
Ab-Initio Molecular Dynamics Simulation of Condensed-Phase Reactivity: The Electrolysis of Ammonia and Ethanimine in Aquatic Carbon Dioxide Solutions


Abstract
:1. Introduction
2. Methods
3. Results and Discussion
3.1. Ammonia and Carbon Dioxide in Aqueous Solution
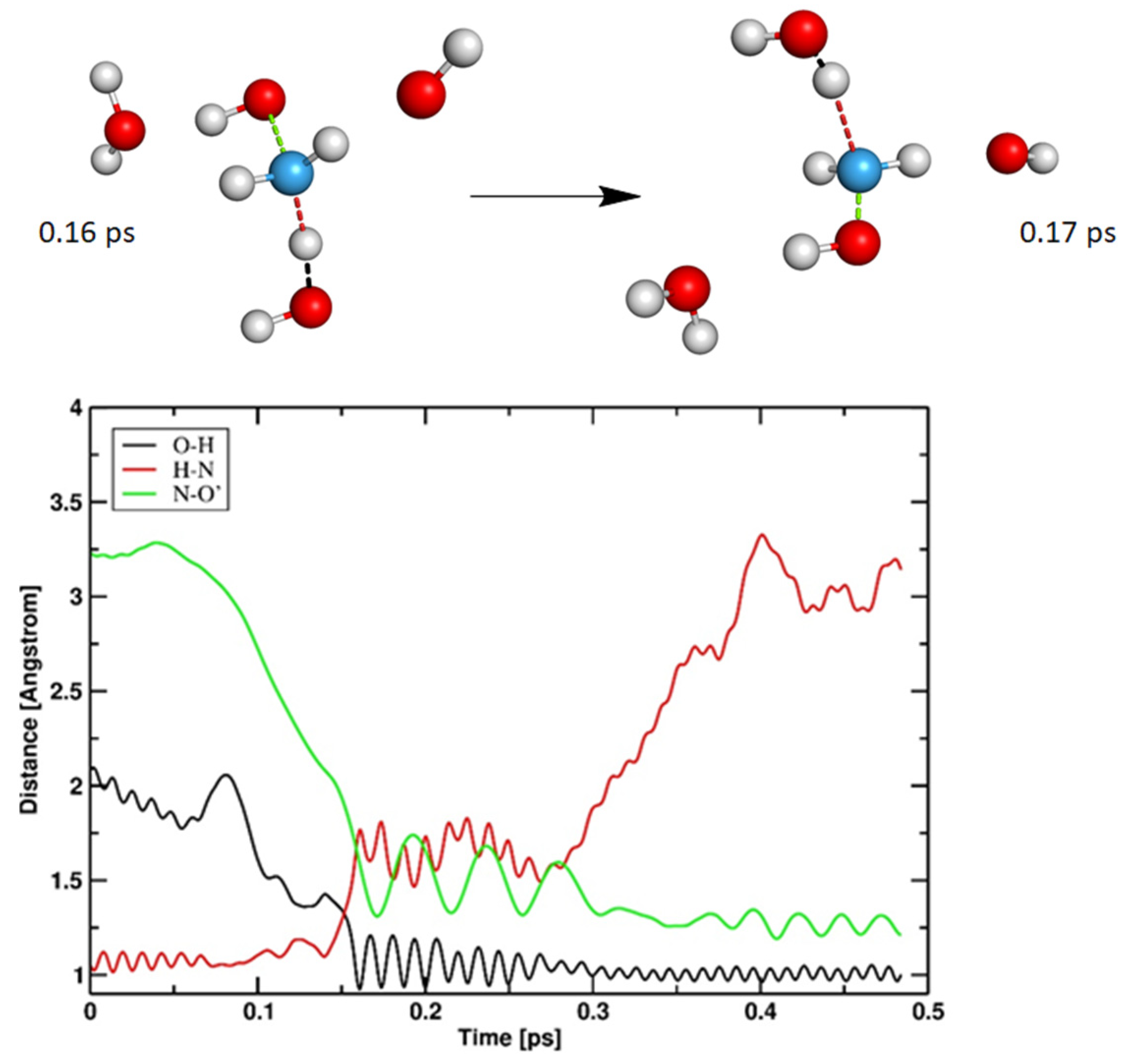
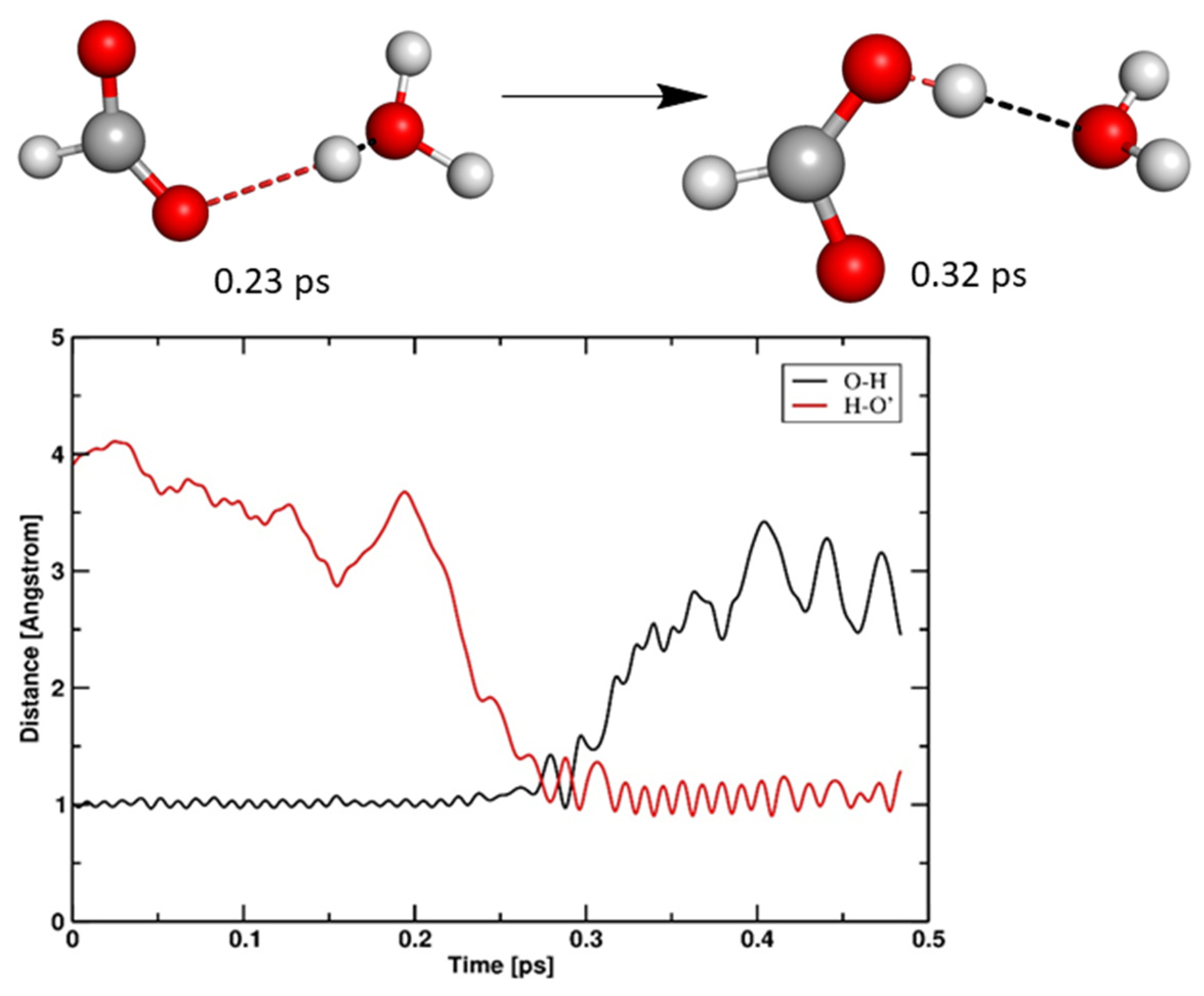
3.1.1. Anodic Reactions
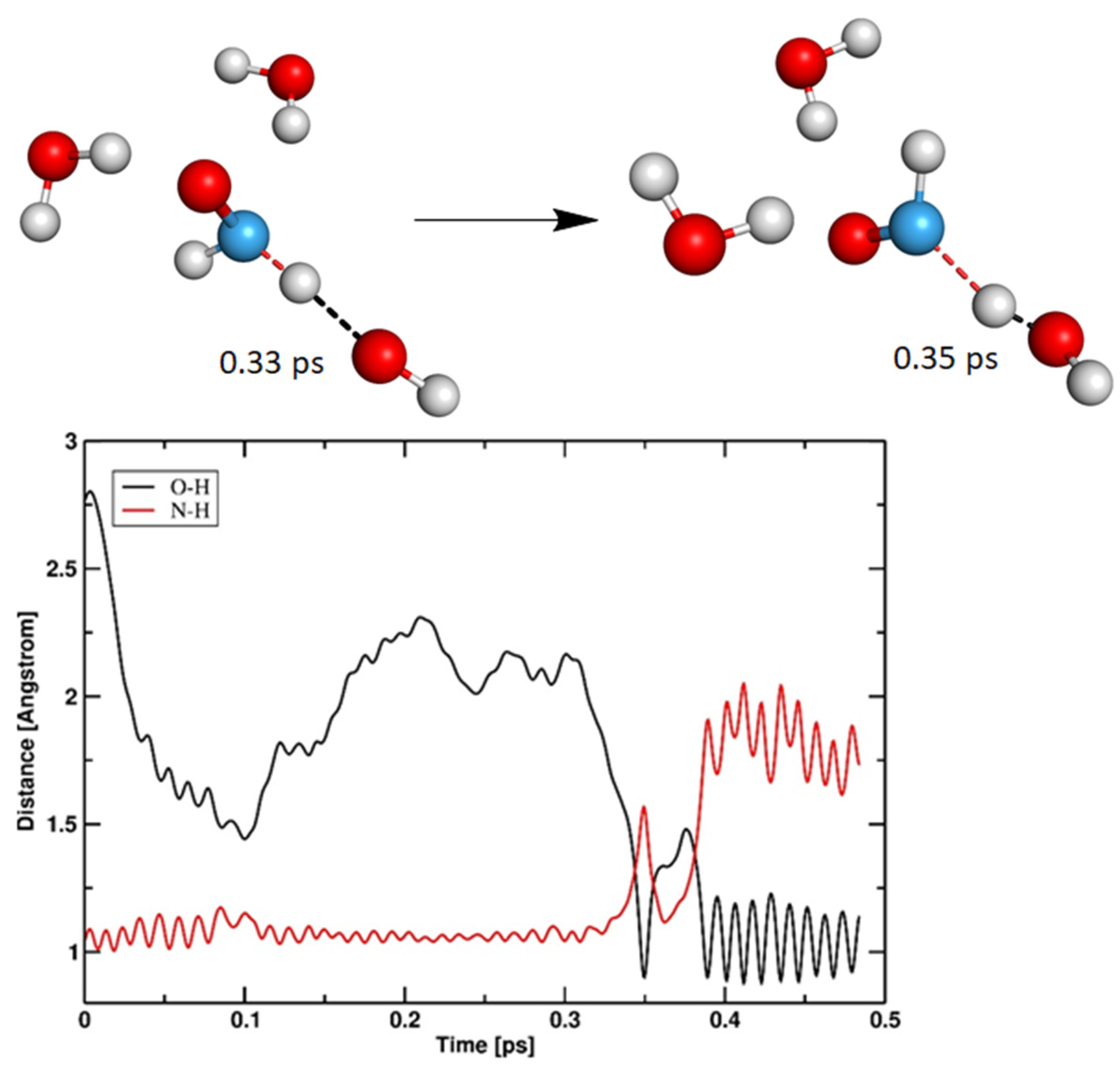
3.1.2. Cathodic Reactions
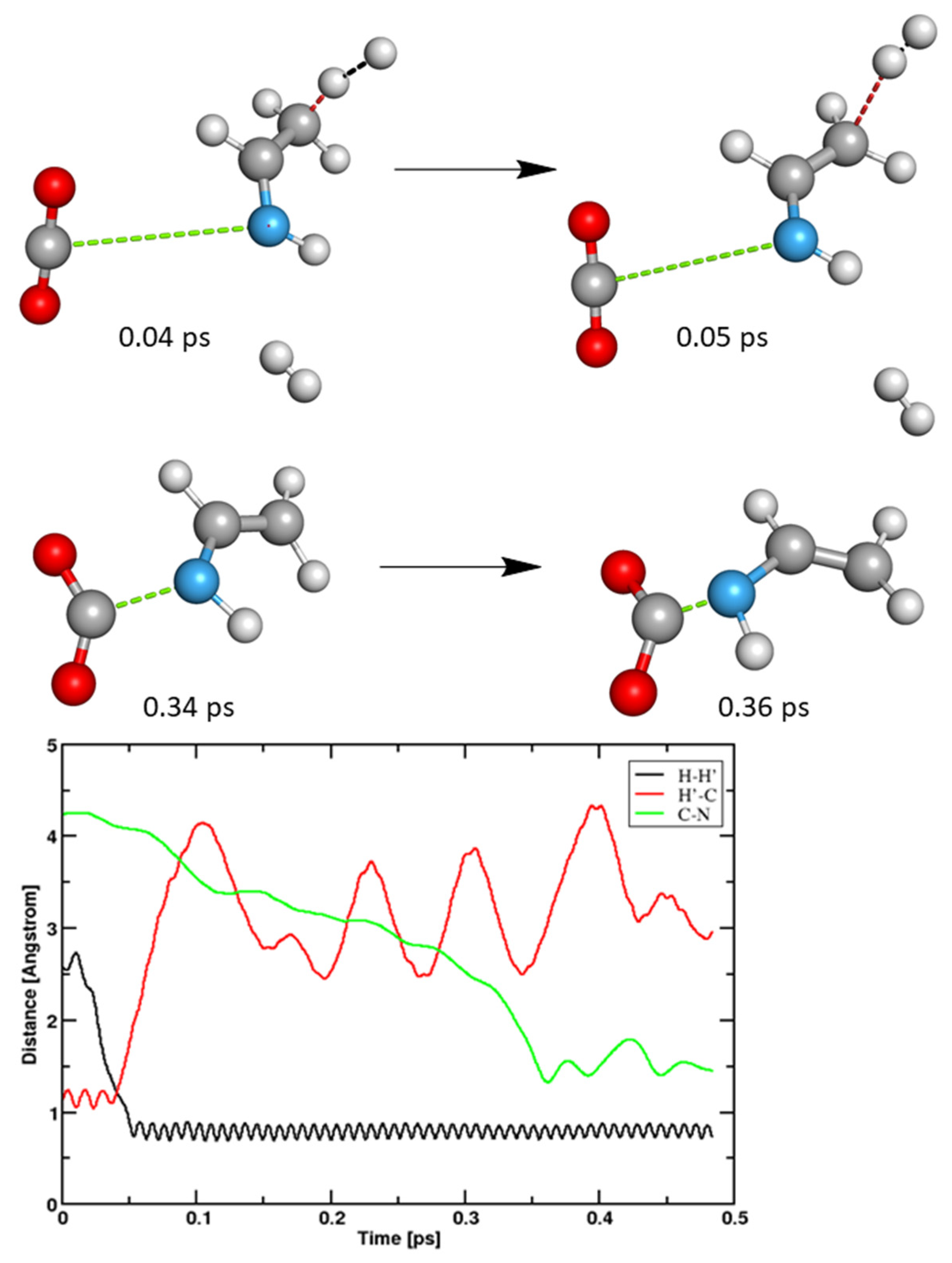
3.2. Ethanimine and Carbon Dioxide in Aqueous Solution
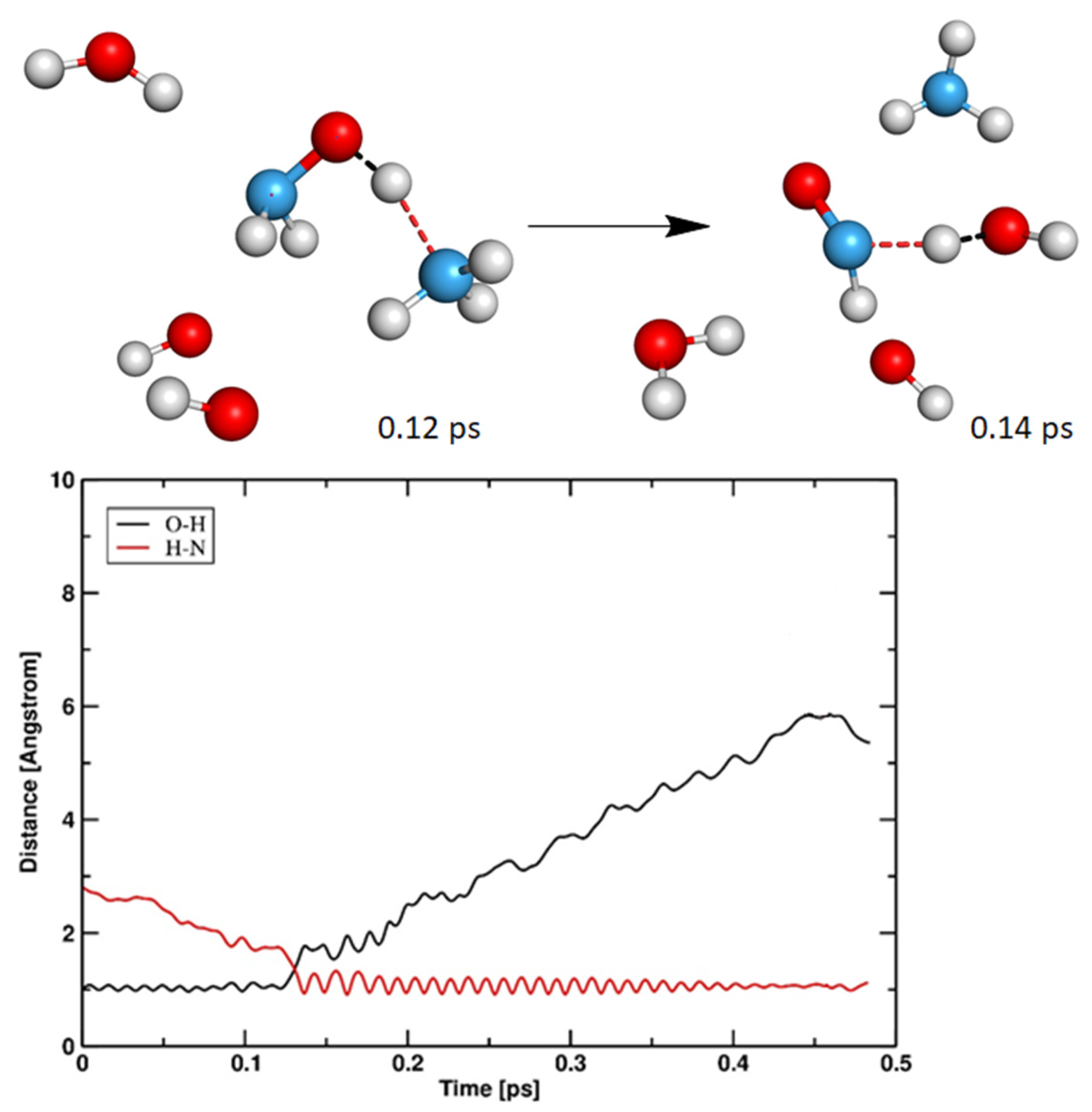
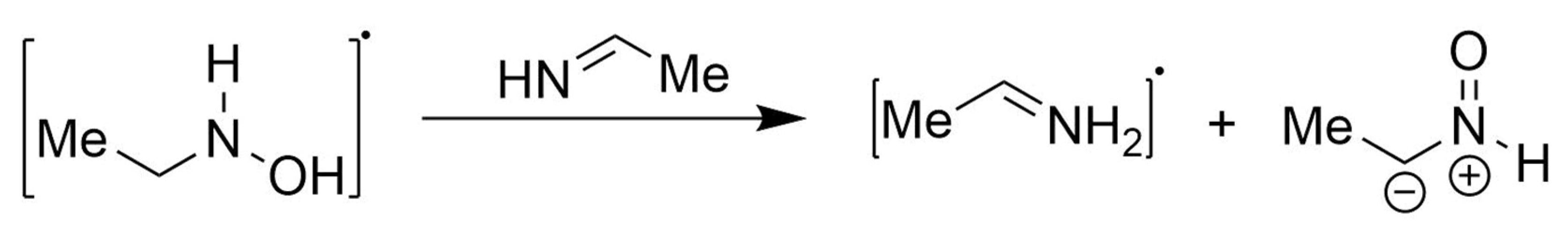
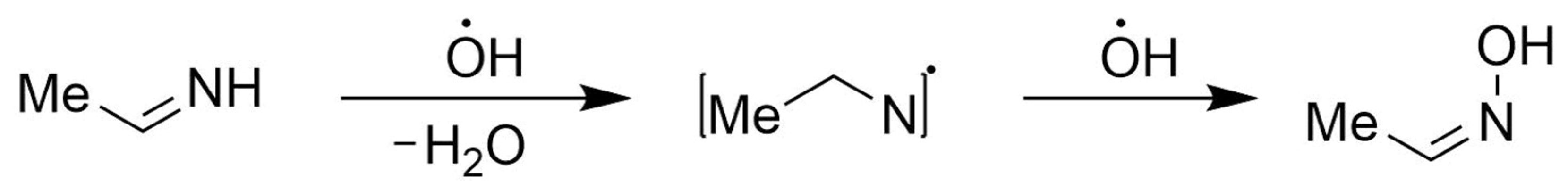
3.2.1. Anodic Reactions
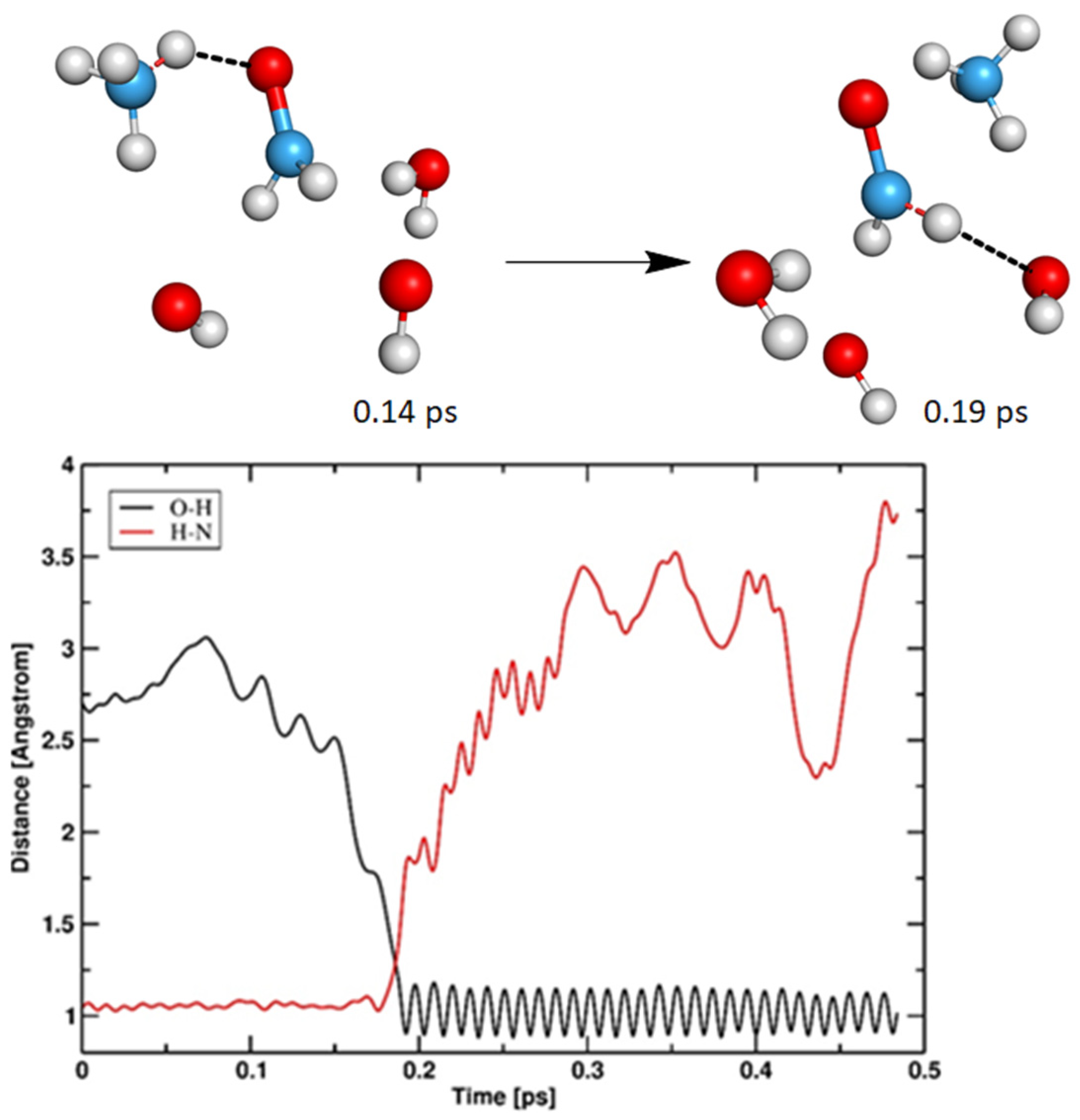
3.2.2. Cathodic Reactions
3.3. Energetics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bismo, S.; Irawan, K.; Karamah, E.F.; Saksono, N. On the Production of OH Radical through Plasma Electrolysis Mechanism for the Processing of Ammonia Waste Water. J. Chem. Chem. Eng. 2013, 7, 6. [Google Scholar]
- Peters, M.; Köhler, B.; Kuckshinrichs, W.; Leitner, W.; Markewitz, P.; Müller, T.E. Chemical technologies for exploiting and recycling carbon dioxide into the value chain. ChemSusChem 2011, 4, 1216–1240. [Google Scholar] [CrossRef]
- Martín, A.J.; Larrazábal, G.O.; Pérez-Ramírez, J. Towards sustainable fuels and chemicals through the electrochemical reduction of CO2: Lessons from water electrolysis. Green Chem. 2015, 17, 5114–5130. [Google Scholar] [CrossRef]
- Sternberg, A.; Bardow, A. Power-to-What?—Environmental assessment of energy storage systems. Energy Environ. Sci. 2015, 8, 389–400. [Google Scholar] [CrossRef]
- Patterson, T.; Savvas, S.; Chong, A.; Law, I.; Dinsdale, R.; Esteves, S. Integration of Power to Methane in a waste water treatment plant–A feasibility study. Bioresour. Technol. 2017, 245, 1049–1057. [Google Scholar] [CrossRef] [PubMed]
- Mebrahtu, C.; Nohl, M.; Dittrich, L.; Foit, S.R.; de Haart, L.B.; Eichel, R.A.; Palkovits, R. Integrated Co-Electrolysis and Syngas Methanation for the Direct Production of Synthetic Natural Gas from CO2 and H2O. ChemSusChem 2021, 14, 2295. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, R.; Wall, D.M.; McDonagh, S.; Murphy, J.D. The potential of power to gas to provide green gas utilising existing CO2 sources from industries, distilleries and wastewater treatment facilities. Renew. Energy 2017, 114, 1090–1100. [Google Scholar] [CrossRef]
- Thema, M.; Bauer, F.; Sterner, M. Power-to-Gas: Electrolysis and methanation status review. Renew. Sustain. Energy Rev. 2019, 112, 775–787. [Google Scholar] [CrossRef]
- Klüh, D.; Waldmüller, W.; Gaderer, M. Kolbe Electrolysis for the Conversion of Carboxylic Acids to Valuable Products—A Process Design Study. Clean Technol. 2021, 3, 1–18. [Google Scholar] [CrossRef]
- Kotowicz, J.; Węcel, D.; Brzęczek, M. Analysis of the work of a “renewable” methanol production installation based ON H2 from electrolysis and CO2 from power plants. Energy 2021, 221, 119538. [Google Scholar] [CrossRef]
- Albert, J.; Jess, A.; Kern, C.; Pöhlmann, F.; Glowienka, K.; Wasserscheid, P. formic acid-based fischer–tropsch synthesis for green fuel production from wet waste biomass and renewable excess energy. ACS Sustain. Chem. Eng. 2016, 4, 5078–5086. [Google Scholar] [CrossRef]
- Kreitz, B.; Brauns, J.; Wehinger, G.D.; Turek, T. Modeling the Dynamic Power-to-Gas Process: Coupling Electrolysis with CO2 Methanation. Chem. Ing. Tech. 2020, 92, 1992–1997. [Google Scholar] [CrossRef]
- Lu, X.; Musin, R.N.; Lin, M.C. Gas-phase reactions of HONO with HNO and NH3: An ab initio MO/TST study. J. Phys. Chem. A 2000, 104, 5141–5148. [Google Scholar] [CrossRef]
- Frank, I. Ab-Initio Molecular Dynamics Simulation of the Electrolysis of Waste Water. ChemistrySelect 2019, 4, 4376–4381. [Google Scholar] [CrossRef]
- Kiakojouri, A.; Nadimi, E.; Frank, I. Ab-Initio Molecular Dynamics Simulation of Condensed-Phase Reactivity: The Electrolysis of Amino Acids and Peptides. Molecules 2020, 25, 5415. [Google Scholar] [CrossRef] [PubMed]
- Oswin, H.G.; Salomon, M. The anodic oxidation of ammonia at platinum black electrodes in aqueous KOH electrolyte. Can. J. Chem. 1963, 41, 1686–1694. [Google Scholar] [CrossRef]
- Despić, A.R.; Dražić, D.M.; Rakin, P.M. Kinetics of electrochemical oxidation of ammonia in alkaline solution. Electrochim. Acta 1966, 11, 997–1005. [Google Scholar] [CrossRef]
- Bunce, N.J.; Bejan, D. Mechanism of electrochemical oxidation of ammonia. Electrochim. Acta 2011, 56, 8085–8093. [Google Scholar] [CrossRef]
- Blumberger, J.; Bernasconi, L.; Tavernelli, I.; Vuilleumier, R.; Sprik, M. Electronic structure and solvation of copper and silver ions: A theoretical picture of a model aqueous redox reaction. J. Am. Chem. Soc. 2004, 126, 3928–3938. [Google Scholar] [CrossRef]
- Zhang, C.; Sayer, T.; Hutter, J.; Sprik, M. Modelling electrochemical systems with finite field molecular dynamics. J. Phys. Energy 2020, 2, 032005. [Google Scholar] [CrossRef]
- Vassilev, P.; Louwerse, M.J.; Baerends, E.J. Ab initio molecular dynamics simulation of the OH radical in liquid water. Chem. Phys. Lett. 2004, 398, 212–216. [Google Scholar] [CrossRef]
- Marx, D.; Hutter, J. Ab Initio Molecular Dynamics: Basic Theory and Advanced Methods; Cambridge University Press: Cambridge, UK, 2009. [Google Scholar]
- Car, R.; Parrinello, M. Unified approach for molecular dynamics and density-functional theory. Phys. Rev. Lett. 1985, 55, 2471. [Google Scholar] [CrossRef] [Green Version]
- Hofbauer, F.; Frank, I. CPMD simulation of a bimolecular chemical reaction: Nucleophilic attack of a disulfide bond under mechanical stress. Chem. A Eur. J. 2012, 18, 16332–16338. [Google Scholar] [CrossRef]
- CPMD. Copyright IBM Corp, 1990–2008, Copyright MPI für Festkörperforschung Stuttgart 1997–2001. Available online: http://www.cpmd.org/ (accessed on 4 October 2021).
- Frank, I. Chemische Reaktionen “on the fly”. Angew. Chem. 2003, 115, 1607–1609. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. 1988, 37, 785. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev. C.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Troullier, N.; Martins, J.L. Efficient pseudopotentials for plane-wave calculations. Phys. Rev. B 1991, 43, 1993. [Google Scholar] [CrossRef] [PubMed]
- Boero, M.; Parrinello, M.; Terakura, K.; Weiss, H. Car—Parrinello study of Ziegler—Natta heterogeneous catalysis: Stability and destabilization problems of the active site models. Mol. Phys. 2002, 100, 2935–2940. [Google Scholar] [CrossRef]
- Zahn, S.; MacFarlane, D.R.; Izgorodina, E.I. Assessment of Kohn–Sham density functional theory and Møller–Plesset perturbation theory for ionic liquids. Phys. Chem. Chem. Phys. 2013, 15, 13664–13675. [Google Scholar] [CrossRef] [PubMed]
- Gunnarsson, O.; Lundqvist, B.I. Exchange and correlation in atoms, molecules, and solids by the spin-density-functional formalism. Phys. Rev. B 1976, 13, 4274. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S. Semiempirical hybrid density functional with perturbative second-order correlation. J. Chem. Phys. 2006, 124, 034108. [Google Scholar] [CrossRef] [Green Version]
- Møller, C.; Plesset, S.M. Note on an approximation treatment for many-electron systems. Phys. Rev. 1934, 46, 618. [Google Scholar] [CrossRef] [Green Version]
- Wiberg, K.B. Ab Initio Molecular Orbital Theory; Hehre, W.J., Radom, L., Schleyer, P.V.R., Pople, J.A., Eds.; John Wiley: New York, NY, USA, 1986; 548p. [Google Scholar] [CrossRef]
- Miertuš, S.; Scrocco, E.; Tomasi, J. Electrostatic interaction of a solute with a continuum. A direct utilizaion of AB initio molecular potentials for the prevision of solvent effects. Chem. Phys. 1981, 55, 117–129. [Google Scholar] [CrossRef]
- Chase, M.W., Jr. NIST-JANAF Thermochemical Tables, Monograph 9, Part 1; American Institute of Physics: New York, NY, USA, 1998. Available online: https://www.nist.gov/ (accessed on 4 October 2021).
- Ruscic, B.; Pinzon, R.E.; Von Laszewski, G.; Kodeboyina, D.; Burcat, A.; Leahy, D.; Montoy, D.; Wagner, A.F. Active Thermochemical Tables: Thermochemistry for the 21st century. In Proceedings of the SciDAC 2005, SCIENTIFIC DISCOVERY THROUGH ADVANCED COMPUTING, San Francisco, CA, USA, 26–30 June 2005; IOP Publishing: Bristol, UK; Volume 16, p. 078.
- Ruscic, B.; Pinzon, R.E.; Morton, M.L.; von Laszevski, G.; Bittner, S.J.; Nijsure, S.G.; Amin, K.A.; Minkoff, M.; Wagner, A.F. Introduction to active thermochemical tables: Several “key” enthalpies of formation revisited. J. Phys. Chem. A 2004, 108, 9979–9997. [Google Scholar] [CrossRef]






























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
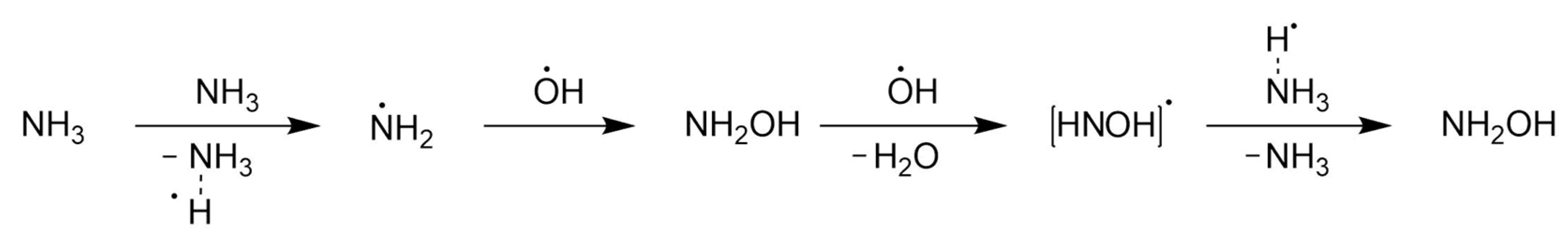{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Deviation (theo-exptl)/kcal · mol−1 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Reaction | ΔrH (exptl)/kcal · mol−1 | Source | BLYP | B3LYP | B2PLYP | HF | MP2 | CCSD | CCSD/aug |
| 4 ⋅OH + NH3 → HNO + 3 H2O | −175.90 | NIST [35] | 7.86 | 18.81 | 16.41 | 110.71 | 1.36 | 25.25 | 18.19 |
| 2 NH3 + 4 ⋅OH → NO⋅ + NH4 + 3 H2O | −124.27 | NIST [35], ACTC [36,37] | 7.91 | 22.36 | 20.99 | 109.99 | 15.59 | 35.70 | 20.16 |
| NH3 + 2 ⋅OH → NH2OH + H2O | −75.86 | NIST [35], ACTC [36,37] | 1.44 | 6.55 | 6.91 | 55.78 | 1.90 | 13.04 | 8.86 |
| 2 NH3 + 3 ⋅OH → HNO + NH4 + 2 H2O | −54.93 | NIST [35], ACTC [36,37] | 5.73 | 19.71 | 20.22 | 97.45 | 14.91 | 32.52 | 18.24 |
| 2 ⋅OH + imin → H2O + imOH | −87.56 | NIST [35], ACTC [36,37] | −2.22 | 3.64 | 3.90 | 56.48 | −1.26 | 11.22 | - |
| Deviation (ΔrE(theo) − ΔHr(exptl))/kcal · mol−1 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Reaction | ΔrH (exptl)/kcal · mol−1 | BLYP | B3LYP | B2PLYP | HF | MP2 | CCSD | CCSD/aug | |
| 4 ⋅OH + NH3 → HNO + 3 H2O | −175.90 | NIST [35] | 3.29 | 13.78 | 11.02 | 104.60 | −3.50 | 19.80 | 12.99 |
| 2 NH3 + 4 ⋅OH → NO⋅ + NH4 + 3 H2O | −124.27 | NIST [35], ACTC [36,37] | 8.40 | 22.32 | 20.14 | 110.56 | 9.10 | 32.73 | 14.65 |
| NH3 + 2 ⋅OH → NH2OH + H2O | −75.86 | NIST [35], ACTC [36,37] | −3.08 | 1.63 | 1.81 | 49.90 | −3.07 | 7.79 | 3.75 |
| 2 NH3 + 3 ⋅OH → HNO + NH4 + 2 H2O | −54.93 | NIST [35], ACTC [36,37] | 8.42 | 21.72 | 21.51 | 99.72 | 12.61 | 31.49 | 14.58 |
| 2 ⋅OH + imin → H2O + imOH | −87.56 | NIST [35], ACTC [36,37] | −11.53 | −5.91 | −5.74 | 46.42 | −10.71 | 1.48 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gordiy, I.; Steinbach, L.; Frank, I. Ab-Initio Molecular Dynamics Simulation of Condensed-Phase Reactivity: The Electrolysis of Ammonia and Ethanimine in Aquatic Carbon Dioxide Solutions. Energies 2021, 14, 6510. https://doi.org/10.3390/en14206510
Gordiy I, Steinbach L, Frank I. Ab-Initio Molecular Dynamics Simulation of Condensed-Phase Reactivity: The Electrolysis of Ammonia and Ethanimine in Aquatic Carbon Dioxide Solutions. Energies. 2021; 14(20):6510. https://doi.org/10.3390/en14206510
Chicago/Turabian StyleGordiy, Igor, Lukas Steinbach, and Irmgard Frank. 2021. "Ab-Initio Molecular Dynamics Simulation of Condensed-Phase Reactivity: The Electrolysis of Ammonia and Ethanimine in Aquatic Carbon Dioxide Solutions" Energies 14, no. 20: 6510. https://doi.org/10.3390/en14206510
APA StyleGordiy, I., Steinbach, L., & Frank, I. (2021). Ab-Initio Molecular Dynamics Simulation of Condensed-Phase Reactivity: The Electrolysis of Ammonia and Ethanimine in Aquatic Carbon Dioxide Solutions. Energies, 14(20), 6510. https://doi.org/10.3390/en14206510