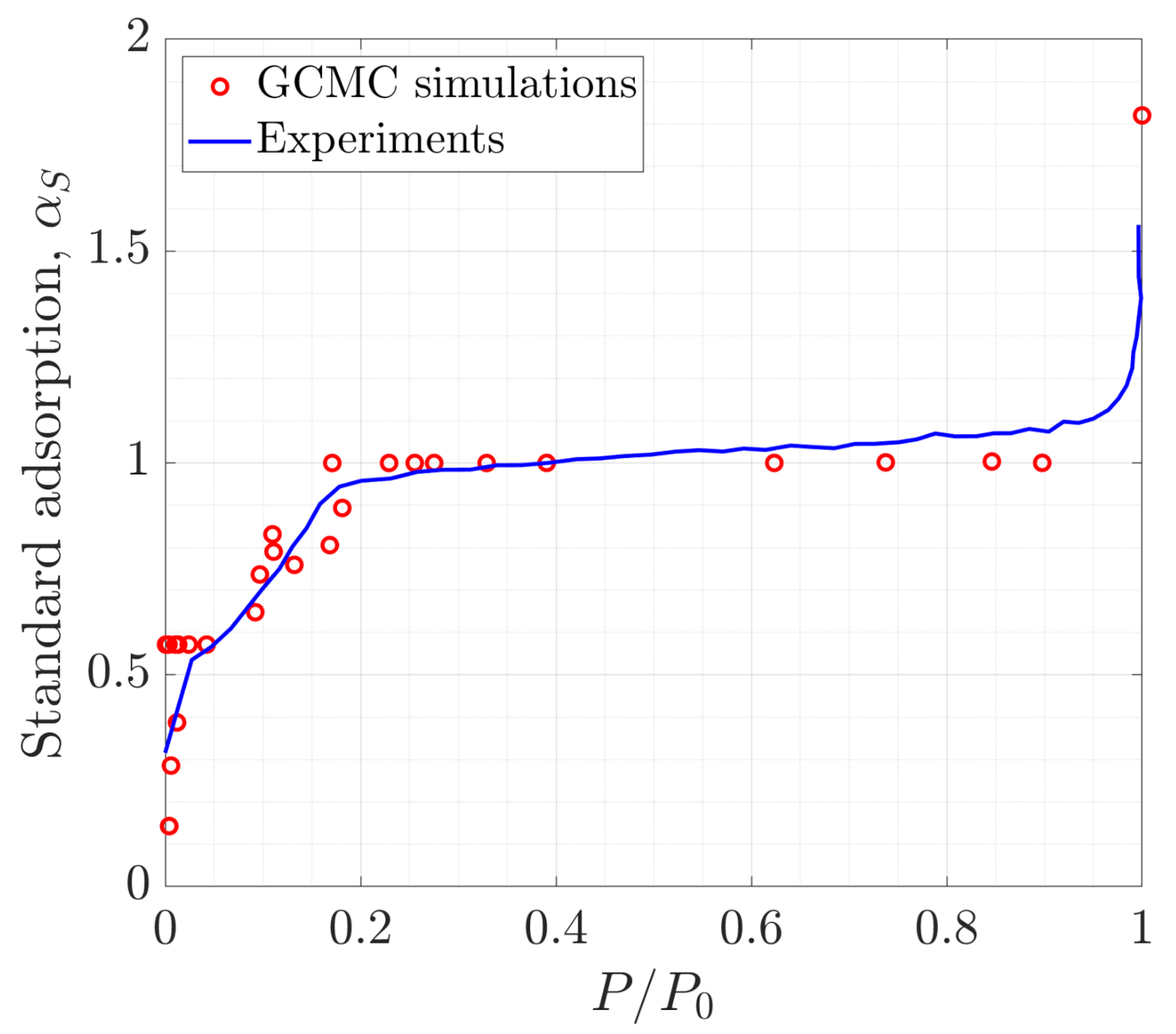
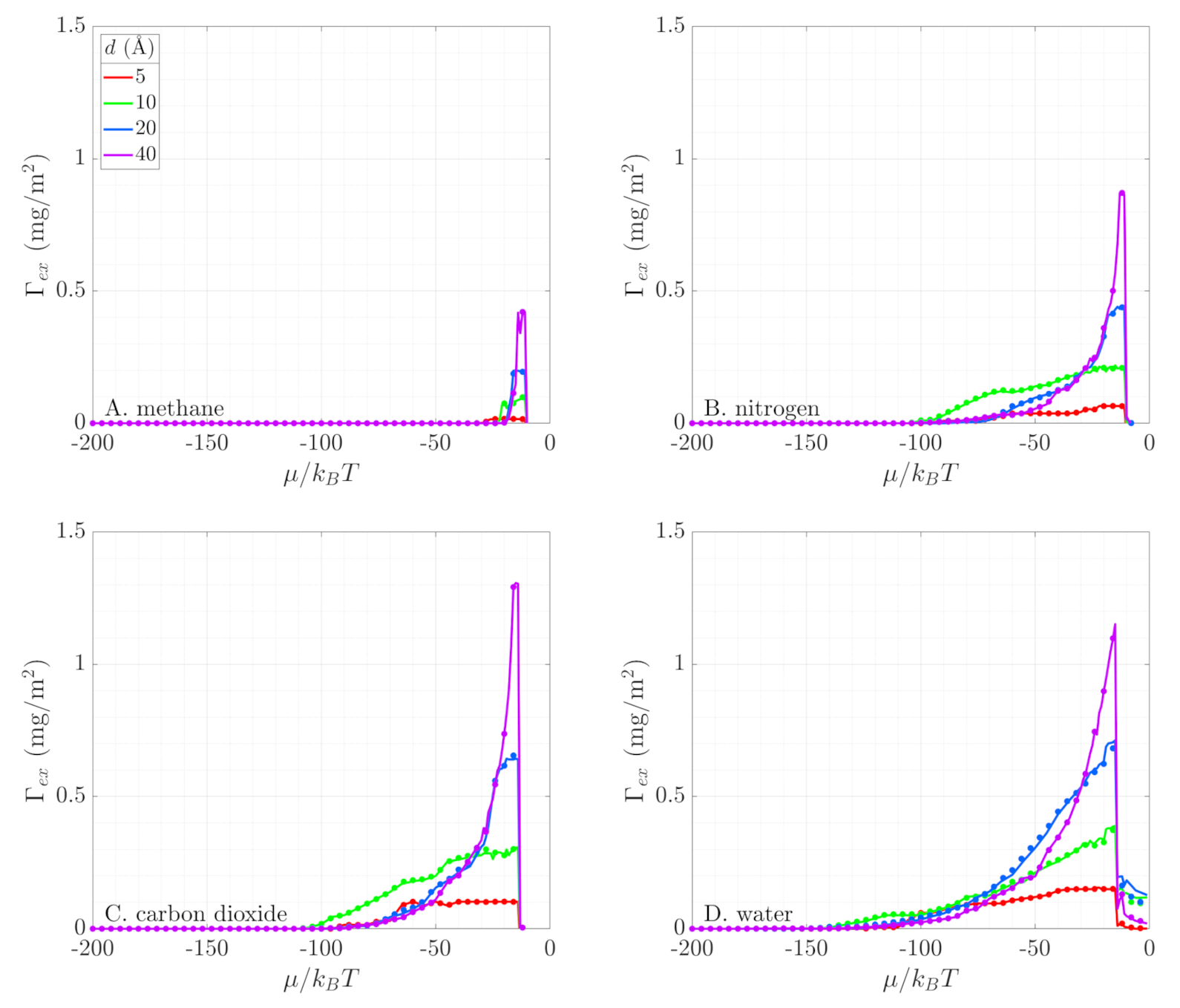
3.3. Excess Adsorption
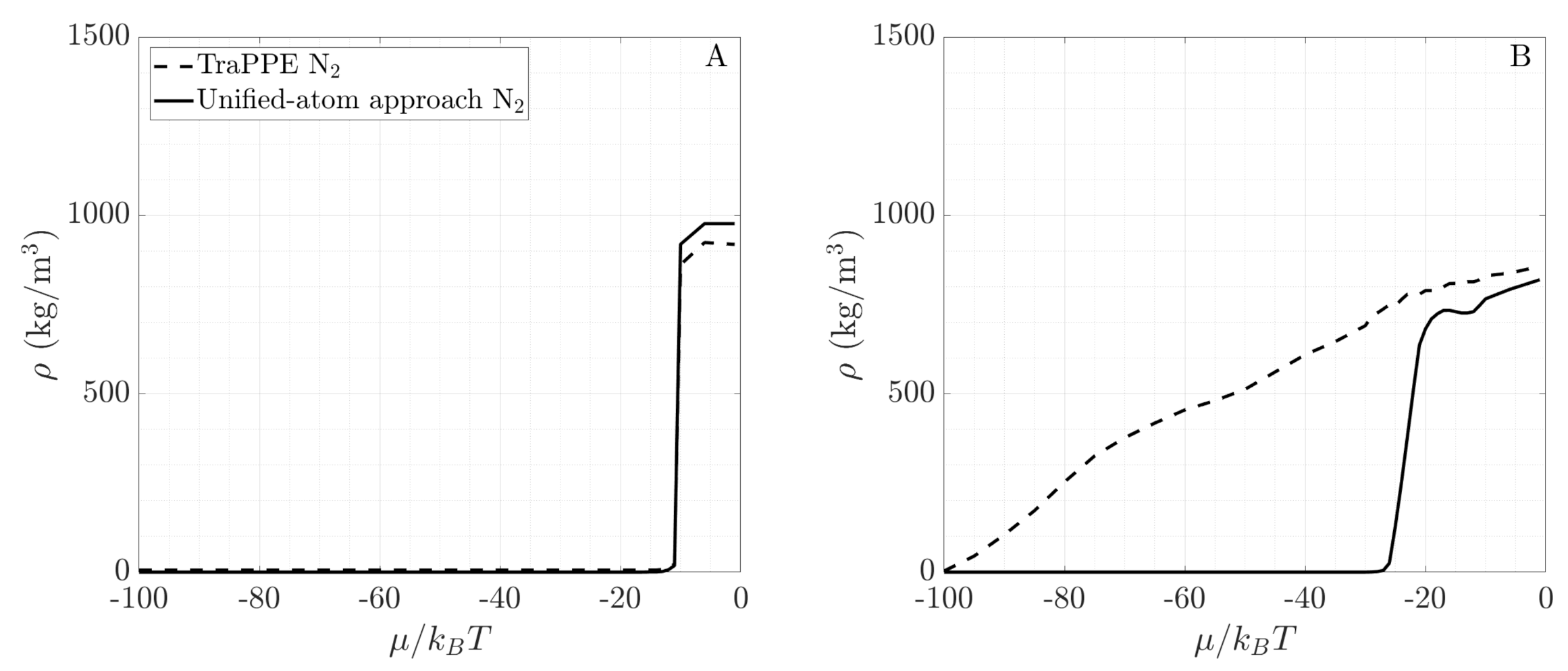
3.4. Toward Real Systems
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Barsotti, E.; Tan, S.P.; Saraji, S.; Piri, M.; Chen, J.H. A review on capillary condensation in nanoporous media: Implications for hydrocarbon recovery from tight reservoirs. Fuel 2016, 184, 344–361. [Google Scholar] [CrossRef]
- Dewers, T.; Eichhubl, P.; Ganis, B.; Gomez, S.; Heath, J.; Jammoul, M.; Kobos, P.; Liu, R.; Major, J.; Matteo, E.; et al. Heterogeneity, pore pressure, and injectate chemistry: Control measures for geologic carbon storage. Int. J. Greenh. Gas Control 2018, 68, 203–215. [Google Scholar] [CrossRef]
- Grotberg, J.B. Respiratory fluid mechanics. Phys. Fluids 2011, 23, 021301. [Google Scholar] [CrossRef] [Green Version]
- David, R.O.; Marcolli, C.; Fahrni, J.; Qiu, Y.; Perez Sirkin, Y.A.; Molinero, V.; Mahrt, F.; Brühwiler, D.; Lohmann, U.; Kanji, Z.A. Pore condensation and freezing is responsible for ice formation below water saturation for porous particles. Proc. Natl. Acad. Sci. USA 2019, 116, 8184–8189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scholes, C.A.; Stevens, G.W.; Kentish, S.E. Membrane gas separation applications in natural gas processing. Fuel 2012, 96, 15–28. [Google Scholar] [CrossRef]
- Nosonovsky, M.; Bhushan, B. Phase behavior of capillary bridges: Towards nanoscale water phase diagram. Phys. Chem. Chem. Phys. 2008, 10, 2137–2144. [Google Scholar] [CrossRef]
- Cai, Y.; Wang, H.E.; Huang, S.Z.; Yuen, M.F.; Cai, H.H.; Wang, C.; Yu, Y.; Li, Y.; Zhang, W.J.; Su, B.L. Porous TiO2 urchins for high performance Li-ion battery electrode: Facile synthesis, characterization and structural evolution. Electrochim. Acta 2016, 210, 206–214. [Google Scholar] [CrossRef]
- Hansen, J.P.; McDonald, I.R. Chapter 6—Inhomogeneous Fluids, 3rd ed.; Academic Press: Burlington, NJ, USA, 2006; pp. 147–177. [Google Scholar] [CrossRef]
- Chen, J.H.; Mehmani, A.; Li, B.; Georgi, D.; Jin, G. Estimation of Total Hydrocarbon in the Presence of Capillary Condensation for Unconventional Shale Reservoirs. In Proceedings of the SPE Middle East Oil and Gas Show and Conference, Manama, Bahrain, 10–13 March 2013. [Google Scholar] [CrossRef]
- Yang, G.; Chai, D.; Fan, Z.; Li, X. Capillary Condensation of Single- and Multicomponent Fluids in Nanopores. Ind. Eng. Chem. Res. 2019, 58, 19302–19315. [Google Scholar] [CrossRef]
- Coasne, B.; Galarneau, A.; Pellenq, R.J.M.; Di Renzo, F. Adsorption, intrusion and freezing in porous silica: The view from the nanoscale. Chem. Soc. Rev. 2013, 42, 4141–4171. [Google Scholar] [CrossRef]
- Burgess, C.G.V.; Everett, D.H.; Nuttall, S. Adsorption hysteresis in porous materials. Pure Appl. Chem. 1989, 61, 1845–1852. [Google Scholar] [CrossRef] [Green Version]
- Desgranges, C.; Delhommelle, J. Nucleation of Capillary Bridges and Bubbles in Nanoconfined CO2. Langmuir 2019, 35, 15401–15409. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Mehta, A. Corresponding state behaviour of capillary condensation of confined alkanes. Mol. Simul. 2019, 45, 1014–1028. [Google Scholar] [CrossRef]
- Aljamaan, H.; Al Ismail, M.; Kovscek, A.R. Experimental investigation and Grand Canonical Monte Carlo simulation of gas shale adsorption from the macro to the nano scale. J. Nat. Gas Sci. Eng. 2017, 48, 119–137. [Google Scholar] [CrossRef]
- Matamoros-Veloza, A.; Newton, R.J.; Benning, L.G. What controls selenium release during shale weathering? Appl. Geochem. 2011, 26, S222–S226. [Google Scholar] [CrossRef]
- Morishige, K.; Fujii, H.; Uga, M.; Kinukawa, D. Capillary Critical Point of Argon, Nitrogen, Oxygen, Ethylene, and Carbon Dioxide in MCM-41. Langmuir 1997, 13, 3494–3498. [Google Scholar] [CrossRef]
- Morishige, K.; Ito, M. Capillary condensation of nitrogen in MCM-41 and SBA-15. J. Chem. Phys. 2002, 117, 8036–8041. [Google Scholar] [CrossRef]
- Wang, H.; Qu, Z.; Yin, Y.; Bai, J.; Yu, B. Review of Molecular Simulation Method for Gas Adsorption/desorption and Diffusion in Shale Matrix. J. Therm. Sci. 2019, 28, 1–16. [Google Scholar] [CrossRef]
- Sun, H.; Sun, W.; Zhao, H.; Sun, Y.; Zhang, D.; Qi, X.; Li, Y. Adsorption properties of CH4 and CO2 in quartz nanopores studied by molecular simulation. RSC Adv. 2016, 6, 32770–32778. [Google Scholar] [CrossRef]
- Zhao, J.; Wang, Z.; Guo, P.; Luo, Q. Molecular level investigation of methane and carbon dioxide adsorption on SiO2 surface. Comput. Mater. Sci. 2019, 168, 2130220. [Google Scholar] [CrossRef]
- Fisher, L.R.; Gamble, R.A.; Middlehurst, J. The Kelvin equation and the capillary condensation of water. Nature 1981, 290, 575–576. [Google Scholar] [CrossRef]
- Puibasset, J.; Pellenq, R.J.M. Water adsorption on hydrophilic mesoporous and plane silica substrates: A grand canonical Monte Carlo simulation study. J. Chem. Phys. 2003, 118, 5613–5622. [Google Scholar] [CrossRef]
- Bonnaud, P.A.; Coasne, B.; Pellenq, R.J.M. Molecular simulation of water confined in nanoporous silica. J. Phys. Condens. Matter 2010, 22, 284110. [Google Scholar] [CrossRef] [PubMed]
- Boelens, A.M.P.; Tchelepi, H.A. Minkowski Functionals for Phase Behavior under Confinement. arXiv 2020, arXiv:2004.01344. [Google Scholar]
- Raju, M.; van Duin, A.; Ihme, M. Phase transitions of ordered ice in graphene nanocapillaries and carbon nanotubes. Sci. Rep. 2018, 8, 2045–2322. [Google Scholar] [CrossRef]
- Kim, S.; Kim, D.; Kim, J.; An, S.; Jhe, W. Direct Evidence for Curvature-Dependent Surface Tension in Capillary Condensation: Kelvin Equation at Molecular Scale. Phys. Rev. X 2018, 8, 041046. [Google Scholar] [CrossRef] [Green Version]
- Jin, Z.; Firoozabadi, A. Effect of water on methane and carbon dioxide sorption in clay minerals by Monte Carlo simulations. Fluid Phase Equilibria 2014, 382, 10–20. [Google Scholar] [CrossRef]
- Mecke, K.R. Additivity, Convexity, and Beyond: Applications of Minkowski Functionals in Statistical Physics. In Statistical Physics and Spatial Statistics; Mecke, K.R., Stoyan, D., Eds.; Springer: Berlin/Heidelberg, Germany, 2000; pp. 111–184. [Google Scholar]
- Gommes, C.J. Stochastic models of disordered mesoporous materials for small-angle scattering analysis and more. Microporous Mesoporous Mater. 2018, 257, 62–78. [Google Scholar] [CrossRef]
- Gommes, C.J.; Roberts, A.P. Stochastic analysis of capillary condensation in disordered mesopores. Phys. Chem. Chem. Phys. 2018, 20, 13646–13659. [Google Scholar] [CrossRef] [Green Version]
- Spagnolo, B.; Valenti, D.; Guarcello, C.; Carollo, A.; Persano Adorno, D.; Spezia, S.; Pizzolato, N.; Di Paola, B. Noise-induced effects in nonlinear relaxation of condensed matter systems. Chaos Solitons Fractals 2015, 81, 412–424. [Google Scholar] [CrossRef]
- Schneider, R. Convex Bodies: The Brunn–Minkowski Theory; Encyclopedia of Mathematics and Its Applications; Cambridge University Press: Cambridge, UK, 2014. [Google Scholar]
- Mecke, K.; Arns, C.H. Fluids in porous media: A morphometric approach. J. Phys. Condens. Matter 2005, 17, S503–S534. [Google Scholar] [CrossRef]
- Goodstein, D.L. States of Matter; Prentice-Hall: Upper Saddle River, NJ, USA, 1975. [Google Scholar]
- König, P.M.; Roth, R.; Mecke, K.R. Morphological Thermodynamics of Fluids: Shape Dependence of Free Energies. Physial Rev. Lett. 2004, 93, 160601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puibasset, J. Grand Potential, Helmholtz Free Energy, and Entropy Calculation in Heterogeneous Cylindrical Pores by the Grand Canonical Monte Carlo Simulation Method. J. Phys. Chem. B 2005, 109, 480–487. [Google Scholar] [CrossRef] [PubMed]
- Plimpton, S. Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef] [Green Version]
- LAMMPS Documentation. 2020. Available online: https://lammps.sandia.gov/ (accessed on 5 October 2020).
- Sing, K. The use of nitrogen adsorption for the characterisation of porous materials. Colloids Surf. A Physicochem. Eng. Asp. 2001, 187–188, 3–9. [Google Scholar] [CrossRef]
- Martin, M.G.; Siepmann, J.I. Transferable Potentials for Phase Equilibria. 1. United-Atom Description of n-Alkanes. J. Phys. Chem. B 1998, 102, 2569–2577. [Google Scholar] [CrossRef]
- Potoff, J.J.; Siepmann, J.I. Vapor–liquid equilibria of mixtures containing alkanes, carbon dioxide, and nitrogen. AIChE J. 2001, 47, 1676–1682. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Grigera, J.R.; Straatsma, T.P. The missing term in effective pair potentials. J. Phys. Chem. 1987, 91, 6269–6271. [Google Scholar] [CrossRef]
- Cygan, R.T.; Liang, J.J.; Kalinichev, A.G. Molecular models of hydroxide, oxyhydroxide, and clay phases and the development of a general force field. J. Phys. Chem. B 2004, 108, 1255–1266. [Google Scholar] [CrossRef]
- Bourg, I.C.; Steefel, C.I. Molecular dynamics simulations of water structure and diffusion in silica nanopores. J. Phys. Chem. C 2012, 116, 11556–11564. [Google Scholar] [CrossRef] [Green Version]
- Bui, T.; Phan, A.; Cole, D.R.; Striolo, A. Transport Mechanism of Guest Methane in Water-Filled Nanopores. J. Phys. Chem. C 2017, 121, 15675–15686. [Google Scholar] [CrossRef] [Green Version]
- Fang, H.; Kulkarni, A.; Kamakoti, P.; Awati, R.; Ravikovitch, P.I.; Sholl, D.S. Identification of high-CO2-capacity cationic zeolites by accurate computational screening. Chem. Mater. 2016, 28, 3887–3896. [Google Scholar] [CrossRef]
- Javanbakht, G.; Sedghi, M.; Welch, W.; Goual, L. Molecular Dynamics Simulations of CO2/Water/Quartz Interfacial Properties: Impact of CO2 Dissolution in Water. Langmuir 2015, 31, 5812–5819. [Google Scholar] [CrossRef] [PubMed]
- Buckingham, A.; Disch, R. The quadrupole moment of the carbon dioxide molecule. In Optical, Electric and Magnetic Properties of Molecules; Clary, D.C., Orr, B.J., Eds.; Elsevier Science B.V.: Amsterdam, The Netherlands, 1997; pp. 81–95. [Google Scholar] [CrossRef]
- Huot, J.; Bose, T.K. Determination of the quadrupole moment of nitrogen from the dielectric second virial coefficient. J. Chem. Phys. 1991, 94, 3849–3854. [Google Scholar] [CrossRef]
- Niu, S.; Tan, M.L.; Ichiye, T. The large quadrupole of water molecules. J. Chem. Phys. 2011, 134, 134501. [Google Scholar] [CrossRef] [Green Version]
- Peterson, B.K.; Gubbins, K.E. Phase transitions in a cylindrical pore. Mol. Phys. 1987, 62, 215–226. [Google Scholar] [CrossRef]
- Shampine, L.F.; Reichelt, M.W. The MATLAB ODE Suite. SIAM J. Sci. Comput. 1997, 18, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Pini, R. Interpretation of net and excess adsorption isotherms in microporous adsorbents. Microporous Mesoporous Mater. 2014, 187, 40–52. [Google Scholar] [CrossRef]
- Hackett, C.; Hammond, K.D. Simulating the effect of the quadrupole moment on the adsorption of nitrogen in siliceous zeolites. Microporous Mesoporous Mater. 2018, 263, 231–235. [Google Scholar] [CrossRef]
- Qajar, A.; Daigle, H.; Prodanović, M. The effects of pore geometry on adsorption equilibrium in shale formations and coal-beds: Lattice density functional theory study. Fuel 2016, 163, 205–213. [Google Scholar] [CrossRef]
- Miele, F.; de Anna, P.; Dentz, M. Stochastic model for filtration by porous materials. Phys. Rev. Fluids 2019, 4, 094101. [Google Scholar] [CrossRef]
- Chiang, W.S.; Fratini, E.; Baglioni, P.; Georgi, D.; Chen, J.H.; Liu, Y. Methane Adsorption in Model Mesoporous Material, SBA-15, Studied by Small-Angle Neutron Scattering. J. Phys. Chem. C 2016, 120, 4354–4363. [Google Scholar] [CrossRef]
- Dantas, S.; Struckhoff, K.C.; Thommes, M.; Neimark, A.V. Phase Behavior and Capillary Condensation Hysteresis of Carbon Dioxide in Mesopores. Langmuir 2019, 35, 11291–11298. [Google Scholar] [CrossRef] [PubMed]
- Frenkel, D.; Smit, B. Understanding Molecular Simulation: From Algorithms to Applications; Computational Science, Elsevier Science: Amsterdam, The Netherlands, 2001. [Google Scholar]
- Tanaka, H.; Hiratsuka, T.; Nishiyama, N.; Mori, K.; Miyahara, M.T. Capillary condensation in mesoporous silica with surface roughness. Adsorption 2013, 19, 631–641. [Google Scholar] [CrossRef]
- Tan, S.P.; Piri, M. Equation-of-state modeling of confined-fluid phase equilibria in nanopores. Fluid Phase Equilibria 2015, 393, 48–63. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule | Particle Type | (K) | (Å) | q (e) | (kcal/mol) | (kcal/mol) |
|---|---|---|---|---|---|---|
| CH4 | CH4 | 148 | 3.730 | - | - | - |
| CO2 | C | 27 | 2.800 | 0.7000 | 5000.0 | 500.0 |
| O | 79 | 3.050 | −0.3500 | |||
| H2O | H | 0.0 | 0.000 | 0.4236 | 1000.0 | 100.0 |
| O | 78.2 | 3.166 | −0.8472 | |||
| N2 | N | 36 | 3.310 | −0.4820 | 5000.0 | 500.0 |
| Ghost | 0.0 | 0.000 | 0.9640 | |||
| SiO2 | Si | 3.302 | 2.1000 | - | - | |
| O | 78.2 | 3.166 | −1.0500 | - | - |
| Molecule | Force Field | Electrostatic Dipole (D) | Electrostatic Quadrupole (DÅ) | Reference |
|---|---|---|---|---|
| CH4 | TraPPE-UA | 0.0 | 0.0 | - |
| N2 | TraPPE | 0.0 | −1.47 | [49] |
| CO2 | TraPPE | 0.0 | −4.1 | [50] |
| H2O | SPC/E | 1.86 | 0.11 | [51] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simeski, F.; Boelens, A.M.P.; Ihme, M. Modeling Adsorption in Silica Pores via Minkowski Functionals and Molecular Electrostatic Moments. Energies 2020, 13, 5976. https://doi.org/10.3390/en13225976
Simeski F, Boelens AMP, Ihme M. Modeling Adsorption in Silica Pores via Minkowski Functionals and Molecular Electrostatic Moments. Energies. 2020; 13(22):5976. https://doi.org/10.3390/en13225976
Chicago/Turabian StyleSimeski, Filip, Arnout M. P. Boelens, and Matthias Ihme. 2020. "Modeling Adsorption in Silica Pores via Minkowski Functionals and Molecular Electrostatic Moments" Energies 13, no. 22: 5976. https://doi.org/10.3390/en13225976
APA StyleSimeski, F., Boelens, A. M. P., & Ihme, M. (2020). Modeling Adsorption in Silica Pores via Minkowski Functionals and Molecular Electrostatic Moments. Energies, 13(22), 5976. https://doi.org/10.3390/en13225976