Effect of Salt Concentration, Solvent Donor Number and Coordination Structure on the Variation of the Li/Li+ Potential in Aprotic Electrolytes


Abstract
:1. Introduction
2. Materials and Methods
3. Results
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yamada, Y.; Furukawa, K.; Sodeyama, K.; Kikuchi, K.; Yaegashi, M.; Tateyama, Y.; Yamada, A. Unusual stability of acetonitrile-based superconcentrated electrolytes for fast-charging lithium-ion batteries. J. Am. Chem. Soc. 2014, 136, 5039–5046. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Yamada, A. Review—Superconcentrated Electrolytes for Lithium Batteries. J. Electrochem. Soc. 2015, 162, A2406–A2423. [Google Scholar] [CrossRef]
- Reddy, K.P.K.; Fischer, P.; Marinaro, M.; Wohlfahrt-Mehrens, M. Improved Li-Metal Cycling Performance in High Concentrated Electrolytes for Li-O 2 Batteries. ChemElectroChem 2018, 5, 1–10. [Google Scholar]
- Kottam, P.K.R.; Kalkan, D.; Wohlfahrt-Mehrens, M.; Marinaro, M. Influence of Li-Salt Concentration on Redox Potential of Lithium Metal and Electrochemistry of Ferrocene in DMSO-Based Electrolytes. J. Electrochem. Soc. 2019, 166, 1574–1579. [Google Scholar] [CrossRef]
- Ernould, B.; Sieuw, L.; Barozzino, G.; Gohy, J.; Vlad, A.; Ernould, B.; Sieuw, L.; Barozzino-consiglio, G.; Gohy, J.; Vlad, A. Negative Redox Potential Shift in Fire-Retardant Electrolytes and Consequences for High-Energy Hybrid Batteries. ACS Appl. Energy Mater. 2019, 2, 11. [Google Scholar] [CrossRef]
- Mozhzhukhina, N.; Longinotti, M.P.; Corti, H.R.; Calvo, E.J. Electrochimica Acta A conductivity study of preferential solvation of lithium ion in acetonitrile-dimethyl sulfoxide mixtures. Electrochim. Acta 2015, 154, 456–461. [Google Scholar] [CrossRef]
- Lutz, L.; Yin, W.; Grimaud, A.; Alves Dalla Corte, D.; Tang, M.; Johnson, L.; Azaceta, E.; Sarou-Kanian, V.; Naylor, A.J.; Hamad, S.; et al. High capacity Na-O2 batteries: Key parameters for solution-mediated discharge. J. Phys. Chem. C 2016, 120, 20068–20076. [Google Scholar] [CrossRef]
- Cataldo, F. A revision of the gutmann donor numbers of a series of phosphoramides including tepa. Eur. Chem. Bull. 2015, 4, 92–97. [Google Scholar]
- Bondue, C.J.; Hegemann, M.; Molls, C.; Thome, E.; Baltruschat, H. A Comprehensive Study on Oxygen Reduction and Evolution from Lithium Containing DMSO Based Electrolytes at Gold Electrodes. J. Electrochem. Soc. 2016, 163, A1765–A1775. [Google Scholar] [CrossRef]
- Bondue, C.J.; Reinsberg, P.; Abd-El-Latif, A.A.; Baltruschat, H. Oxygen reduction and oxygen evolution in DMSO based electrolytes: The role of the electrocatalyst. Phys. Chem. Chem. Phys. 2015, 17, 25593–25606. [Google Scholar] [CrossRef]
- Marinaro, M.; Riek, U.; Eswara Moorthy, S.K.; Bernhard, J.; Kaiser, U.; Wohlfahrt-Mehrens, M.; Jörissen, L. Au-coated carbon cathodes for improved oxygen reduction and evolution kinetics in aprotic Li-O2batteries. Electrochem. Commun. 2013, 37, 53–56. [Google Scholar] [CrossRef]
- Marinaro, M.; Theil, S.; Jörissen, L.; Wohlfahrt-Mehrens, M. New insights about the stability of lithiumbis(trifluoromethane) sulfonimide-tetraglyme as electrolyte for Li-O2 batteries. Electrochim. Acta 2013, 108, 795–800. [Google Scholar] [CrossRef]
- Balasubramanian, P.; Marinaro, M.; Theil, S.; Wohlfahrt-Mehrens, M.; Jrissen, L. Au-coated carbon electrodes for aprotic Li-O2 batteries with extended cycle life: The key issue of the Li-ion source. J. Power Sources 2015, 278, 156–162. [Google Scholar] [CrossRef]
- Marinaro, M.; Balasubramanian, P.; Gucciardi, E.; Theil, S.; Joerissen, L.; Wohlfahrt-Mehrens, M. Importance of Reaction Kinetics and Oxygen Crossover in aprotic Li-O-2 Batteries Based on a Dimethyl Sulfoxide Electrolyte. ChemSusChem 2015, 8, 3139–3145. [Google Scholar] [CrossRef]
- Jung, H.G.; Hassoun, J.; Park, J.B.; Sun, Y.K.; Scrosati, B. An improved high-performance lithium-air battery. Nat. Chem. 2012, 4, 579–585. [Google Scholar] [CrossRef]
- Peng, Z.; Freunberger, S.A.; Chen, Y.; Bruce, P.G. A reversible and higher-rate Li-O2 battery. Science 2012, 337, 563–566. [Google Scholar] [CrossRef]
- Mozhzhukhina, N.; Calvo, E.J. The Correct Assessment of Standard Potentials of Reference Electrodes in Non-Aqueous Solution. J. Electrochem. Soc. 2017, 164, 2295–2297. [Google Scholar] [CrossRef] [Green Version]
- Ueno, K.; Tatara, R.; Tsuzuki, S.; Saito, S.; Doi, H.; Yoshida, K.; Mandai, T.; Matsugami, M.; Umebayashi, Y.; Dokk, K.; et al. Li+ solvation in glyme–Li salt solvate ionic liquids. Phys. Chem. Chem. Phys. 2015, 17, 8248–8257. [Google Scholar] [CrossRef]
- Brouillette, D.; Irish, D.E.; Taylor, N.J.; Perron, G.; Odziemkowski, M.; Desnoyers, J.E. Stable solvates in solution of lithium bis(trifluoromethylsulfone)imide in glymes and other aprotic solvents: Phase diagrams, crystallography and Raman spectroscopy. Phys. Chem. Chem. Phys. 2002, 4, 6063–6071. [Google Scholar] [CrossRef]
- Tsuzuki, S.; Shinoda, W.; Matsugami, M.; Umebayashi, Y.; Ueno, K.; Mandai, T.; Seki, S.; Dokko, K.; Watanabe, M. interactions with anions in equimolar mixtures of glymes and Li [TFSA]: Analysis by molecular dynamics simulations †. Phys. Chem. Chem. Phys. 2014, 17, 126–129. [Google Scholar] [CrossRef] [PubMed]
- Tatara, R.; Kwabi, D.G.; Batcho, T.P.; Tulodziecki, M.; Watanabe, K.; Kwon, H.M.; Thomas, M.L.; Ueno, K.; Thompson, C.V.; Dokko, K.; et al. Oxygen Reduction Reaction in Highly Concentrated Electrolyte Solutions of Lithium Bis(trifluoromethanesulfonyl)amide/Dimethyl Sulfoxide. J. Phys. Chem. C 2017, 121, 9162–9172. [Google Scholar] [CrossRef]
- Sun, Y.; Hamada, I. Insight into the Solvation Structure of Tetraglyme-Based Electrolytes via First-Principles Molecular Dynamics Simulation. J. Phys. Chem. B 2018, 122, 10014–10022. [Google Scholar] [CrossRef] [PubMed]
- Mandai, T.; Yoshida, K.; Ueno, K. Criteria for solvate ionic liquids †. Phys. Chem. Chem. Phys. 2014, 16, 8761–8772. [Google Scholar] [CrossRef] [PubMed]
- Ueno, K.; Yoshida, K.; Tsuchiya, M.; Tachikawa, N.; Dokko, K.; Watanabe, M. Glyme—Lithium Salt Equimolar Molten Mixtures: Concentrated Solutions or Solvate Ionic Liquids? J. Phys. Chem. B 2012, 116, 11323. [Google Scholar] [CrossRef] [PubMed]
- Semino, R.; Zaldívar, G.; Calvo, E.J.; Laria, D.; Semino, R.; Zaldívar, G.; Calvo, E.J.; Laria, D. Lithium solvation in dimethyl sulfoxide-acetonitrile mixtures Lithium solvation in dimethyl sulfoxide-acetonitrile mixtures. J. Chem. Phys. 2014, 141, 214509. [Google Scholar] [CrossRef] [PubMed]
- Laoire, C.O.; Plichta, E.; Hendrickson, M.; Mukerjee, S.; Abraham, K.M. Electrochimica Acta Electrochemical studies of ferrocene in a lithium ion conducting organic carbonate electrolyte. Electrochim. Acta 2009, 54, 6560–6564. [Google Scholar] [CrossRef]
- Li, Z.; Borodin, O.; Smith, G.D.; Bedrov, D. Effect of Organic Solvents on Li+ Ion Solvation and Transport in Ionic Liquid Electrolytes: A Molecular Dynamics Simulation Study. J. Phys. Chem. B 2015, 119, 3085–3096. [Google Scholar] [CrossRef]
- Borodin, O.; Smith, G.D. Li + Transport Mechanism in Oligo (Ethylene Oxide) s Compared to Carbonates. J. Solut. Chem. 2007, 36, 803–813. [Google Scholar] [CrossRef]




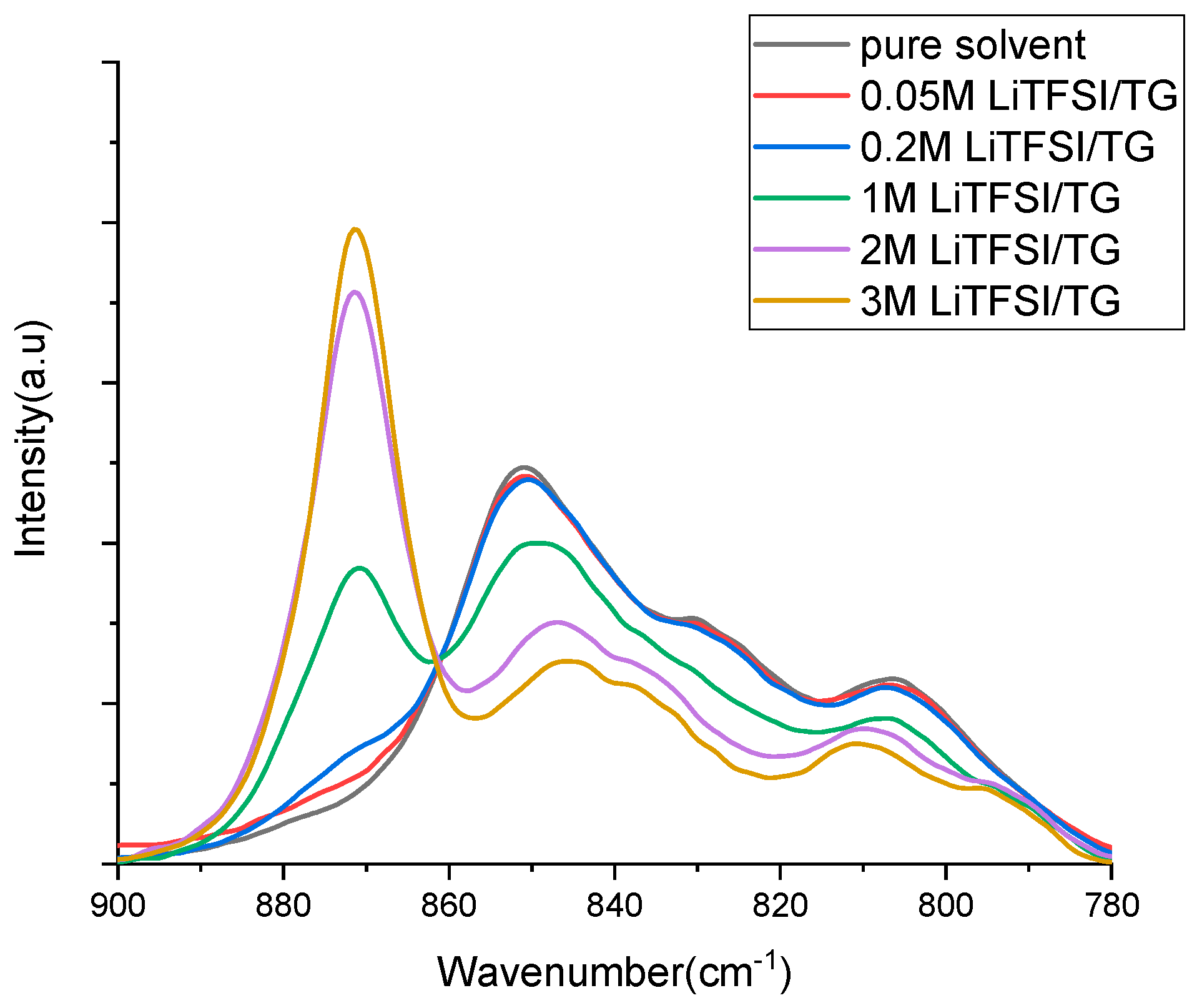{kind=link}
{kind=link}
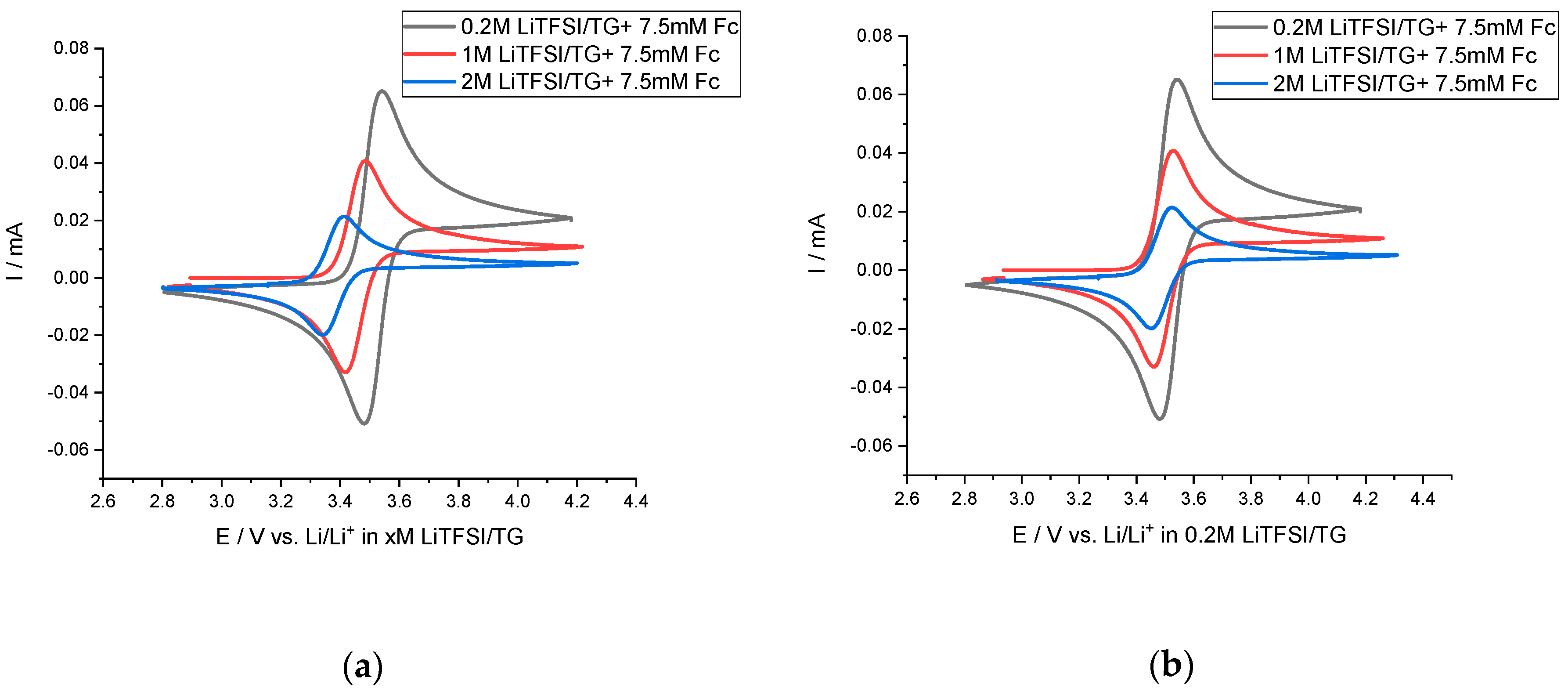{kind=link}
| Solution | n [TG]/[LiTFSI] | ([TG]coor) mol/L | ([TG]free) mol/L | n |
|---|---|---|---|---|
| Free solvent | - | - | 4.53 | - |
| 0.05 M LiTFSI/TG | 87.14 | 0.043 | 4.313 | 0.81 |
| 0.2 M LiTFSI/ TG | 21.15 | 0.152 | 4.077 | 0.76 |
| 1 M LiTFSI/ TG | 3.79 | 0.995 | 2.796 | 0.99 |
| 2 M LiTFSI/ TG | 1.58 | 2.217 | 0.937 | 1.11 |
| 3 M LiTFSI/ TG | 0.83 | 2.502 | - | 0.83 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kottam, P.K.R.; Dongmo, S.; Wohlfahrt-Mehrens, M.; Marinaro, M. Effect of Salt Concentration, Solvent Donor Number and Coordination Structure on the Variation of the Li/Li+ Potential in Aprotic Electrolytes. Energies 2020, 13, 1470. https://doi.org/10.3390/en13061470
Kottam PKR, Dongmo S, Wohlfahrt-Mehrens M, Marinaro M. Effect of Salt Concentration, Solvent Donor Number and Coordination Structure on the Variation of the Li/Li+ Potential in Aprotic Electrolytes. Energies. 2020; 13(6):1470. https://doi.org/10.3390/en13061470
Chicago/Turabian StyleKottam, P. K. R., S. Dongmo, M. Wohlfahrt-Mehrens, and M. Marinaro. 2020. "Effect of Salt Concentration, Solvent Donor Number and Coordination Structure on the Variation of the Li/Li+ Potential in Aprotic Electrolytes" Energies 13, no. 6: 1470. https://doi.org/10.3390/en13061470
APA StyleKottam, P. K. R., Dongmo, S., Wohlfahrt-Mehrens, M., & Marinaro, M. (2020). Effect of Salt Concentration, Solvent Donor Number and Coordination Structure on the Variation of the Li/Li+ Potential in Aprotic Electrolytes. Energies, 13(6), 1470. https://doi.org/10.3390/en13061470