
XPS Study on the Stability and Transformation of Hydrate and Carbonate Phases within MgO Systems

Abstract
:1. Introduction
2. Materials and Methods
3. Results and Discussion
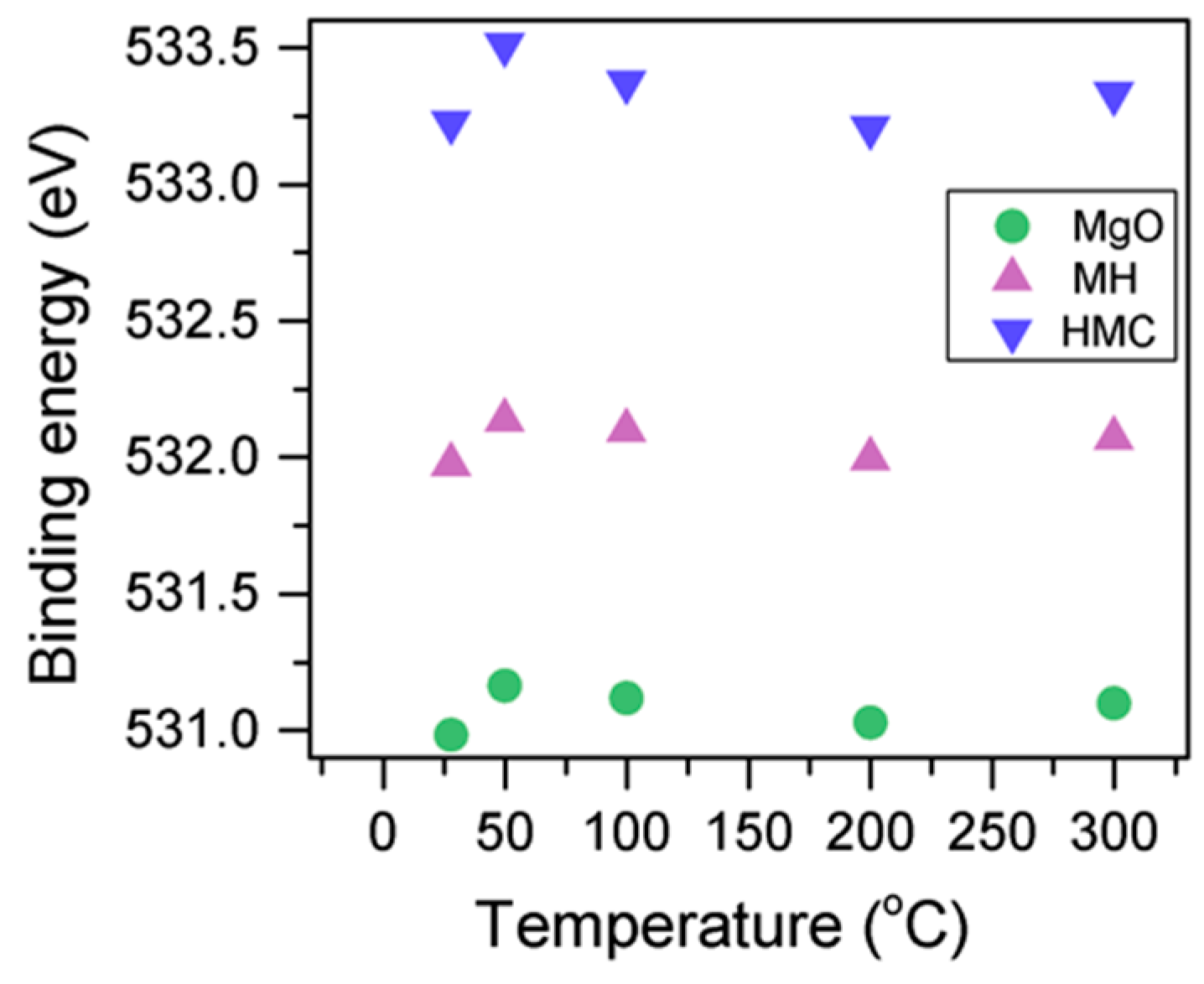
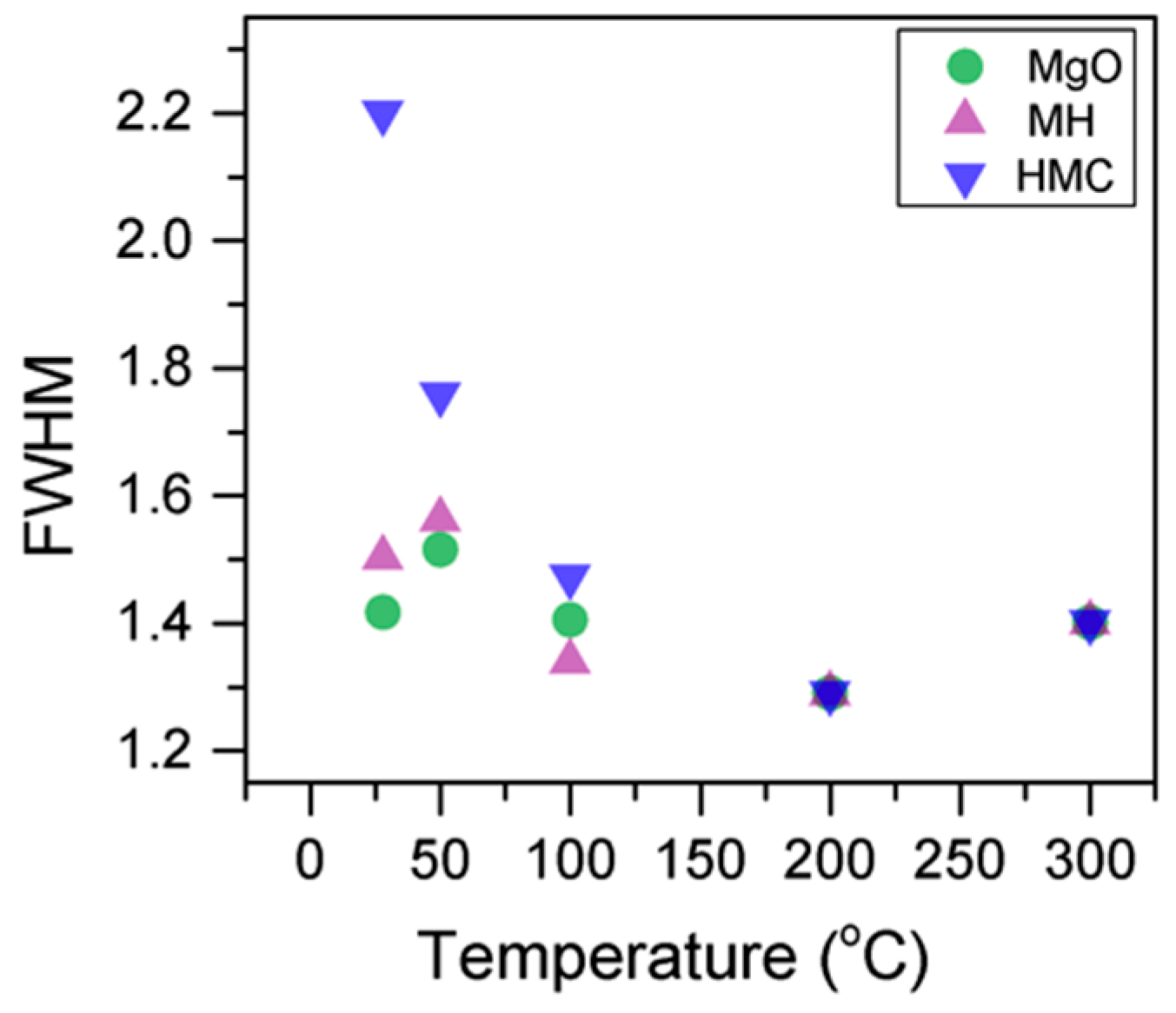
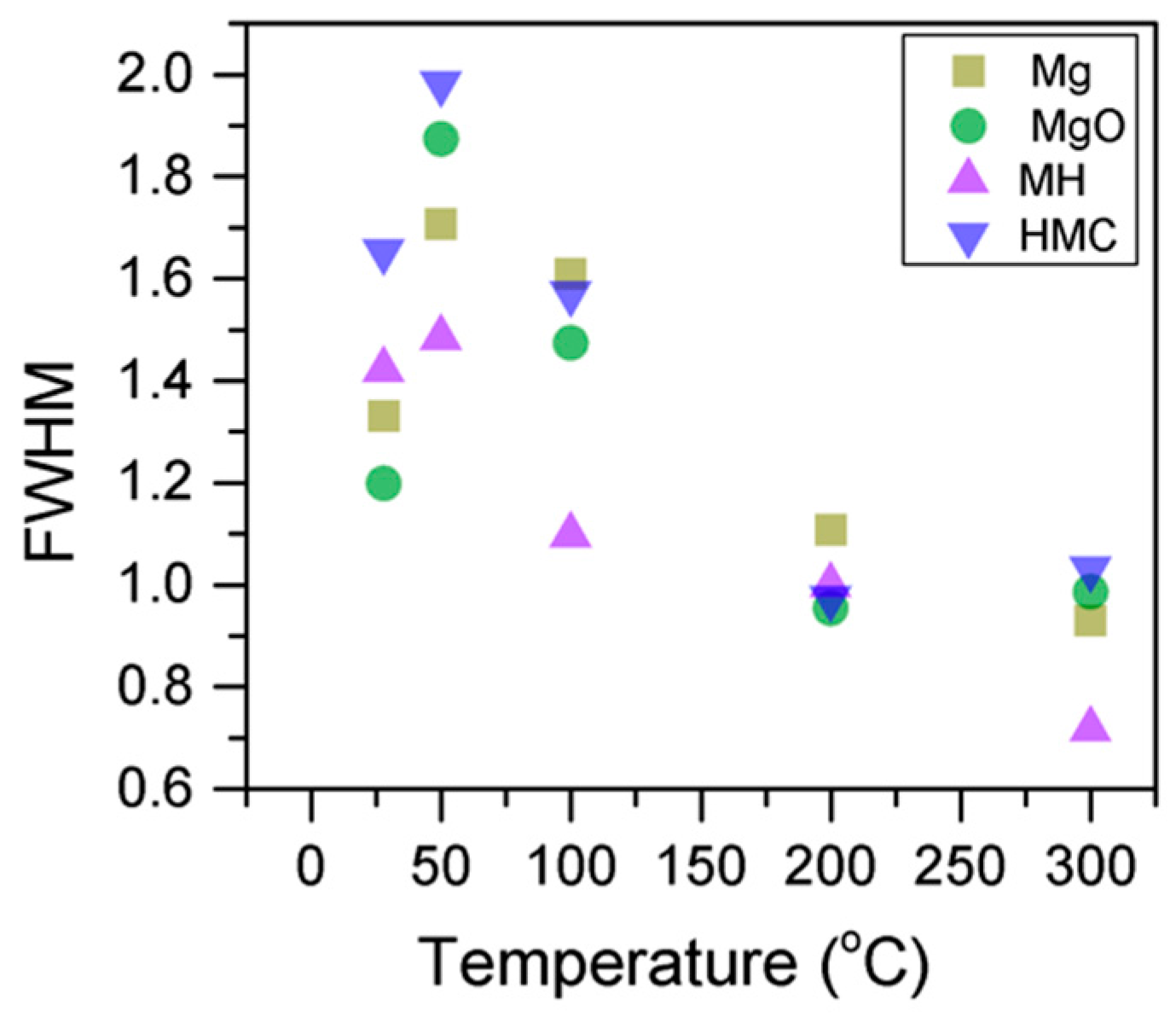
3.1. Samples Exposed to High Temperatures
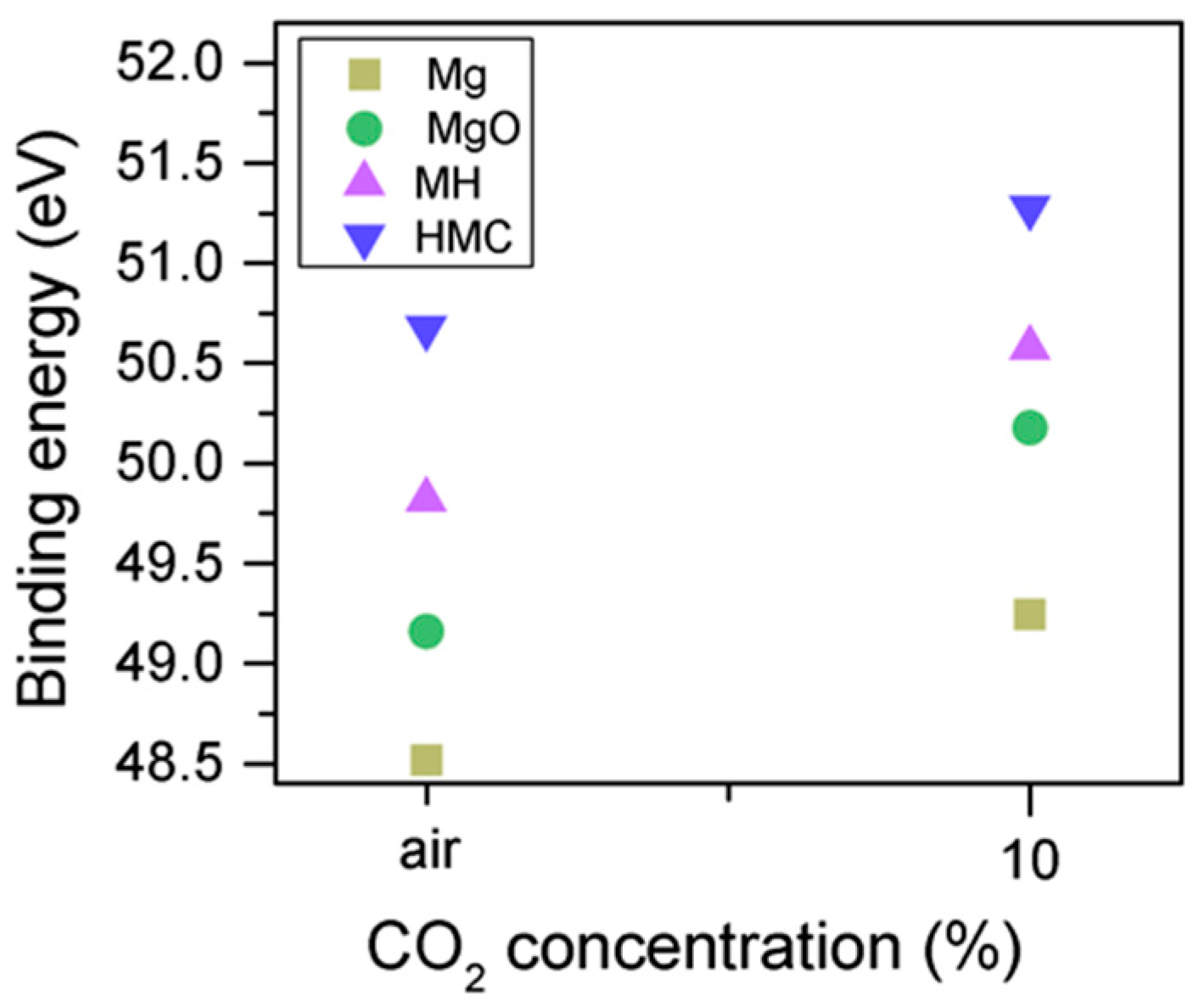
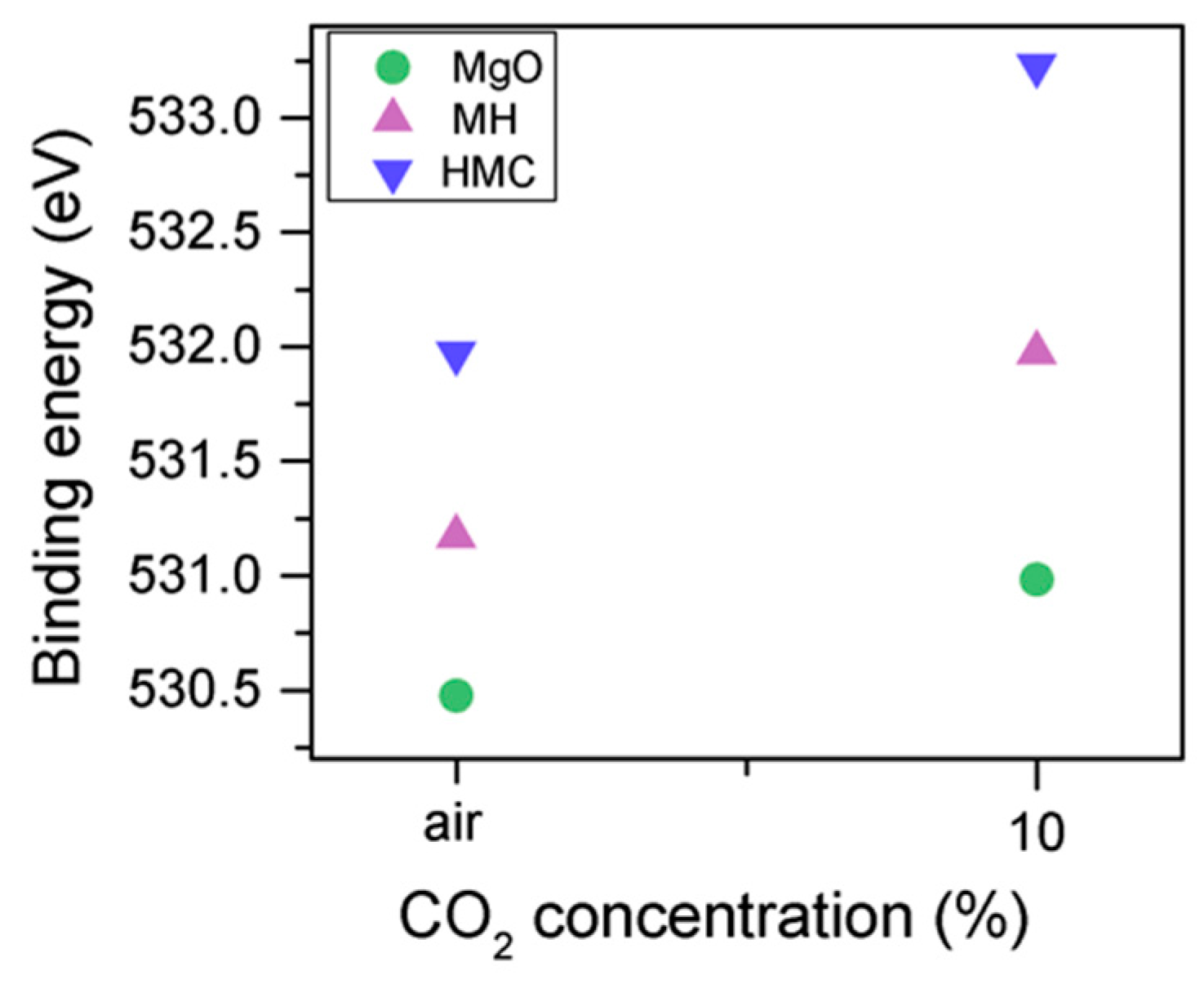
3.2. Effect of Exposure to Air or CO2
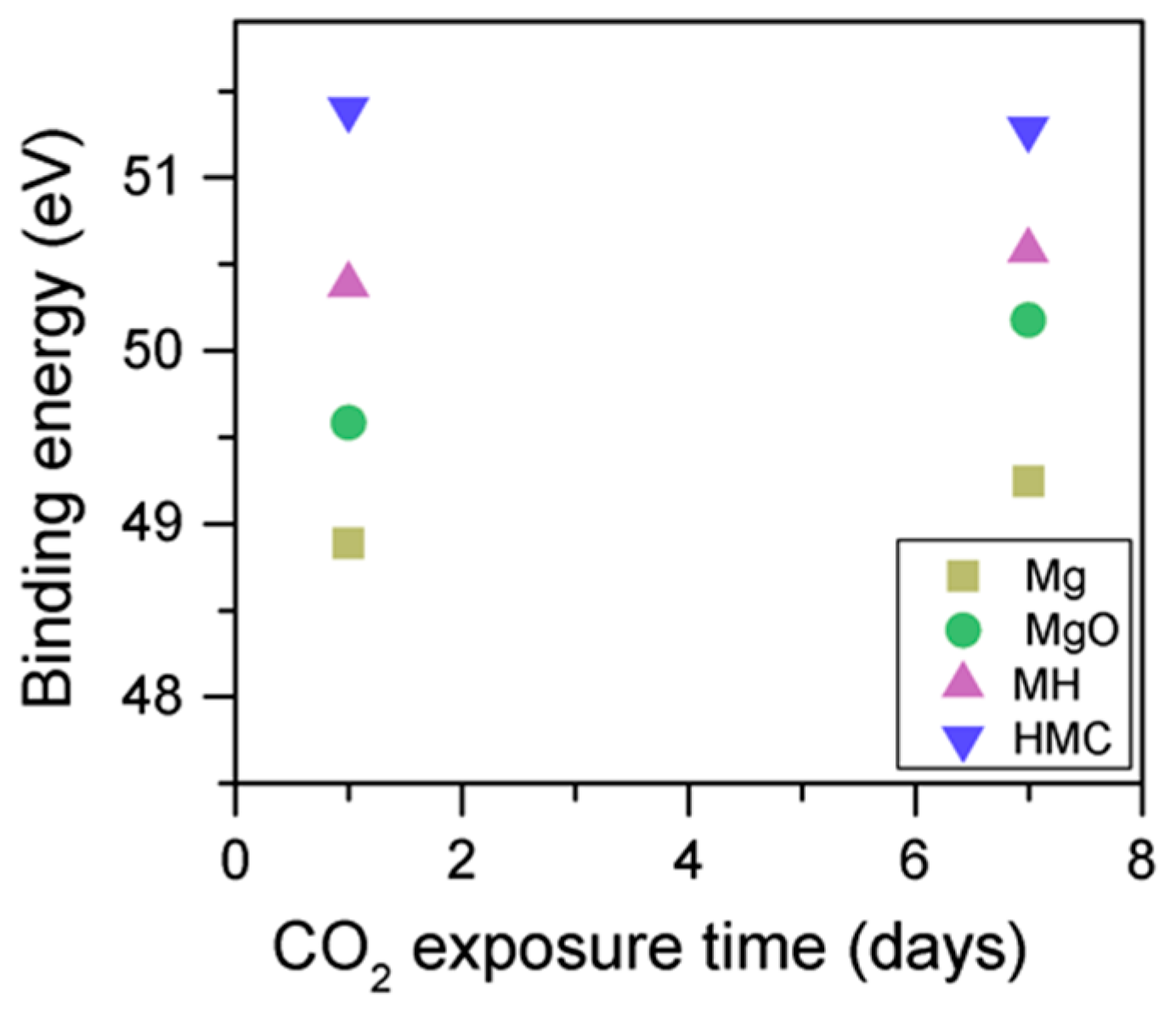
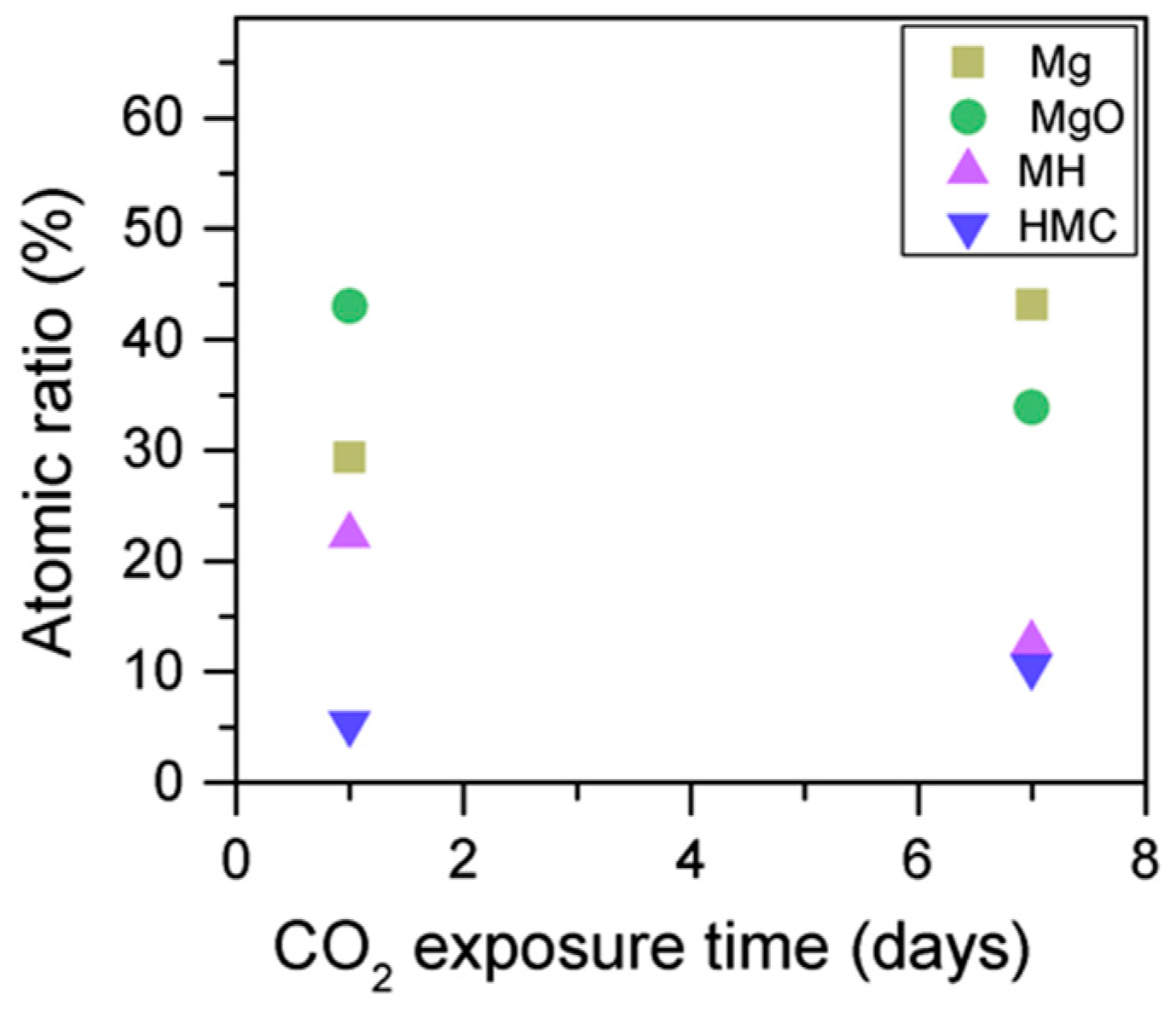
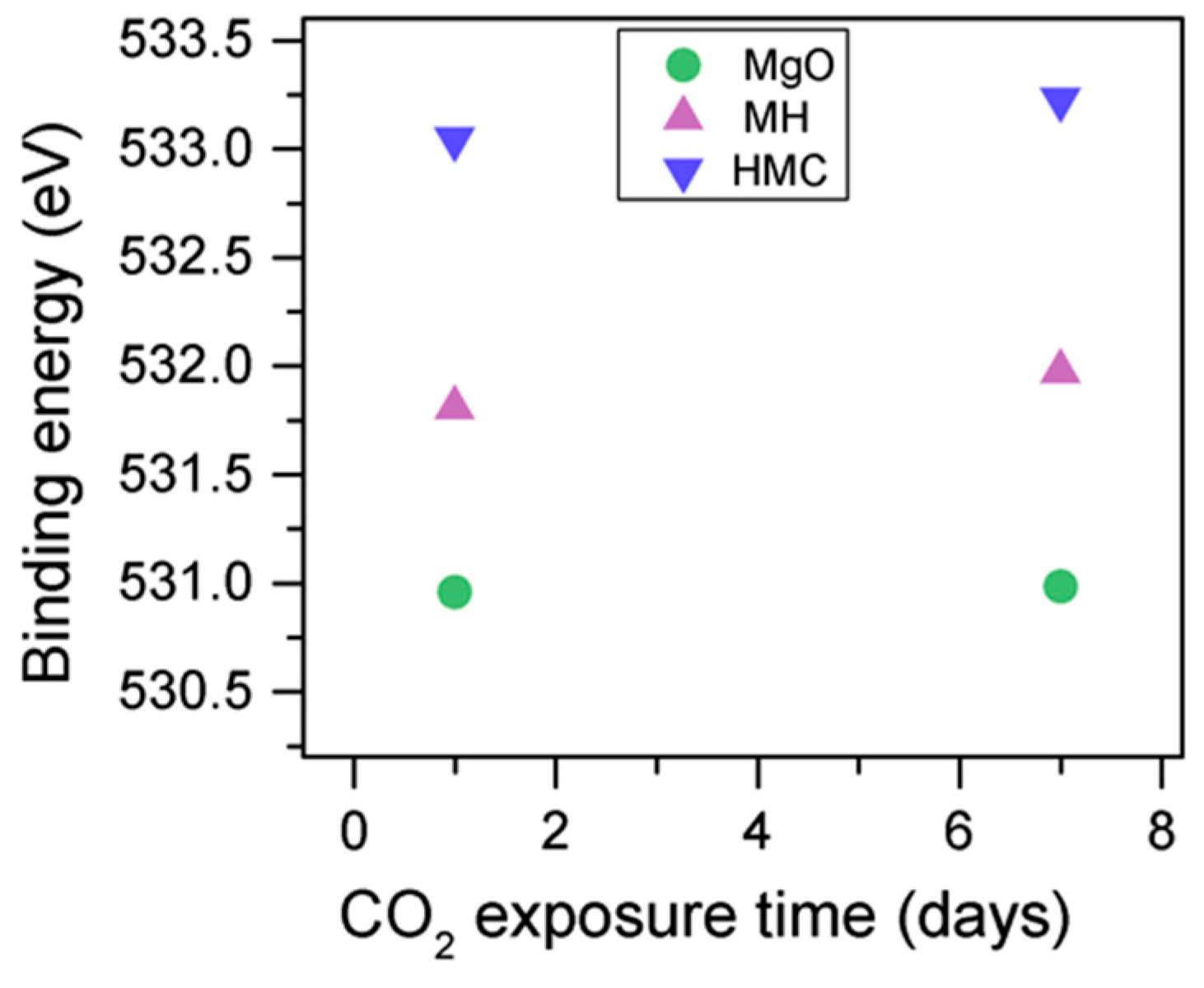
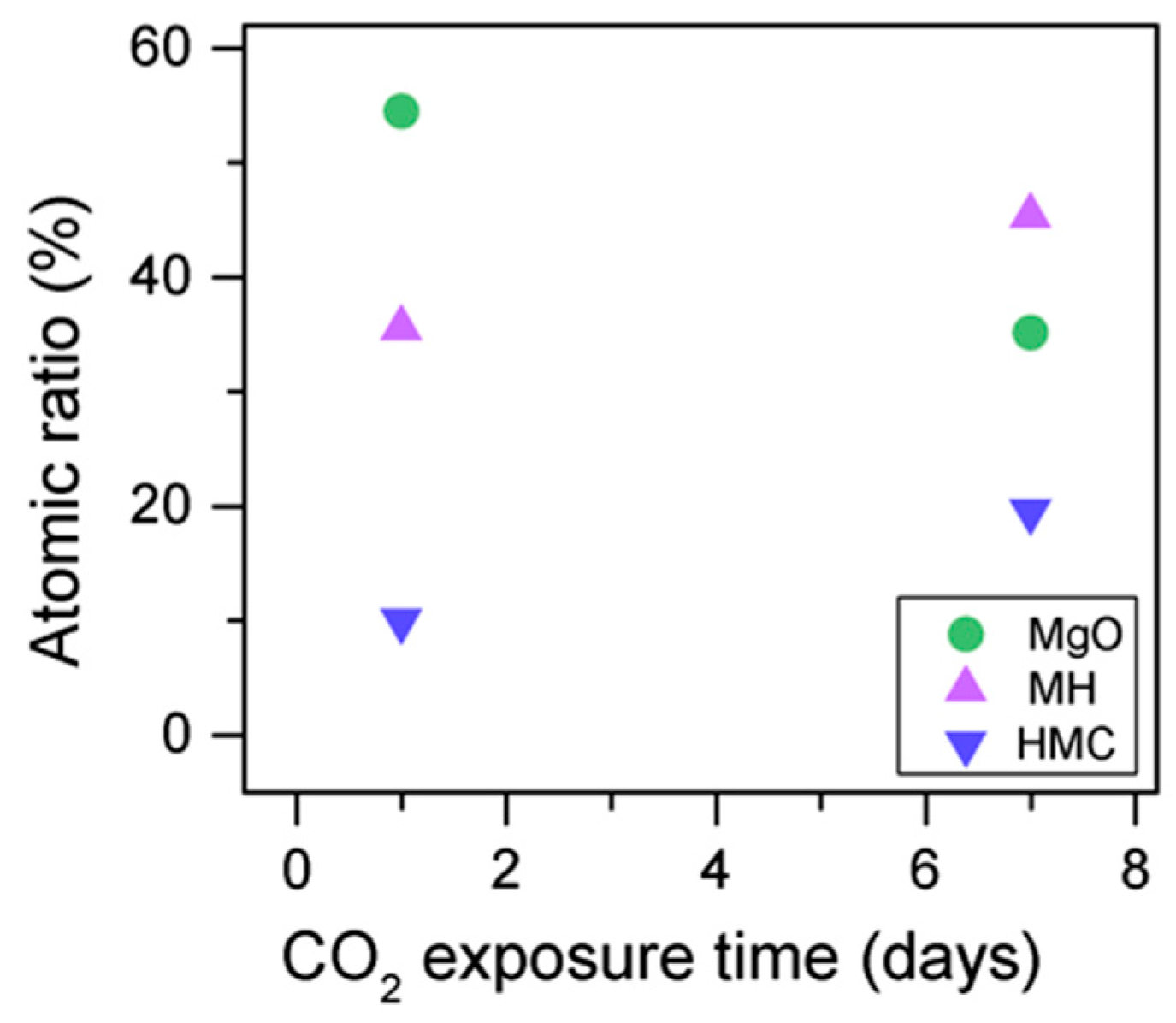
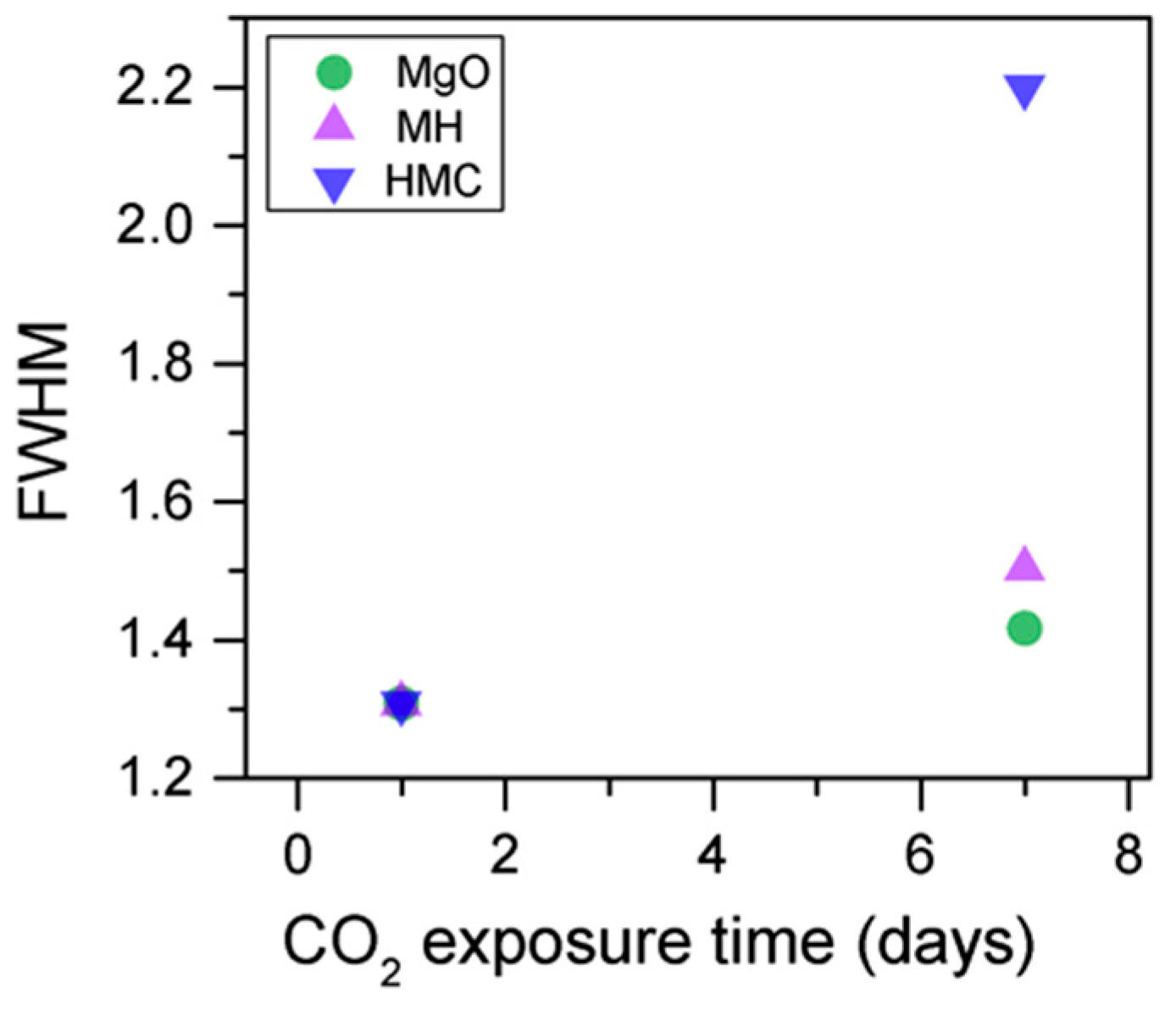
3.3. Effect of the Length of CO2 Exposure
4. Conclusions
- Increases in temperature led to the transformation of nesquehonite into hydromagnesite, as indicated by the changes in the BE of the O1s peak. This was eventually followed by the conversion of hydromagnesite into magnesite. The atomic ratio of the carbonates fluctuated without significant changes, as decarbonation is expected to only start at higher temperatures. Decreases in the FWHM indicated changes in the oxygen bonding. Four components were observed for Mg2p in all samples, which are related to metallic Mg (which can be trapped inside particles from the precursor), MgO, MH and HMCs. Their BEs could not be evaluated alone due to abnormalities in the adventitious carbon peak observed in the literature for samples heated to elevated temperatures, and instead, the BEs were analyzed together with the concentration, FWHM, and the other components present in the sample.
- Increases in the BE of C1s and O1s as the CO2 concentration increased indicated the formation of carbonate phases. At high energies, the Mg2p peak (related to the carbonates) was extremely small for the sample exposed to only air, which demonstrated the effect of the carbon concentration on the formation of HMCs. Agreements between the C1s and O1s peaks were observed via the increases in BE with the concentration (from air to 10% CO2), while the magnesium hydrates peak shifts and the amount of carbonates increase significantly, together with an increase in the FWHM of the carbonated species.
- An increase in the CO2 exposure time from one to seven days was demonstrated via shifts of all components to higher BEs. This was accompanied with the decrease of the concentrations of periclase and the hydrated components and the increase in the concentration of carbonates, which indicated the continuous formation of hydrate and carbonate phases for longer exposure times.
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Shand, M.A. The Chemistry and Technology of Magnesia; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2006. [Google Scholar]
- Marini, L. Geological Sequestration of Carbon Dioxide: Thermodynamics, Kinetics, and Reaction Path Modeling; Elsevier: Amsterdam, The Netherlands, 2007. [Google Scholar]
- Langmuir, D. Stability of carbonates in the system MgO–CO2–H2O. J. Geol. 1965, 73, 730–754. [Google Scholar] [CrossRef]
- Chaka, A.M.; Felmy, A.R. Ab initio thermodynamic model for magnesium carbonates and hydrates. J. Phys. Chem. A 2014, 118, 7469–7488. [Google Scholar] [CrossRef] [PubMed]
- Mineralogy Database. Available online: http://webmineral.com/ (accessed on 22 December 2016).
- Dell, R.; Weller, S.W. The thermal decomposition of nesquehonite MgCO3·H2O and magnesium ammonium carbonate MgCO3·(NH4)2CO3·4H2O. Trans. Faraday Soc. 1959, 55, 2203–2220. [Google Scholar] [CrossRef]
- Ming, D.W.; Franklin, W.T. Synthesis and characterization of lansfordite and nesquehonite. Soil Sci. Soc. Am. J. 1985, 49, 1303–1308. [Google Scholar] [CrossRef]
- Morgan, B.; Wilson, S.A.; Madsen, I.C.; Gozukara, Y.M.; Habsuda, J. Increased thermal stability of nesquehonite (MgCO3·3H2O) in the presence of humidity and CO2: Implications for low-temperature CO2 storage. Int. J. Greenh. Gas Control 2015, 39, 366–376. [Google Scholar] [CrossRef]
- Davies, P.J.; Bubela, B. The transformation of nesquehonite into hydromagnesite. Chem. Geol. 1973, 12, 289–300. [Google Scholar] [CrossRef]
- Botha, A.; Strydom, C.A. Preparation of a magnesium hydroxy carbonate from magnesium hydroxide. Hydrometallurgy 2001, 62, 175–183. [Google Scholar] [CrossRef]
- Power, I.M.; Wilson, S.A.; Thom, J.M.; Dipple, G.M.; Southam, G. Biologically induced mineralization of dypingite by cyanobacteria from an alkaline wetland near Atlin, British Columbia, Canada. Geochem. Trans. 2007, 8, 13. [Google Scholar] [CrossRef] [PubMed]
- Canterford, J.H. Some observations on the properties of dypingite, Mg5(CO3)4(OH)2·5H2O, and related minerals. Mineral. Mag. 1984, 48, 437–442. [Google Scholar] [CrossRef]
- Sayles, F.L.; Fyfe, W.S. The crystallization of magnesite from aqueous solution. Geochim. Cosmochim. Acta 1973, 37, 87–99. [Google Scholar] [CrossRef]
- Chaka, A.M.; Felmy, A.R.; Qafoku, O. Ab initio thermodynamics of magnesium carbonates and hydrates in water-saturated supercritical CO2 and CO2-rich regions. Chem. Geol. 2016, 434, 1–11. [Google Scholar] [CrossRef]
- Hollingbery, L.A.; Hull, T.R. The thermal decomposition of huntite and hydromagnesite—A review. Thermochim. Acta 2010, 509, 1–11. [Google Scholar] [CrossRef]
- Bhattacharjya, D.; Selvamani, T.; Mukhopadhyay, I. Thermal decomposition of hydromagnesite. J. Therm. Anal. Calorim. 2012, 107, 439–445. [Google Scholar] [CrossRef]
- Ballirano, P.; De Vito, C.; Ferrini, V.; Mignardi, S. The thermal behaviour and structural stability of nesquehonite, MgCO3·3H2O, evaluated by in situ laboratory parallel-beam X-ray powder diffraction: New constraints on CO2 sequestration within minerals. J. Hazard. Mater. 2010, 178, 522–528. [Google Scholar] [CrossRef] [PubMed]
- Ballirano, P.; de Vito, C.; Mignardi, S.; Ferrini, V. Phase transitions in the MgCO2H2O system and the thermal decomposition of dypingite, Mg5(CO3)4(OH)2·5H2O: Implications for geosequestration of carbon dioxide. Chem. Geol. 2013, 340, 59–67. [Google Scholar] [CrossRef]
- Santamaria, M.; di Quarto, F.; Zanna, S.; Marcus, P. Initial surface film on magnesium metal: A characterization by X-ray photoelectron spectroscopy (XPS) and photocurrent spectroscopy (PCS). Electrochim. Acta 2007, 53, 1314–1324. [Google Scholar] [CrossRef] [Green Version]
- Fournier, V.; Marcus, P.; Olefjord, I. Oxidation of magnesium. Surf. Interface Anal. 2002, 34, 494–497. [Google Scholar] [CrossRef]
- Feliu, S., Jr.; Merino, M.C.; Arrabal, R.; Coy, A.E.; Matykina, E. XPS study of the effect of aluminium on the atmospheric corrosion of the AZ31 magnesium alloy. Surf. Interface Anal. 2009, 41, 143–150. [Google Scholar] [CrossRef]
- Fotea, C.; Callaway, J.; Alexander, M.R. Characterisation of the surface chemistry of magnesium exposed to the ambient atmosphere. Surf. Interface Anal. 2006, 38, 1578–1587. [Google Scholar] [CrossRef]
- Forsgren, J.; Frykstrand, S.; Grandfield, K.; Mihranyan, A.; Strømme, M. A template-free, ultra-adsorbing, high surface area carbonate nanostructure. PLoS ONE 2013, 8, e68486. [Google Scholar] [CrossRef] [PubMed]
- Vasquez, R.P. MgO(100) by XPS. Surf. Sci. Spectra 1993, 2, 13–19. [Google Scholar] [CrossRef]
- Nefedov, V.I.; Firsov, M.N.; Shaplygin, I.S. Electronic structures of MRhO2, MRh2O4, RhMO4 and Rh2MO6 on the basis of X-ray spectroscopy and ESCA data. J. Electron Spectrosc. Relat. Phenom. 1982, 26, 65–78. [Google Scholar] [CrossRef]
- Lanas, J.; Alvarez, J.I. Dolomitic lime: Thermal decomposition of nesquehonite. Thermochim. Acta 2004, 421, 123–132. [Google Scholar] [CrossRef]
- Jauffret, G.; Morrison, J.; Glasser, F.P.; Yoon, S.; Imbabi, M.S. Low Carbon Cement Based on Hydrated Magnesium Carbonate. University of Aberdeen. Available online: http://159.226.251.229/videoplayer/C05_J_Morrison_Poster.pdf?ich_u_r_i=fa6feed05f87cf7e7617df3a3b544088&ich_s_t_a_r_t=0&ich_e_n_d=0&ich_k_e_y=1745018913750763222400&ich_t_y_p_e=1&ich_d_i_s_k_i_d=1&ich_u_n_i_t=1 (accessed on 7 November 2016).
- Xiong, Y.; Lord, A.S. Experimental investigations of the reaction path in the MgO–CO2–H2O system in solutions with various ionic strengths, and their applications to nuclear waste isolation. Appl. Geochem. 2008, 23, 1634–1659. [Google Scholar] [CrossRef]
- Hänchen, M.; Prigiobbe, V.; Baciocchi, R.; Mazzotti, M. Precipitation in the Mg-carbonate system-effects of temperature and CO2 pressure. Chem. Eng. Sci. 2008, 63, 1012–1028. [Google Scholar] [CrossRef]
- Königsberger, E.; Königsberger, L.-C.; Gamsjäger, H. Low-temperature thermodynamic model for the system Na2CO3–MgCO3–CaCO3–H2O. Geochim. Cosmochim. Acta 1999, 63, 3105–3119. [Google Scholar] [CrossRef]
- Zhao, L.; Sang, L.; Chen, J.; Ji, J.; Teng, H.H. Aqueous carbonation of natural brucite: relevance to CO2 sequestration. Environ. Sci. Technol. 2010, 44, 406–411. [Google Scholar] [CrossRef] [PubMed]
- Hopkinson, L.; Kristova, P.; Rutt, K.; Cressey, G. Phase transitions in the system MgO–CO2–H2O during CO2 degassing of Mg-bearing solutions. Geochim. Cosmochim. Acta 2012, 76, 1–13. [Google Scholar] [CrossRef]
- Jauffret, G.; Morrison, J.; Glasser, F.P. On the thermal decomposition of nesquehonite. J. Therm. Anal. Calorim. 2015, 122, 5–10. [Google Scholar] [CrossRef]
- Frost, R.L.; Palmer, S.J. Infrared and infrared emission spectroscopy of nesquehonite Mg(OH)(HCO3)·2H2O-implications for the formula of nesquehonite. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2011, 78, 1255–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hopkinson, L.; Rutt, K.; Cressey, G. The transformation of nesquehonite to hydromagnesite in the system CaO–MgO–H2O–CO2: An experimental spectroscopic study. 2008, 116, 387–400. [Google Scholar] [CrossRef] [Green Version]
- Vágvölgyi, V.; Frost, R.L.; Hales, M.; Locke, A.; Kristóf, J.; Horváth, E. Controlled rate thermal analysis of hydromagnesite. J. Therm. Anal. Calorim. 2008, 92, 893–897. [Google Scholar] [CrossRef]
- Montes-Hernandez, G.; Renard, F.; Chiriac, R.; Findling, N.; Toche, F. Rapid precipitation of magnesite microcrystals from Mg(OH)2–H2O–CO2 slurry enhanced by NaOH and a heat-aging step (from ~20 to 90 °C). Cryst. Growth Des. 2012, 12, 5233–5240. [Google Scholar] [CrossRef] [Green Version]
- Jerome, R.; Teyssie, P.; Pireaux, J.J.; Verbist, J.J. Surface analysis of polymers end-capped with metal carboxylates using X-ray photoelectron spectroscopy. Appl. Surf. Sci. 1986, 27, 93–105. [Google Scholar] [CrossRef]
- Yao, H.; Li, Y.; Wee, A.T. An XPS investigation of the oxidation/corrosion of melt-spun Mg. Appl. Surf. Sci. 2000, 158, 112–119. [Google Scholar] [CrossRef]
- Corneille, J.S.; He, J.-W.; Goodman, D.W. XPS characterization of ultra-thin MgO films on a Mo(100) surface. Surf. Sci. 1994, 306, 269–278. [Google Scholar] [CrossRef]
- Hoogewijs, R.; Fiermans, L.; Vennik, J. Electronic relaxation processes in the KLL′ auger spectra of the free magnesium atom, solid magnesium and MgO. J. Electron Spectrosc. Relat. Phenom. 1977, 11, 171–183. [Google Scholar] [CrossRef]
- Haycock, D.E.; Kasrai, M.; Nicholls, C.J.; Urch, D.S. The Electronic structure of magnesium hydroxide (brucite) using X-ray emission, X-ray photoelectron, and auger spectroscopy. J. Chem. Soc. Daltan Trans. 1978, 12, 1791–1796. [Google Scholar] [CrossRef]
- Larachi, F.; Daldoul, I.; Beaudoin, G. Fixation of CO2 by chrysotile in low-pressure dry and moist carbonation: Ex-situ and in-situ characterizations. Geochim. Cosmochim. Acta 2010, 74, 3051–3075. [Google Scholar] [CrossRef]
- Talapatra, A.; Bandyopadhyay, S.; Pintu, S.; Barat, P.; Mukherjee, S.; Mukherjee, M. X-ray photoelectron spectroscopy studies of MgB2 for valence state of Mg. Phys. C Superconductivity Appl. 2005, 419, 141–147. [Google Scholar] [CrossRef]
- Newberg, J.T.; Starr, D.E.; Yamamoto, S.; Kaya, S.; Kendelewicz, T.; Mysak, E.R.; Porsgaard, S.; Salmeron, M.B.; Brown, G.E.; Nilsson, A.; et al. Formation of hydroxyl and water layers on MgO films studied with ambient pressure XPS. Surf. Sci. 2011, 605, 89–94. [Google Scholar] [CrossRef]
- Inoue, Y.; Yasumori, I. Catalysis by alkaline earth metal oxides. III. X-ray photoelectron spectroscopic study of catalytically active MgO, CaO, and BaO surfaces. Bull. Chem. Soc. Jpn. 1981, 54, 1505–1510. [Google Scholar] [CrossRef]
- Bender, M.; Yakovkin, I.N.; Freund, H.-J. Adsorption and reaction of magnesium on Cr2O3(0001)/Cr(110). Surf. Sci. 1996, 365, 394–402. [Google Scholar] [CrossRef]
- Werrett, C.R.; Bhattacharya, A.K.; Pyke, D.R. The validity of C1s charge referencing in the XPS of oxidised Al-Si alloys. Appl. Surf. Sci. 1996, 103, 403–407. [Google Scholar] [CrossRef]
- Feliu, S.; Samaniego, A.; Violeta, B.; El-Hadad, A.A.; Llorente, I.; Adeva, P. The effect of heat treatment on surface chemistry and corrosion resistance of commercial magnesium alloys AZ31 and AZ61 in 0.6 M NaCl solution. Corros. Sci. 2014, 80, 461–472. [Google Scholar] [CrossRef]
- Pickard, C.J.; Needs, R.J. Structures and stability of calcium and magnesium carbonates at mantle pressures. Phys. Rev. B 2015, 91, 104101. [Google Scholar] [CrossRef]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Number of Mg Ions | Compound | Chemical Formula |
|---|---|---|---|
| Brucite | Mg(OH)2 | ||
| Magnesite | MgCO3 | ||
| Group I | 1 | Barringtonite | MgCO3·2H2O |
| Nesquehonite | MgCO3·3H2O | ||
| Lansfordite | MgCO3·5H2O | ||
| Group II | 2 | Pokrovskite | Mg2(CO3)(OH)2·0.5H2O |
| Artinite | Mg2(CO3)(OH)2·3H2O | ||
| Group III | 5 | Hydromagnesite | Mg5(CO3)4(OH)2·4H2O |
| Dypingite | Mg5(CO3)4(OH)2·5H2O | ||
| Giorgiosite | Mg5(CO3)4(OH)2·5–6H2O | ||
| Group IV | 7 | Shelkovite | Mg7(CO3)5(OH)4·24H2O |
| Chemical Composition (%) | Physical Properties | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| MgO | SiO2 | CaO | R2O3 | K2O | Na2O | LOI | Specific Gravity (g/cm3) | Specific Surface Area (m2/g) | |
| RMC | >91.5 | 2.0 | 1.6 | 1.0 | - | - | 4.0 | 3.0 | 16.3 |
| Sample | Age (Days) | CO2 Exposure | Temperature (°C) |
|---|---|---|---|
| M7-10-50 | 7 | 10% | 50 |
| M7-10-100 | 7 | 10% | 100 |
| M7-10-200 | 7 | 10% | 200 |
| M7-10-300 | 7 | 10% | 300 |
| M1-10-28 | 1 | 10% | 28 |
| M7-10-28 | 7 | 10% | 28 |
| M7-A-28 | 7 | air | 28 |
| Mg2p | O1s | |||||
|---|---|---|---|---|---|---|
| Temperature (°C) Sample ID | Peak Position (eV) | Concentration (%) | FWHM | Peak Position (eV) | Concentration (%) | FWHM |
| 0 M7-10-28 | 49.2 | 43.1 | 1.33 | 531.0 | 35.1 | 1.42 |
| 50.2 | 33.9 | 1.20 | 532.0 | 45.3 | 1.50 | |
| 50.6 | 12.6 | 1.42 | 533.2 | 19.5 | 2.20 | |
| 51.3 | 10.4 | 1.65 | - | - | - | |
| 50 M7-10-50 | 49.4 | 31.6 | 1.71 | 531.2 | 42.6 | 1.51 |
| 50.0 | 42.7 | 1.87 | 532.1 | 43.8 | 1.56 | |
| 50.6 | 14.3 | 1.48 | 533.5 | 13.5 | 1.76 | |
| 51.3 | 11.4 | 1.98 | - | - | - | |
| 100 M7-10-100 | 49.3 | 36.6 | 1.61 | 531.1 | 43.6 | 1.40 |
| 49.9 | 36.0 | 1.47 | 532.1 | 38.9 | 1.34 | |
| 50.6 | 12.8 | 1.10 | 533.4 | 17.5 | 1.47 | |
| 51.3 | 14.6 | 1.57 | - | - | - | |
| 200 M7-10-200 | 49.2 | 37.7 | 1.11 | 531.0 | 39.2 | 1.29 |
| 50.1 | 33.5 | 0.95 | 532.0 | 44.8 | 1.29 | |
| 50.6 | 18.8 | 1.00 | 533.2 | 16.0 | 1.29 | |
| 51.2 | 10.0 | 0.97 | - | - | - | |
| 300 M7-10-300 | 49.2 | 35.1 | 0.93 | 531.1 | 39.0 | 1.40 |
| 50.1 | 37.4 | 0.99 | 532.1 | 45.2 | 1.40 | |
| 50.8 | 15.0 | 0.72 | 533.3 | 15.7 | 1.40 | |
| 51.3 | 12.5 | 1.03 | - | - | - | |
| Mg2p | O1s | |||||
|---|---|---|---|---|---|---|
| CO2 Concentration Sample ID | Peak Position (eV) | Concentration (%) | FWHM | Peak Position (eV) | Concentration (%) | FWHM |
| Air M7-A-28 | 48.5 | 21.4 | 0.8 | 530.5 | 23.2 | 1.1 |
| 49.2 | 44.5 | 0.8 | 531.2 | 57.9 | 1.2 | |
| 49.8 | 27.2 | 0.8 | 532.0 | 18.9 | 1.9 | |
| 50.7 | 6.9 | 0.8 | - | - | - | |
| 10% CO2 M7-10-28 | 49.2 | 43.1 | 1.3 | 531.0 | 35.1 | 1.4 |
| 50.2 | 33.9 | 1.2 | 532.0 | 45.3 | 1.5 | |
| 50.6 | 12.6 | 1.4 | 533.2 | 19.5 | 2.2 | |
| 51.3 | 10.4 | 1.7 | - | - | - | |
| Mg2p | O1s | |||||
|---|---|---|---|---|---|---|
| Exposure Time (Days) Sample ID | Concentration % | Peak Position (eV) | FWHM | Concentration % | Peak Position (eV) | FWHM |
| 1 M1-10-28 | 29.4 | 48.9 | 0.8 | 54.5 | 531.0 | 1.3 |
| 43.0 | 49.6 | 0.8 | 35.5 | 531.8 | 1.3 | |
| 22.3 | 50.4 | 0.8 | 10.0 | 533.0 | 1.3 | |
| 5.3 | 51.4 | 0.8 | ||||
| 7 M7-10-28 | 43.1 | 49.2 | 1.3 | 35.1 | 531.0 | 1.4 |
| 33.9 | 50.2 | 1.2 | 45.3 | 532.0 | 1.5 | |
| 12.6 | 50.6 | 1.4 | 19.5 | 533.2 | 2.2 | |
| 10.4 | 51.3 | 1.7 | ||||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rheinheimer, V.; Unluer, C.; Liu, J.; Ruan, S.; Pan, J.; Monteiro, P.J.M. XPS Study on the Stability and Transformation of Hydrate and Carbonate Phases within MgO Systems. Materials 2017, 10, 75. https://doi.org/10.3390/ma10010075
Rheinheimer V, Unluer C, Liu J, Ruan S, Pan J, Monteiro PJM. XPS Study on the Stability and Transformation of Hydrate and Carbonate Phases within MgO Systems. Materials. 2017; 10(1):75. https://doi.org/10.3390/ma10010075
Chicago/Turabian StyleRheinheimer, Vanessa, Cise Unluer, Jiawei Liu, Shaoqin Ruan, Jisheng Pan, and Paulo J. M. Monteiro. 2017. "XPS Study on the Stability and Transformation of Hydrate and Carbonate Phases within MgO Systems" Materials 10, no. 1: 75. https://doi.org/10.3390/ma10010075
APA StyleRheinheimer, V., Unluer, C., Liu, J., Ruan, S., Pan, J., & Monteiro, P. J. M. (2017). XPS Study on the Stability and Transformation of Hydrate and Carbonate Phases within MgO Systems. Materials, 10(1), 75. https://doi.org/10.3390/ma10010075