Salt-Assisted Ultrasonicated De-Aggregation and Advanced Redox Electrochemistry of Detonation Nanodiamond


Abstract
:1. Introduction
2. Experimental
2.1. Materials and Methods
2.2. Structural and Spectroscopic Characterization
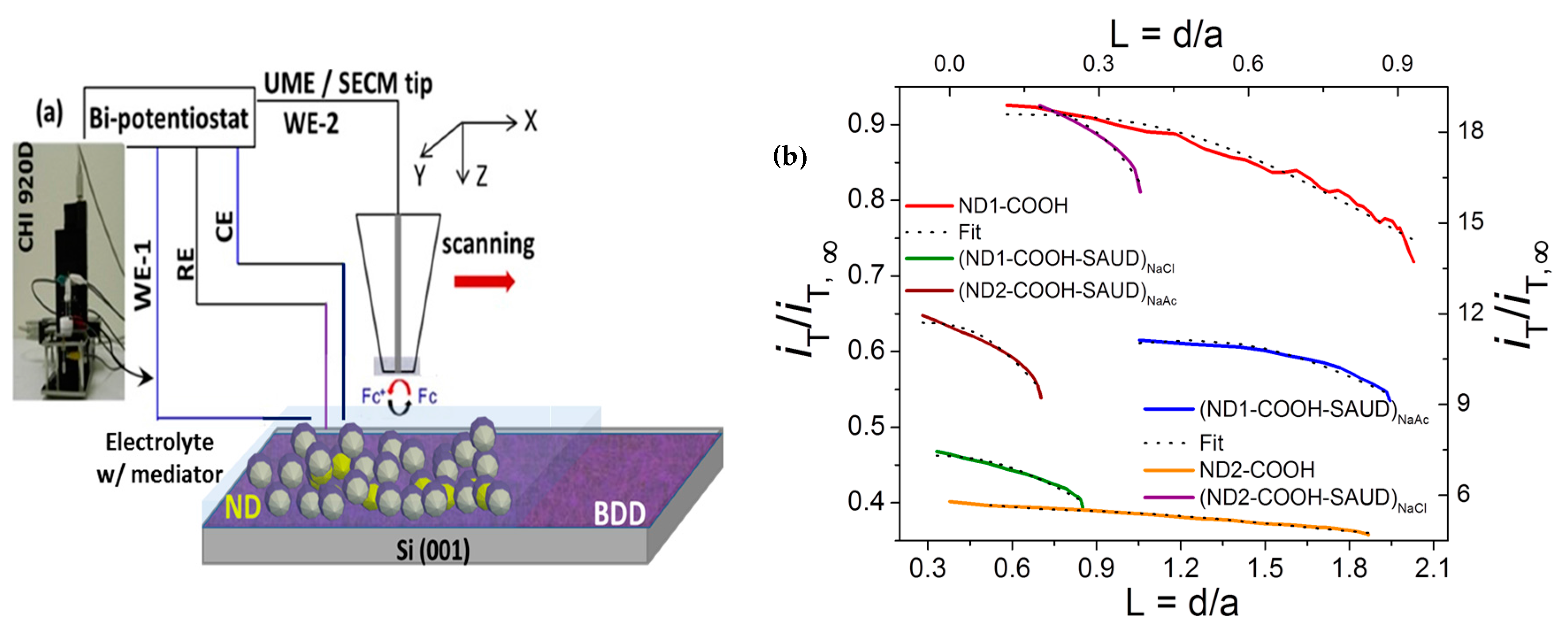
2.3. Electrochemical Properties and Scanning Electrochemical Microscopy
3. Results and Discussion
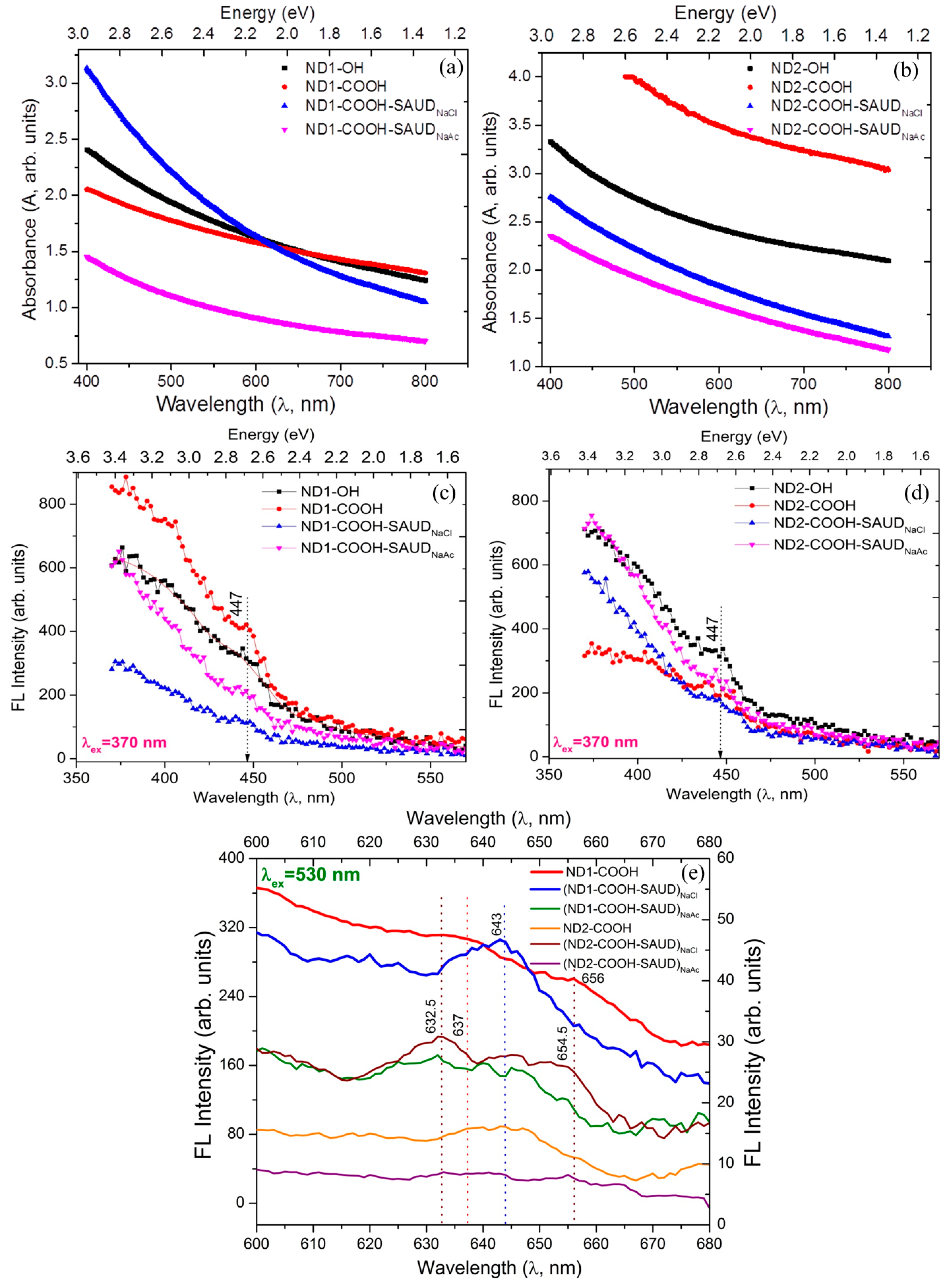
3.1. Microstructural Properties
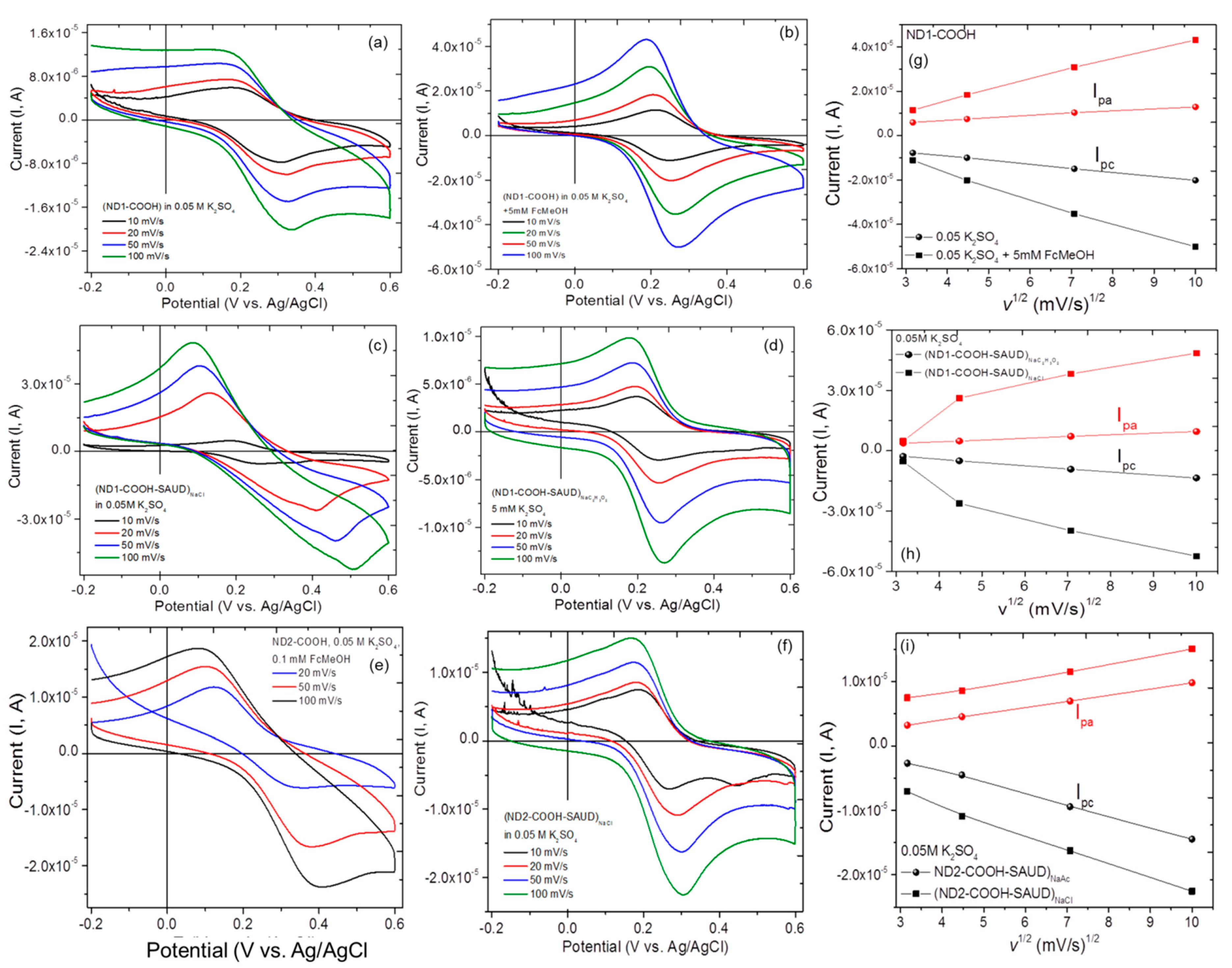
3.2. Electrochemical Measurements Cyclic Voltammetry
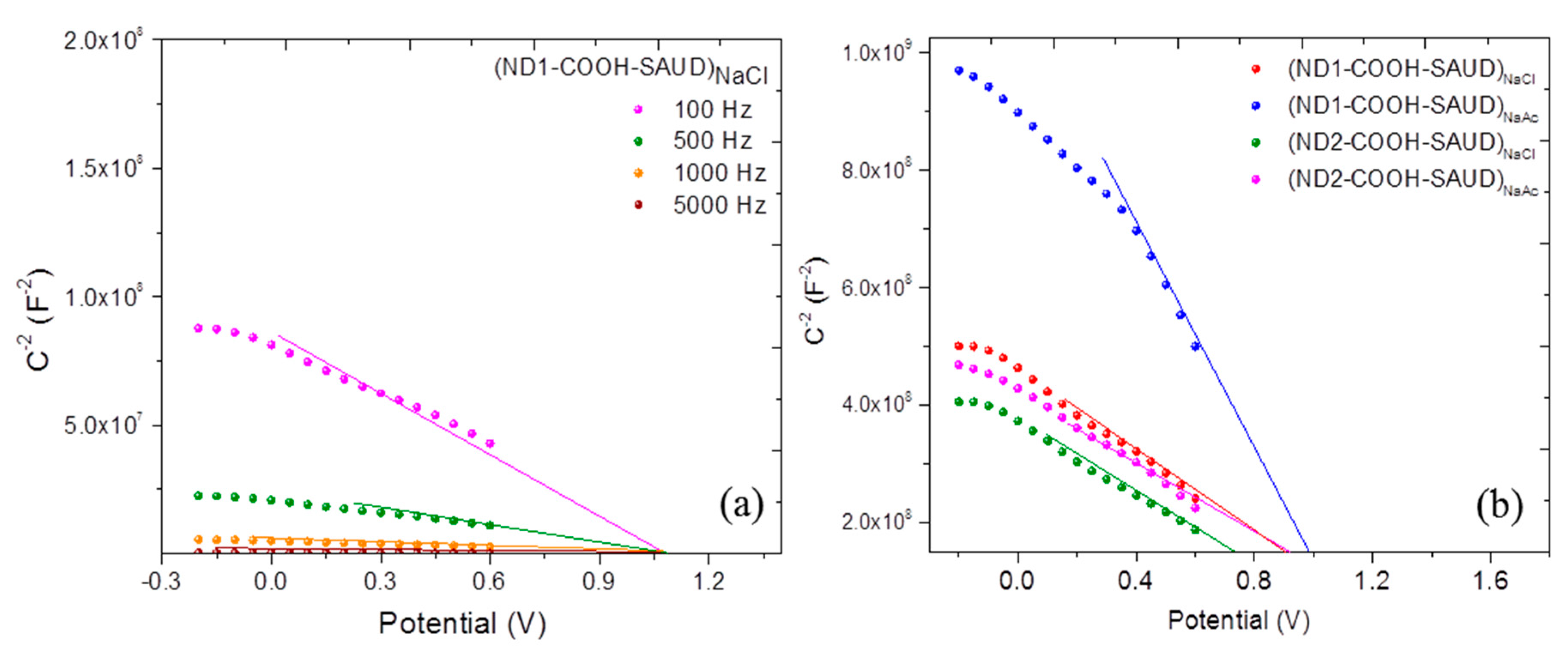
3.2.1. AC Impedance versus Potential
3.2.2. Scanning Electrochemical Microscopy (SECM)
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Prawer, S.; Nemanich, R.J. Raman spectroscopy of diamond and doped diamond. Philos. Trans. R. Soc. Lond. A 2004, 362, 2537–2565. [Google Scholar] [CrossRef] [PubMed]
- Mochalin, V.N.; Shenderova, O.; Ho, D.; Gogotsi, Y. The properties and applications of nanodiamonds. Nat. Nanotechnol. 2012, 7, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Danilenko, V.V. On the history of the discovery of nanodiamond synthesis. Phys. Solid State 2004, 46, 595–599. [Google Scholar] [CrossRef]
- Baidakova, M.; Vul’, A. New prospects and frontiers of nanodiamond clusters. Phys. D Appl. Phys. 2007, 40, 6300–6311. [Google Scholar] [CrossRef]
- Raty, J.-Y.; Galli, G. Ultradispersity of diamond at the nanoscale. Nat. Mater. 2003, 2, 792–795. [Google Scholar] [CrossRef] [PubMed]
- Raty, J.-Y.; Galli, G.; Bostedt, C.; van Buuren, T.W.; Terminello, L.J. Quantum Confinement and Fullerenelike Surface Reconstructions in Nanodiamonds. Phys. Rev. Lett. 2003, 90, 037401. [Google Scholar] [CrossRef] [PubMed]
- Foord, J.; Hu, J.P. Electrochemical oxidation and reduction processes at diamond electrodes of varying phase purity. Phys. Status Solidi 2006, 203, 3121–3127. [Google Scholar] [CrossRef]
- Paci, J.T.; Man, H.B.; Saha, B.; Ho, D.; Schatz, G.C. Understanding the Surfaces of Nanodiamonds. J. Phys. Chem. C 2013, 117, 17256–17267. [Google Scholar] [CrossRef]
- Krüger, A.; Kataoka, F.; Ozawa, M.; Fujino, T.; Suzuki, Y.; Aleksenskii, A.; Vúl, A.Y.; Osawa, E. Unusually tight aggregation in detonation nanodiamond: Identification and disintegration. Carbon 2005, 43, 1722–1730. [Google Scholar] [CrossRef]
- Holt, K.B. Undoped diamond nanoparticles: Origins of surface redox chemistry. Phys. Chem. Chem. Phys. 2010, 12, 2048–2058. [Google Scholar] [CrossRef] [PubMed]
- Brenneis, A.; Gaudreau, L.; Seifert, M.; Karl, H.; Brandt, M.S.; Huebl, H.; Garrido, J.A.; Koppens, F.H.L.; Holleitner, A.W. Ultrafast electronic readout of diamond nitrogen–vacancy centres coupled to graphene. Nat. Nanotechnol. 2014, 10, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; McDonald, B.; Carrizosa, S.B. Surface Redox Chemistry of Immobilized Nanodiamond: Effects of Particle Size and Electrochemical Environment. J. Electron. Mater. 2017. [Google Scholar] [CrossRef]
- Osawa, E. Monodisperse single nanodiamond particulates. Pure Appl. Chem. 2008, 80, 1365–1379. [Google Scholar] [CrossRef]
- Krüger, A.; Liang, Y.; Harre, G.; Stegk, J. Surface functionalisation of detonation diamond suitable for biological applications. J. Mater. Chem. 2006, 16, 2322–2328. [Google Scholar] [CrossRef]
- Turcheniuk, K.; Trecazzi, C.; Deeleepojanan, C.; Mochalin, V.N. Salt-Assisted Ultrasonic Deaggregation of Nanodiamond. ACS Appl. Mater. Interfaces 2016, 8, 25461–25468. [Google Scholar] [CrossRef] [PubMed]
- Pentecost, A.; Gour, S.; Mochalin, V.; Knoke, L.; Gogotsi, Y. Deaggregation of Nanodiamond Powders Using Salt- and Sugar-Assisted Milling. ACS Appl. Mater. Interfaces 2010, 2, 3289–3294. [Google Scholar] [CrossRef] [PubMed]
- Shnederova, O.A.; McGuire, G.E. Science and engineering of nanodiamond particle surfaces for biological applications (Review). Biointerphases 2015, 10, 030802. [Google Scholar] [CrossRef] [PubMed]
- Osawa, E.; Ho, D. Nanodiamond and its application to drug delivery. J. Med. Allied Sci. 2012, 2, 31–40. [Google Scholar]
- Barnard, A.S. Diamond standard in diagnostics: Nanodiamond biolabels make their mark. Analyst 2009, 134, 1751–1764. [Google Scholar] [CrossRef] [PubMed]
- Williams, O.A.; Hees, J.; Dieker, C.; Jager, W.; Kirste, L.; Nebel, C.E. Size-Dependent Reactivity of Diamond Nanoparticles. ACS Nano 2010, 4, 4824–4830. [Google Scholar] [CrossRef] [PubMed]
- Schrand, A.M.; Hens, S.A.C.; Shenderova, O.A. Nanodiamond Particles: Properties and Perspectives for Bioapplications. Crit. Rev. Solid State Mater. Sci. 2009, 34, 18–74. [Google Scholar] [CrossRef]
- Gupta, S.; Irihamye, A. Probing the nature of electron transfer in metalloproteins on graphene-family materials as nanobiocatalytic scaffold using electrochemistry. AIP Adv. 2015, 5, 037106. [Google Scholar] [CrossRef]
- Wang, X.; Low, X.C.; Hou, W.; Abdullah, L.N.; Toh, T.B.; Rashid, M.M.A.; Ho, D.; Chow, E.K.-H. Epirubicin-Adsorbed Nanodiamonds Kill Chemoresistant Hepatic Cancer Stem Cells. ACS Nano 2014, 8, 12151–12166. [Google Scholar] [CrossRef] [PubMed]
- Kreuger, A. New Carbon Materials: Biological Applications of Functionalized Nanodiamond Materials. Chemistry 2008, 145, 1382–1390. [Google Scholar] [CrossRef] [PubMed]
- Holt, K.B. Diamond at the nanoscale: Applications of diamond nanoparticles from cellular biomarkers to quantum computing. Philos. Trans. A 2007, 365, 2845–2861. [Google Scholar] [CrossRef] [PubMed]
- Varley, T.S.; Hirani, M.; Harrison, G.; Holt, K.B. Nanodiamond surface redox chemistry: Influence of physicochemical properties on catalytic processes. Faraday Discuss. 2014, 172, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.E.; Show, Y.; Swain, G.M. Electrochemical Performance of Diamond Thin-Film Electrodes from Different Commercial Sources. Anal. Chem. 2004, 76, 2553–2560. [Google Scholar] [CrossRef] [PubMed]
- Holt, K.B.; Ziegler, C.; Caruana, D.J.; Zang, J.; Mill´an-Barrios, E.J.; Hu, J.; Foord, J.S. Redox properties of undoped 5 nm diamond nanoparticles. Phys. Chem. Chem. Phys. 2008, 10, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Rodrigo, M.A.; Michaud, P.A.; Duo, I.; Panizza, M.; Cerisola, G.; Cominellis, C. Oxidation of 4-Chlorophenol at Boron-Doped Diamond Electrode for Wastewater Treatment. J. Electrochem. Soc. 2001, 148, D60–D64. [Google Scholar] [CrossRef]
- Tryk, D.A.; Tachibana, H.; Inoue, H.; Fujishima, A. Boron-doped diamond electrodes: The role of surface termination in the oxidation of dopamine and ascorbic acid. Diam. Relat. Mater. 2007, 16, 881–887. [Google Scholar] [CrossRef]
- Bennet, K.E.; Lee, K.H.; Kruchowski, J.N.; Chang, S.-Y.; Marsh, M.P.; van Orsow, A.A.; Paez, A.; Manciu, F.S. Development of Conductive Boron-Doped Diamond Electrode: A microscopic, Spectroscopic, and Voltammetric Study. Materials 2013, 6, 5726–5741. [Google Scholar] [CrossRef] [PubMed]
- Ramamurti, R.; Becker, M.; Schuelke, T.; Grotjohn, T.; Reinhard, D.; Swain, G.; Asmussen, J. Boron doped diamond deposited by microwave plasma-assisted CVD at low and high pressures. Diam. Relat. Mater. 2008, 17, 481–485. [Google Scholar] [CrossRef]
- Yang, N.; Hoffmann, R.; Smirnov, W.; Nebel, C.E. Direct electrochemistry of cytochrome c on nanotextured diamond surface. Electrochem. Commun. 2010, 12, 1218. [Google Scholar] [CrossRef]
- Bondar, V.; Puzyr’, A. Nanodiamonds for biological investigations. Phys. Solid State 2004, 46, 716–719. [Google Scholar] [CrossRef]
- Novoselova, I.A.; Fedoryshena, E.N.; Panov, E.V.; Bochechka, A.A.; Romanko, L.A. Electrochemical properties of compacts of nano-and microdisperse diamond powders in aqueous electrolytes. Phys. Solid State 2004, 46, 748–750. [Google Scholar] [CrossRef]
- Landstrass, M.I.; Ravi, K.V. Resistivity of chemical vapor deposited diamond films. Appl. Phys. Lett. 1989, 55, 975–978. [Google Scholar] [CrossRef]
- Ristein, J.; Zhang, W.; Ley, L. Hydrogen-terminated diamond electrodes. I. Charges, potentials, and energies. Phys. Rev. E 2008, 78, 041602. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Ristein, J.; Ley, L. Hydrogen-terminated diamond electrodes. II. Redox activity. Phys. Rev. E 2008, 78, 041603. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.; Watanabe, H.; Nebel, C.E. Redox-couple interactions with undoped single crystalline CVD diamond. Diam. Relat. Mater. 2006, 15, 121–128. [Google Scholar] [CrossRef]
- Chakrapani, V.; Angus, J.C.; Anderson, A.B.; Wolter, S.D.; Stoner, B.R.; Sumanasekera, G.U. Charge transfer equilibria between diamond and an aqueous oxygen electrochemical redox couple. Science 2007, 318, 1424–1430. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.R.; Lee, H.-Y.; Chen, K.; Chang, C.-C.; Tsai, D.-S.; Fu, C.-C.; Lim, T.-S.; Tzen, Y.-K.; Fang, C.-Y.; Han, C.-C.; et al. Mass production and dynamic imaging of fluorescent nanodiamonds. Nat. Nanotechnol. 2008, 3, 284–288. [Google Scholar] [CrossRef] [PubMed]
- Osswald, S.; Yushin, G.; Mochalin, V.; Kucheyev, S.O.; Gogotsi, Y. Control of sp2/sp3 Carbon Ratio and Surface Chemistry of Nanodiamond Powders by Selective Oxidation in Air. J. Am. Chem. Soc. 2006, 128, 11635–11642. [Google Scholar] [CrossRef] [PubMed]
- Reich, K. Optical properties of nanodiamond suspensions. JETP Lett. 2011, 94, 22–26. [Google Scholar] [CrossRef]
- Alekenskii, A.; Vul’, A.; Konyakhin, Y.; Reich, K.; Sharanova, L.; Eidel’man, E. Optical properties of detonation nanodiamond hydrosols. Phys. Status Solidi 2012, 54, 578–581. [Google Scholar]
- Krueger, A.; Stegk, J.; Liang, Y.J.; Barre, G. Biotinylated nanodiamond: Simple and efficient functionalization of detonation diamond. Langmuir 2008, 24, 4200–4204. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Li, W.; Zhang, Y.; Li, J.; Liang, L.; Zhang, X.; Chen, N.; Sun, Y.; Chen, W.; Tai, R.; et al. Excessive sodium ions delivered into cells by nanodiamonds: Implications for tumor therapy. Small 2012, 8, 1771–1779. [Google Scholar] [CrossRef] [PubMed]
- Walker, J. Optical absorption and luminescence in diamond. Rep. Prog. Phys. 1979, 42, 1605–1659. [Google Scholar] [CrossRef]
- Mochalin, V.N.; Penetcrost, A.; Li, X.-M.; Neitzel, I.; Nelson, M.; Wei, C.; He, T.; Guo, F.; Gogotsi, Y. Adsorption of Drugs on Nanodiamond: Toward Development of a Drug Delivery Platform. Mol. Pharm. 2013, 10, 3728–3735. [Google Scholar] [CrossRef] [PubMed]
- Mona, J.; Tu, J.S.; Kang, T.Y.; Tsai, C.Y.; Perevedentseva, E.; Cheng, C.L. Surface modification of nanodiamond: Photoluminescence and Raman Studies. Diam. Relat. Mater. 2012, 24, 134–138. [Google Scholar] [CrossRef]
- Gupta, S.; Weiss, B.L.; Weiner, B.R.; Pilione, L.; Badzian, A.; Morell, G. Electron field emission properties of gamma irradiated microcrystalline diamond and nanocrystalline carbon thin films. J. Appl. Phys. 2002, 92, 3311–3317. [Google Scholar] [CrossRef]
- Osswald, S.; Mochalin, V.N.; Havel, M.; Yushin, G.; Gogotsi, Y. Phonon confinement effects in the Raman spectrum of nanodiamond. Phys. Rev. B 2009, 80, 075419. [Google Scholar] [CrossRef]
- Ager, J.W., III; Veirs, D.K.; Rosenblatt, G.M. Spatially resolved Raman studies of diamond films grown by chemical vapor deposition. Phys. Rev. B 2009, 43, 6491–6499. [Google Scholar] [CrossRef]
- Gao, Y.; Zhao, Z.; Yin, P.; Gao, F. Size-Dependent Raman Shifts for nanocrystals. Sci. Rep. 2016. [Google Scholar] [CrossRef] [PubMed]
- Chaigneau, M.; Picardi, G.; Girard, H.A.; Arnault, J.-C.; Ossikovski, R. Effect of particle size and laser power on the Raman spectra of CuAlO2 delafossite nanoparticles. J. Nanopart. Res. 2012, 14, 955–962. [Google Scholar] [CrossRef]
- Prawer, S.; Nugent, K.W.; Jamieson, D.N.; Orwa, J.O.; Bursill, L.A.; Peng, J.L. The Raman spectrum of nanocrystalline diamond. Chem. Phys. Lett. 2000, 332, 93–97. [Google Scholar] [CrossRef]
- Tu, J.-S.; Perenvedentseva, E.; Chung, P.-H.; Cheng, C.-L. Size-dependent surface CO stretching frequency investigations on nanodiamond particles. J. Chem. Phys. 2006, 125, 174713–174717. [Google Scholar] [CrossRef] [PubMed]
- Derjaguin, B.; Landau, L. Theory of the Stability of Strongly Charged Lyophobic Sols and of the Adhesion of Strongly Charged Particles in Solutions of Electrolytes. Acta Phys. Chem. URSS 1941, 14, 633–662. [Google Scholar] [CrossRef]
- Israelacvili, J.N. Intermolecular and Surface Forces; Academic Press: London, UK, 2007. [Google Scholar]
- Bhattacharjee, S.; Elimelech, M.; Borkovec, M. DLVO Interaction between Colloidal Particles: Beyond Derjaguin’s Approximation. Croat. Chim. Acta 1998, 71, 883–903. [Google Scholar]
- Bard, A.; Faulkner, L.R. Electrochemical Methods: Fundamentals and Applications, 2nd ed.; Wiley: Hoboken, NJ, USA, 2001. [Google Scholar]
- Pleskov, Y.V.; Krotova, M.D.; Elkin, V.V.; Varnin, V.P.; Teremetskaya, I.G. Characterization of CVD Diamond Thin Film Electrodes in Terms of their Semiconductivity. Electrocatalysis 2013, 4, 241–244. [Google Scholar] [CrossRef]
- Latto, M.N.; Riley, D.J.; May, P.W. Impedance studies of boron-doped CVD diamond electrodes. Diam. Relat. Mater. 2000, 9, 1181–1183. [Google Scholar] [CrossRef]
- Ramesham, R. Determination of flatband potential for boron doped diamond electrode in 0.5 M NaCl by AC impedance spectroscopy. Thin Solid Films 1998, 322, 158–166. [Google Scholar] [CrossRef]
- Gomes, W.P.; Vanmaekelbergh, D. Impedance spectroscopy at semiconductor electrodes: Review and recent developments. Electrochim. Acta 1996, 41, 967–973. [Google Scholar] [CrossRef]
- Bott, A.W. Electrochemistry of semiconductors. Curr. Sep. 1998, 17, 87–91. [Google Scholar]
- Sarswat, P.K.; Bhattacharyya, D.; Free, M.L.; Misra, M. Augmented Z scheme blueprint for efficient solar water splitting system using quaternary chalcogenide absorber material. Phys. Chem. Chem. Phys. 2016, 18, 3788–3803. [Google Scholar] [CrossRef] [PubMed]
- Pelskov, Y.V.; Sakharova, A.Y.; Krotova, M.D.; Bouilov, L.L.; Spitsyn, B.V. Photoelectrochemical properties of semiconductor diamond. J. Electroanal. Chem. Interfacial Electrochem. 1987, 228, 19–27. [Google Scholar] [CrossRef]
- Sakharova, A.Y.; Pleskov, Y.V.; Quarto, F.D.; Piazza, S.; Sunseri, C.; Teremetskaya, I.G.; Varnin, V.P. Synthetic Diamond Electrodes: Photoelectrochemical Investigation of Undoped and Boron-Doped Polycrystalline Thin Films. J. Electrochem. Soc. 1995, 142, 2704–2709. [Google Scholar] [CrossRef]
- Wang, Y.; Kececi, K.; Velmurugan, J.; Mirkin, M.V. Electron transfer/ion transfer mode of scanning electrochemical microscopy (SECM): A new tool for imaging and kinetic studies. Chem. Sci. 2013, 4, 3606–3616. [Google Scholar] [CrossRef]
- Mcreery, R.L. Advanced Carbon Electrode Materials for Molecular Electrochemistry. Chem. Rev. 2008, 108, 2646–2687. [Google Scholar] [CrossRef] [PubMed]
- Mareček, V.; Samec, Z.; Weber, J. The dependence of the electrochemical charge-transfer coefficient on the electrode potential: Study of the Fe(CN)63−/Fe(CN)64− redox reaction on polycrystalline Au electrode in KF solutions. J. Electroanal. Chem. 1978, 94, 169–185. [Google Scholar]
- Bourdilion, C.; Demaille, C.; Moiroux, J.; Saveant, J.-M. Catalysis and Mass Transport in Spatially Ordered Enzyme Assemblies on Electrodes. J. Am. Chem. Soc. 1995, 117, 11499–11506. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Ef (V vs. Ag/AgCl) | N (cm−3) |
|---|---|---|
| ND1–COOH | 1.02 | 1.2 × 1020 |
| ND2–COOH | 1.00 | 3.5 × 1021 |
| (ND1–COOH–SAUD)NaCl | 0.91 | 0.5 × 1021 |
| (ND1–COOH–SAUD)NaAc | 0.99 | 4.07 × 1021 |
| (ND2–COOH–SAUD)NaCl | 0.90 | 5.1 × 1021 |
| (ND2–COOH–SAUD)NaAc | 0.71 | 6.2 × 1021 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gupta, S.; Evans, B.; Henson, A.; Carrizosa, S.B. Salt-Assisted Ultrasonicated De-Aggregation and Advanced Redox Electrochemistry of Detonation Nanodiamond. Materials 2017, 10, 1292. https://doi.org/10.3390/ma10111292
Gupta S, Evans B, Henson A, Carrizosa SB. Salt-Assisted Ultrasonicated De-Aggregation and Advanced Redox Electrochemistry of Detonation Nanodiamond. Materials. 2017; 10(11):1292. https://doi.org/10.3390/ma10111292
Chicago/Turabian StyleGupta, Sanju, Brendan Evans, Alex Henson, and Sara B. Carrizosa. 2017. "Salt-Assisted Ultrasonicated De-Aggregation and Advanced Redox Electrochemistry of Detonation Nanodiamond" Materials 10, no. 11: 1292. https://doi.org/10.3390/ma10111292
APA StyleGupta, S., Evans, B., Henson, A., & Carrizosa, S. B. (2017). Salt-Assisted Ultrasonicated De-Aggregation and Advanced Redox Electrochemistry of Detonation Nanodiamond. Materials, 10(11), 1292. https://doi.org/10.3390/ma10111292