1. Introduction
Solid olefin polymerization catalysts represent a very important class of materials. Information about the nature of active sites and spectroscopic information about the Ziegler–Natta polymerization catalysts are scarce. Far more information is available about the kinetics, preparation, and chemistry of the catalysts, including the mechanism of activation of the catalysts by aluminum-containing organic agents and the use of internal donors. Mixed oxides under study represent hybrid materials, which are known to be important for catalytic applications [
1]. Several groups have demonstrated various techniques to unravel the state of titanium active species in these catalysts. Spectroscopic investigation of the TiCl
4–MgCl
2 systems was done [
2]. Carbon monoxide was used as a probe molecule to test the surface sites in the MgCl
2 support and in the TiCl
4–MgCl
2 catalysts of ethylene polymerization. It has been shown that a few types of Lewis acid sites (low-coordinated Mg ions) are present at the surface of the carrier, but they disappear after deposition of TiCl
4 interacting with the surface sites of MgCl
2.
The organic component of the Ziegler–Natta catalysts, i.e., internal donors at the surface of the Ti–Mg stereospecific propylene polymerization catalysts, was also studied by diffuse-reflectance Fourier-transform IR spectroscopy (DRIFT) [
3]. Ethyl benzoate (EB) and di-n-butyl phthalate (DBP) were chosen as internal donors (ID). These molecules demonstrated a superposition band in the carbonyl region of the IR spectra that can be ascribed to perturbed ID molecules. Deconvolution of this band allowed the authors to distinguish three types of ID complexes in the case of EB; only one of these three was formed in the case of DBP. The data obtained were interpreted from the standpoint of the competition between ID and TiCl
4 for the surface sites.
A model Ziegler–Natta catalyst on a Pd(111) substrate coated with the MgCl
2(001) layer was prepared in an ultra-high vacuum and studied using the electron-spin resonance (ESR) technique from the point of view of the formation of radical species during the ethylene polymerization process [
4]. Particular attention was paid to the role of AlR
3 moieties in the reaction, for example, via the interaction with TiCl
4:
The authors also studied the effect of the so-called colored sites (defect sites, F-centers) in the development of the most effective catalyst. The formation of methyl radicals from AlMe3 was also detected by the low-temperature ESR spectroscopy. Furthermore, ethyl radicals were detected as a result of the reaction of methyl radicals with AlMe3. However, the authors, surprisingly, did not find any Ti3+ species in the ESR spectra.
One more type of model Ziegler–Natta polymerization catalysts was prepared by spin-coating on a flat SiO
2/Si(100) support [
5]. XPS analysis of such a catalyst showed the presence of Ti
3+ species after the thermal treatment of the system at 723 K, when a significant part of the chlorine is removed.
Yet another approach to study the Ziegler–Natta polymerization catalysts was reported [
6]. The use of high-resolution electron microscopy in the investigation of the ethylene polymerization catalysts allowed the authors to discriminate the α-TiCl
3 phase, which was destroyed rapidly after exposure to ambient air. It should be noted, in view of our further discussion, that such a phase would not reveal any Ti
3+ paramagnetic centers visible in ESR.
Quite a detailed study of the model Mg–Ti catalysts prepared by gas phase deposition of the components onto the Au polycrystalline foil under UHV conditions (pressures around 1.3 × 10
−5 Pa) is reported in [
7]. The authors found the formation of a TiCl
2 layer onto the TiCl
4 layer. Addition of AlEt
3 into the system leads to the formation of mixed TiR
nCl
4−n complexes. Again, no sign of the presence of TiCl
3 was found in the spectra. These model catalysts were tested in ethylene and propylene polymerization. The fraction of Ti
4+ species in the total Ti content after the treatment with AlEt
3 decreased gradually from 0.6 to 0.2 when increasing the AlEt
3 exposure (measured in Langmuir units).
There are a few papers in which studies were carried out using not model, but real olefin polymerization catalysts [
8,
9]. The authors investigated the interaction between the EB/TiCl
4/MgCl
2 catalyst and an AlR
3 co-catalyst with the goal of finding a relationship between the degree of titanium reduction and the activity in the olefin polymerization reaction. They observed the dependence of the Ti
2+ and Ti
3+ contents determined by redox titration on the Al/Ti ratio, activation temperature, and aging time or reaction time. For instance, for the catalytic systems activated at 70 °C for 2 h at the Al/Ti ratio of 200, the following distribution was revealed from the XPS data: Ti
4+:Ti
3+:Ti
2+ = 9:31:60, i.e., even after severe reduction conditions, a portion of titanium remains in the non-reduced state. The conclusion has been drawn that for ethylene polymerization both Ti
3+ and Ti
2+ species seem to contribute to the overall activity, whereas in the case of propylene conversion the major contribution is likely made by the higher oxidation states (Ti
4+ and Ti
3+), with the latter being most active in the title process. Similar conclusions were made in the second paper of the Italian team [
9].
The Japanese group [
10,
11] reported on the interaction of the TiCl
4/MgCl
2 catalyst with an internal donor and found that the position of the Ti XPS peak does not change when varying the nature of the internal donor and the preparation method. The peak position was, however, different for a model TiCl
4-ester complex, which indicated that such a complex is not formed at the surface, and titanium most probably was present in the reduced state. Thus, the authors concluded that the internal donor could only influence the state of titanium via the intermediacy of MgCl
2.
ESR spectroscopy is an informative method in the physicochemical study of Ti catalysts for olefin polymerization. However, in spite of the frequent use of ESR in monitoring the formation and role of paramagnetic sites in polymerization reactions, a lot of conflicting data are reported about the nature of catalytically active sites and active species formed [
12,
13,
14,
15,
16,
17,
18,
19,
20]. Both the influence of the Ti oxidation state on the catalyst properties and the catalytic activity of cationic Ti
3+ complexes are mainly claimed [
12,
13,
17,
18]. In a study of the copolymerization of styrene and ethylene [
14], ESR spectroscopic analysis combined with copolymerization kinetics results suggested the presence of a Ti
4+ active center that is responsible for the formation of polyethylene, a Ti
3+ species active in the syndiospecific polymerization of styrene, and, moreover, the presence of a third intermediate contributing to the promotion of the copolymerization of styrene with ethylene. The formation of paramagnetic Ti
27+ pairs is discussed [
8]. In the ESR study of the Cp*TiMe
3/B(C
6F
5)
3 system, it was shown that a small portion (<0.01%) of titanium is occasionally present during polymerization as a complex of Ti
3+, thereby suggesting that a contribution of the Ti
3+ species to the catalytic process is unlikely. Different Ti
4+ species are identified using
13C- and
1H-NMR spectroscopy, and the portion of ESR-active Ti
3+ species in the Cp*TiCl
3/MAO system is evaluated as <1% [
9].
Thus, most studies published so far are related to the model catalysts and only the XPS method was used in studying the Ziegler–Natta polymerization catalysts. In this paper, we present details of characterization of the Ziegler–Natta polymerization catalysts by a complex of surface- and bulk-sensitive methods (DRIFTS, XPS, ESR, and XAS).
2. Results and Discussion
2.1. DRIFTS
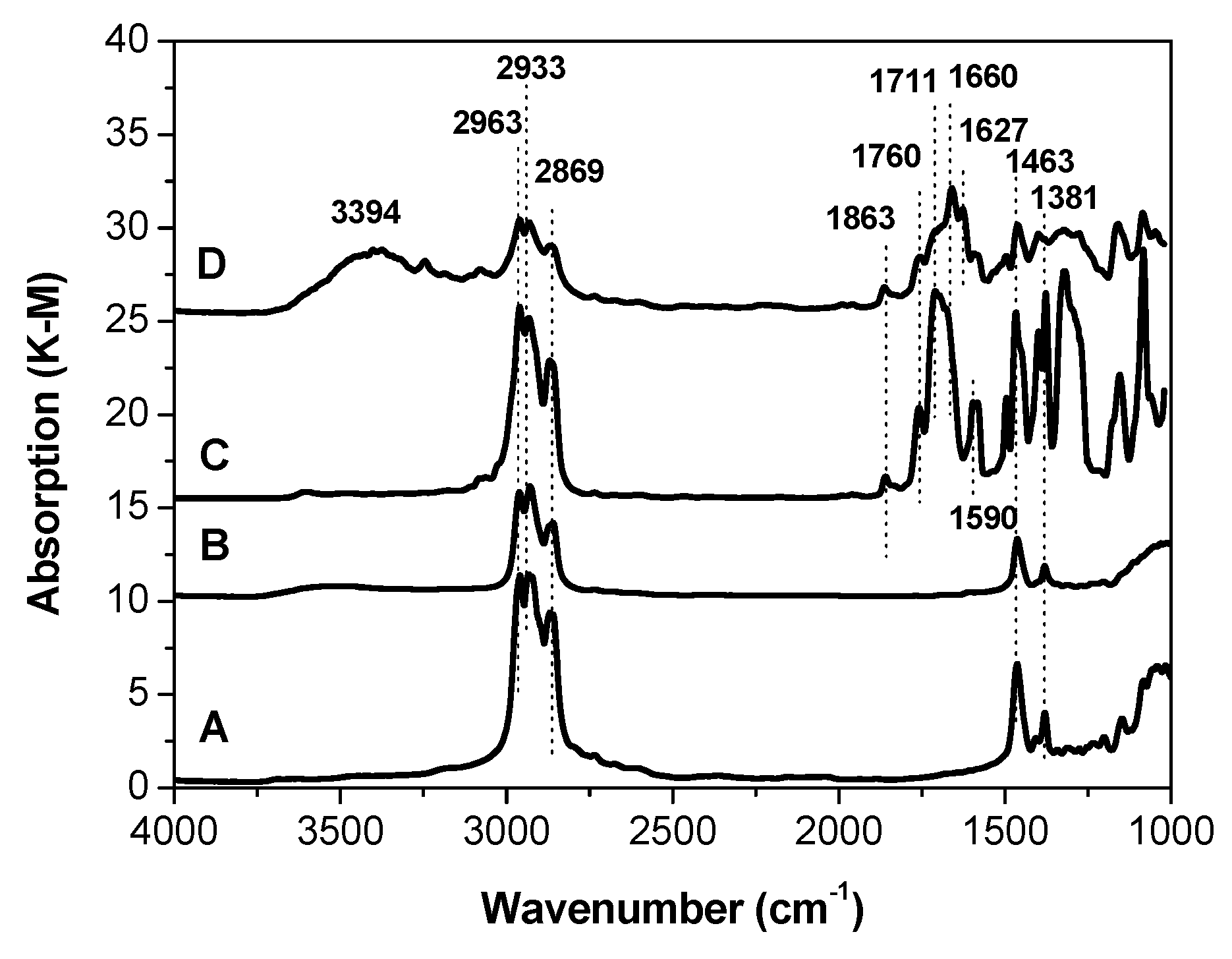
Figure 1 presents the DRIFT spectra of the starting samples after evacuation and before the adsorption of probe molecules. The spectra of A carrier and B catalyst contain absorption bands assigned to the alkyl fragments, presumably formed at the stage of MgCl
2 synthesis. The bands at 2963–2869 cm
−1 can be attributed to C–H stretching vibrations in CH
2 group, whereas the bands at 1463–1381 cm
−1 belong to the bending vibrations of the CH
2 and CH
3 groups [
21].
The IR spectra of the C and D catalysts are more complicated: they contain bands characterizing the presence of absorption due to the modification of the catalysts with internal donors and a reduction agent. In addition, in the spectrum of sample D, a broad absorption band centered at about 3400 cm
−1 is observed [
22]. This band may be assigned to the stretching vibrations of OH groups that are hydrogen-bonded with some ligands.
OH groups, most probably caused by a hydrolysis of the reduction agent (Al(C
2H
5)
2Cl), are seen in sample D exclusively. The absorption band at 1627 cm
−1 can be assigned to the complex of (Al(C
2H
5)
2Cl) with carboxyl group of DEP [
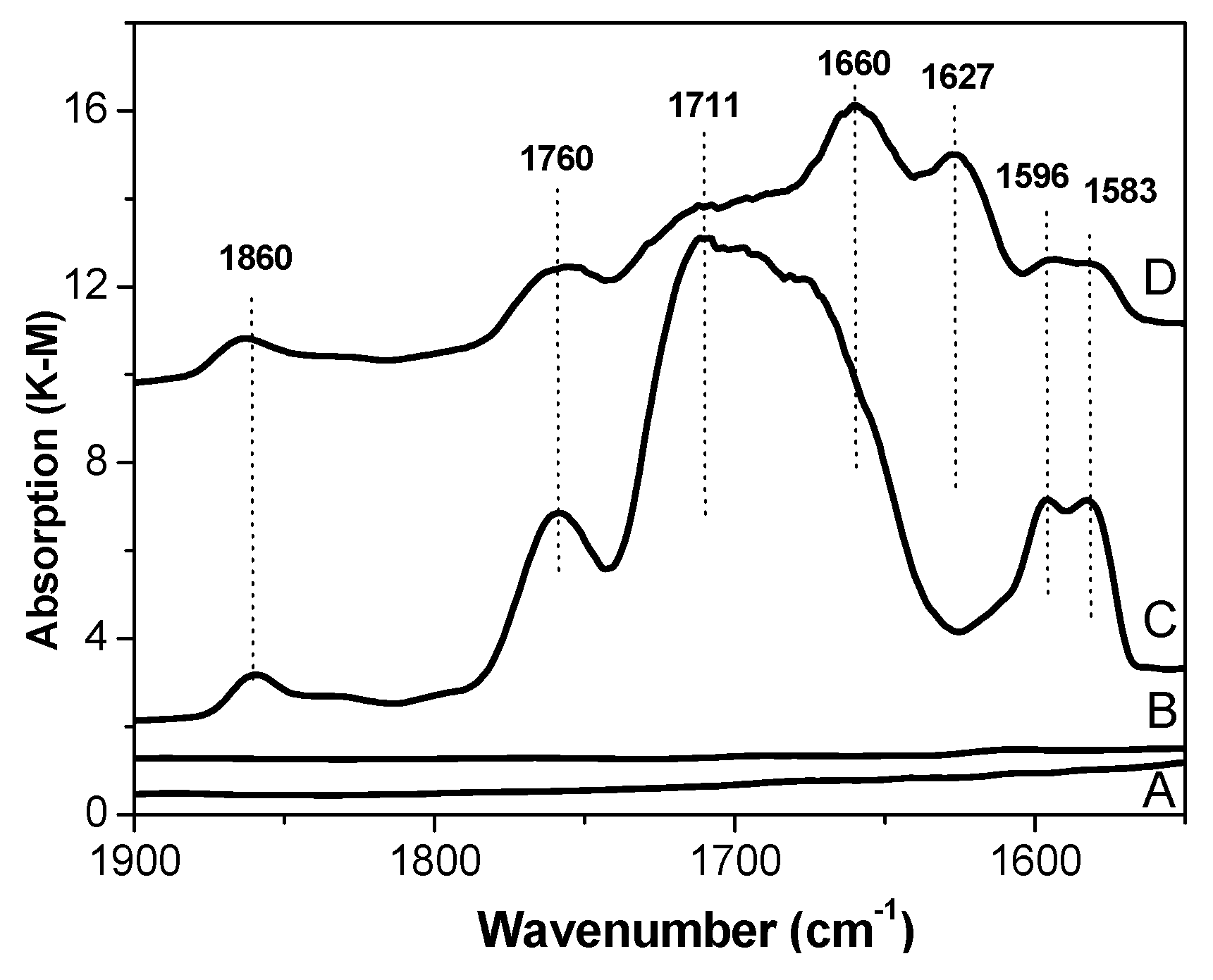
23]. In contrast to support (A) and catalyst (B) in IR spectra of C and D catalysts (
Figure 2), a few absorption bands belonging to stretching vibrations of carbonyl groups were observed [
24]. The doublet band at 1596–1583 cm
−1 most probably belongs to (TiCl
4DEP)
2 and (TiCl
4DEHP)
2 surface complexes. The wide band near 1700 cm
−1 may be assigned to MgCl
2DEP and MgCl
2DEHP, whereas the band at 1660 cm
−1 may be assigned to AlEt
2Cl/DEHP surface complexes. The bands at 1760 and 1860 cm
−1 observed in spectra of catalysts C and D are seemingly characteristic of physisorbed DEP and DEHP and/or their complexes with MgCl
2, as proposed in the literature [
25].
The CO adsorption at room temperature produces no changes in the spectra. This effect was observed for all the samples under study and indicates that strong low-coordinated Lewis acid sites capable of adsorbing a weak base like the CO molecule are not present at the surface of the catalysts and carrier. Heating of the samples at 85 °C in the presence of CO did not produce any changes in the IR spectra either.
The IR spectra of the catalysts measured after adsorption of d
3-acetonitrile are shown in
Figure 3. It is seen that the adsorption of a stronger base (acetonitrile compared to CO) leads to the appearance in the spectra of all the samples of the absorption bands characteristic for С≡N stretching vibrations (2299–2306 cm
−1) due to the formation of complexes with the so-called “shielded” Lewis acid sites (inaccessible to weak bases but accessible to strong bases due to the possibility of the latter changing the coordination sphere of the metal ions) [
26,
27,
28]. In addition, the absorption bands attributed to unperturbed (physical adsorption) С≡N and С–D stretching vibrations (2248–2245 сm
−1and 2109 сm
−1, respectively) are observed.
Comparison of the spectra of adsorbed d3-acetonitrile shows that stronger Lewis acid sites are observed for sample D. The shift of the frequency of the C≡N stretching vibrations for these catalysts is 53 cm−1 relative to the gas phase frequency (2253 cm−1). Somewhat weaker Lewis acid sites are present in the B and C catalysts, with the corresponding shifts being equal to 49–50 cm−1. The weakest Lewis acid sites (only Mg2+ cations) were found at the surface of the carrier (sample A). The С≡N frequency shift for this sample is 46 cm−1. It should be noted that sample C is most likely characterized by a rather broad distribution of the Lewis acid sites of different strengths, since a broadened absorption band is observed in the IR spectrum of this catalyst in the region of the C≡N stretching vibrations. The Lewis acid sites in the B, C, and D samples are Mg2+ together with Tin+ (n = 4, 3, 2) cations.
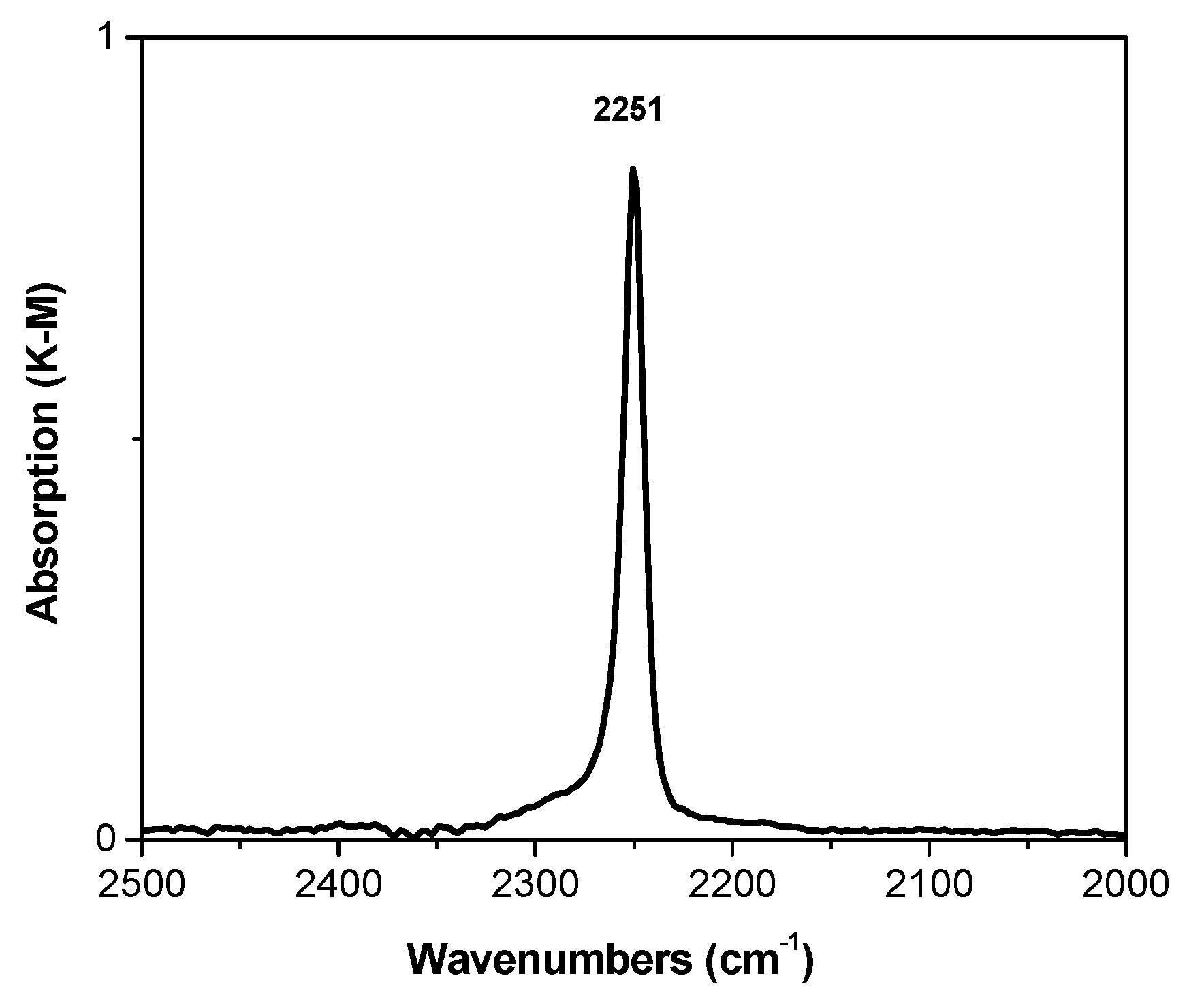
Figure 4 shows the spectrum of sample D, measured after adsorption of CDCl
3 at room temperature. The spectrum contains only one band attributed to the stretching vibrations of the C–D bond at 2251 сm
−1. This band is shifted by some 12 cm
−1 to lower frequencies as compared to the CDCl
3 frequency in the gas phase (2263 cm
−1) [
29]. From these data, it follows that no strong basic sites exist in this propylene polymerization catalyst; only moderate-strength sites are present in a significant concentration.
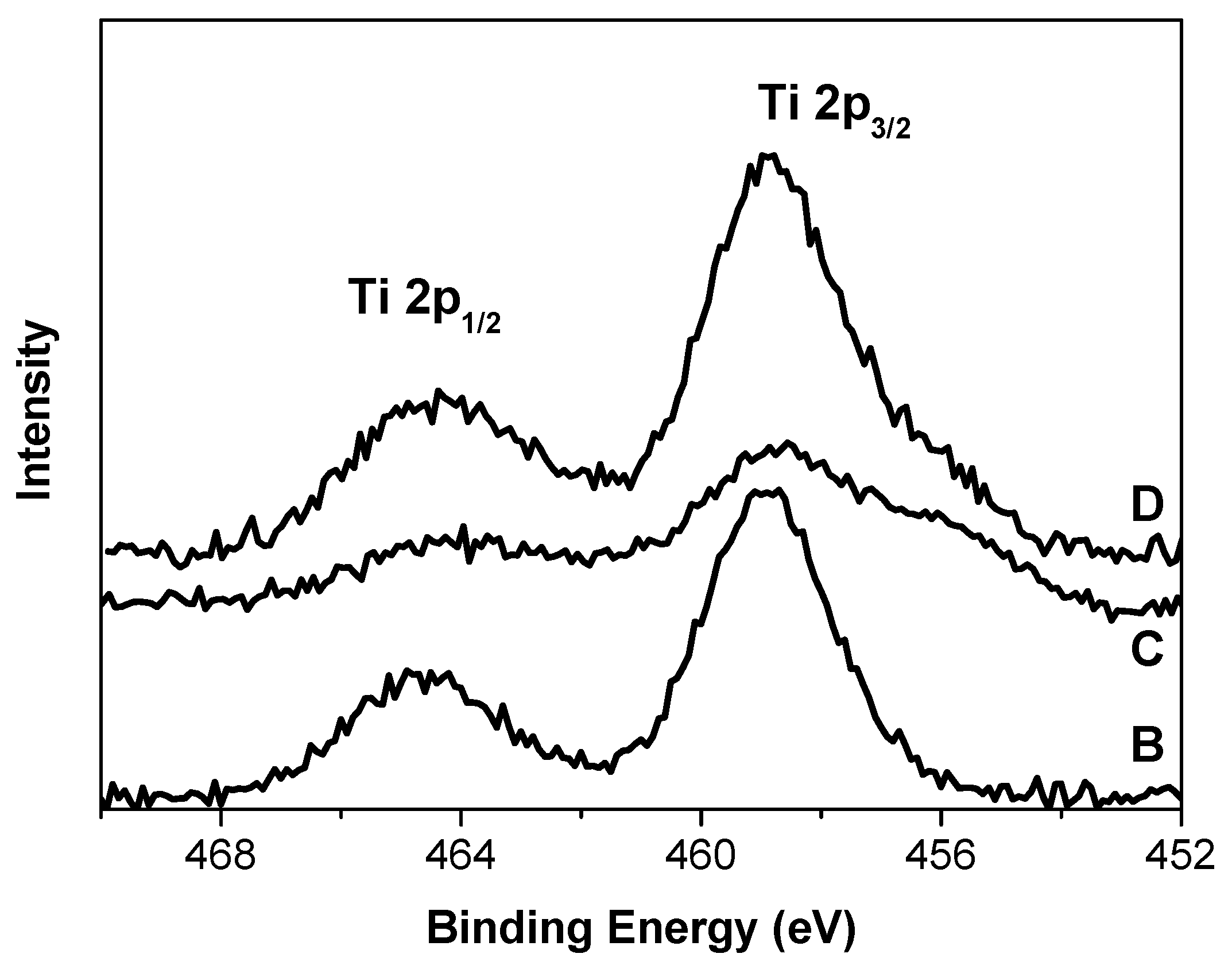
2.2. XPS
X-ray photoelectron spectra of the Ti 2p region observed for the B, C, and D catalysts are shown in
Figure 5. In
Table 1, the positions and full widths at half maximum (FWHM) of the XPS peaks are collected, whereas
Table 2 summarizes the surface atomic ratios.
XPS survey spectra show the presence of O, C, Mg, Cl, Ti, and Al atoms in the surface layers of the B, C, and D catalysts. The electronic state of oxygen in the carrier (A) and catalysts (B and C) is about the same (Е
b O 1s = 532.4–532.7 eV). The FWHM of XPS peaks is almost identical (3.1–3.3 eV) (
Table 1). It should be noted that the O 1s line in the XP spectra of sample D is broadened compared to sample C (3.6 vs. 3.2 eV) and Е
b O 1s is lower (531.8 vs. 532.4 eV). This fact can be accounted for by the presence of different oxygen-containing internal donors at the surface of these two catalysts and the presence of water in sample D (see
Figure 1 and
Figure 2).
The electronic state of chlorine is about the same in samples A and B (Еb Cl 2p = 199.7–199.8 eV). A somewhat higher electron density on the chlorine atoms was found in samples C and D (Еb Cl 2p = 199.2–199.5 eV). The FWHM of the Cl 2p line is the smallest for sample B (3.2 eV), whereas for the other samples it was 3.4 eV.
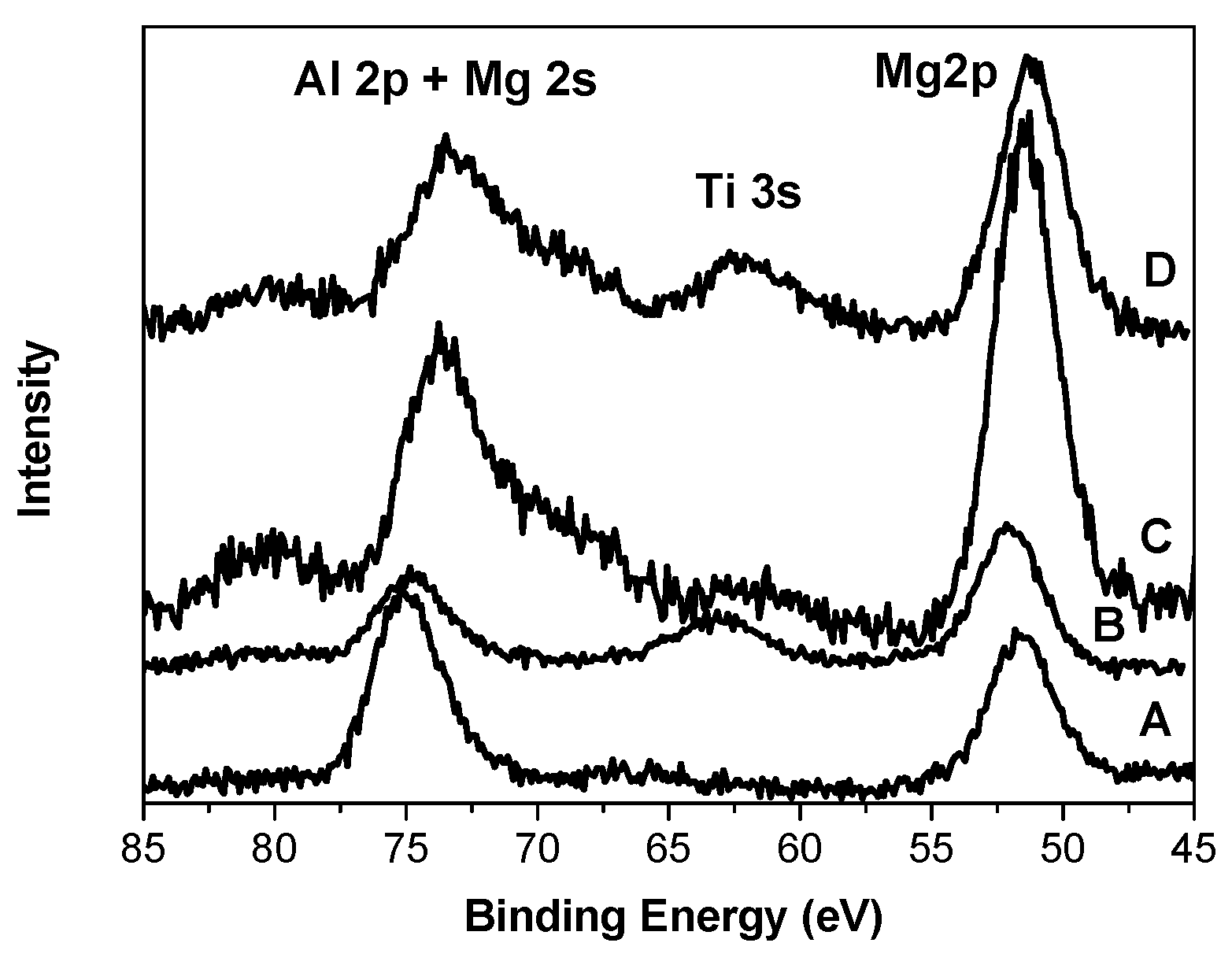
Figure 6 demonstrates XP spectra in the region of Al 2p, Mg 2s, Ti 3s, and Mg 2p electrons. The XPS lines of the Al 2p and Mg 2s electrons overlap. The presence of titanium compounds in sample B leads to a change in the state of magnesium and aluminum compared with the carrier (sample A) containing these two components only. The binding energy of Mg 2p electrons increases, whereas that of Al 2p electrons decreases (
Table 1). The presence of the internal donors in two other samples results in an electron density increase on Mg in the C and D samples (Е
b Mg 2p = 51.4–51.2 eV). The binding energy of Al 2p electrons in both C and D samples decreases significantly as well. In addition, the Al 2p line in the XP spectra of samples C and D is wide (3.6–4.0 eV), while the width of the Mg 2p line is nearly the same (2.8–3.0 eV).
It is seen in
Figure 5 that the Ti 2p line in the XP spectrum of sample B represents a narrow symmetric doublet with symmetrical components. On the contrary, the components of the Ti 2p doublet in the spectra of two other samples are broadened. The Ti 2p spectrum of sample C explicitly demonstrates the additional inhomogeneity as shoulders in the region of lower binding energies. From the position of the maxima of the Ti 2p
3/2 line in the spectra of the catalysts and the width of these lines we assume the presence of one or several states of titanium in the surface layers of the catalysts available for XPS analysis. In the literature, there are no significant differences in the binding energies (B.E.) of the Ti 2p
3/2 lines in the XP spectra of complexes of Ti
4+ (or Ti
3+) and solid compounds containing Ti
4+ (or Ti
3+) [
8,
10,
30,
31,
32]. The position and shape of the Ti 2p
3/2 line in the XP spectrum of sample B (E
b = 458.9 eV) and its FWHM (2.3 eV) (
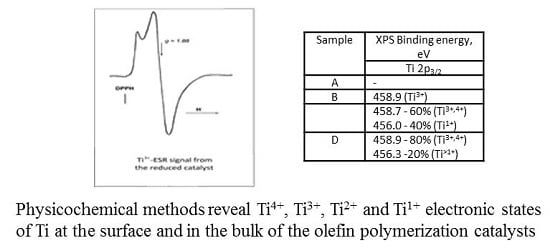
Table 2) show that both Ti states may be possible. Further differentiation by ESR study (see below) allowed us to identify the state of titanium in this sample as Ti
3+. The mathematical treatment of Ti 2p spectra of samples C and D (
Figures S1 and S2, supporting information) shows that titanium in this oxidation state is also present at the surface layers of C (about 60%) and D (about 80%) samples. The rest of titanium in the C and D samples is identified as a Ti species with a lower positive charge compared to sample B. Part of titanium can be identified as Ti
2+ according to [
33]. In addition, the Ti centers in sample C have a large electron density. The higher B.E. of the Ti 2p
3/2 lines found in the XP spectra of sample D might be caused, according to [
9], by the presence of H
2O in this sample. The presence of water at the surface of this sample was confirmed by DRIFT (see
Figure 1).
Table 2 summarizes the atomic ratios of the components with respect to magnesium, which is present in all these samples. There is some chlorine deficit at the surface of the starting MgCl
2 sample (the Cl/Mg ratio is equal to 0.78 instead of 2). It is seen that the atomic ratio Ti/Mg is higher in the surface layers of the MgCl
2 + TiCl
4 sample (B) as compared to the other samples under study.
2.3. ESR
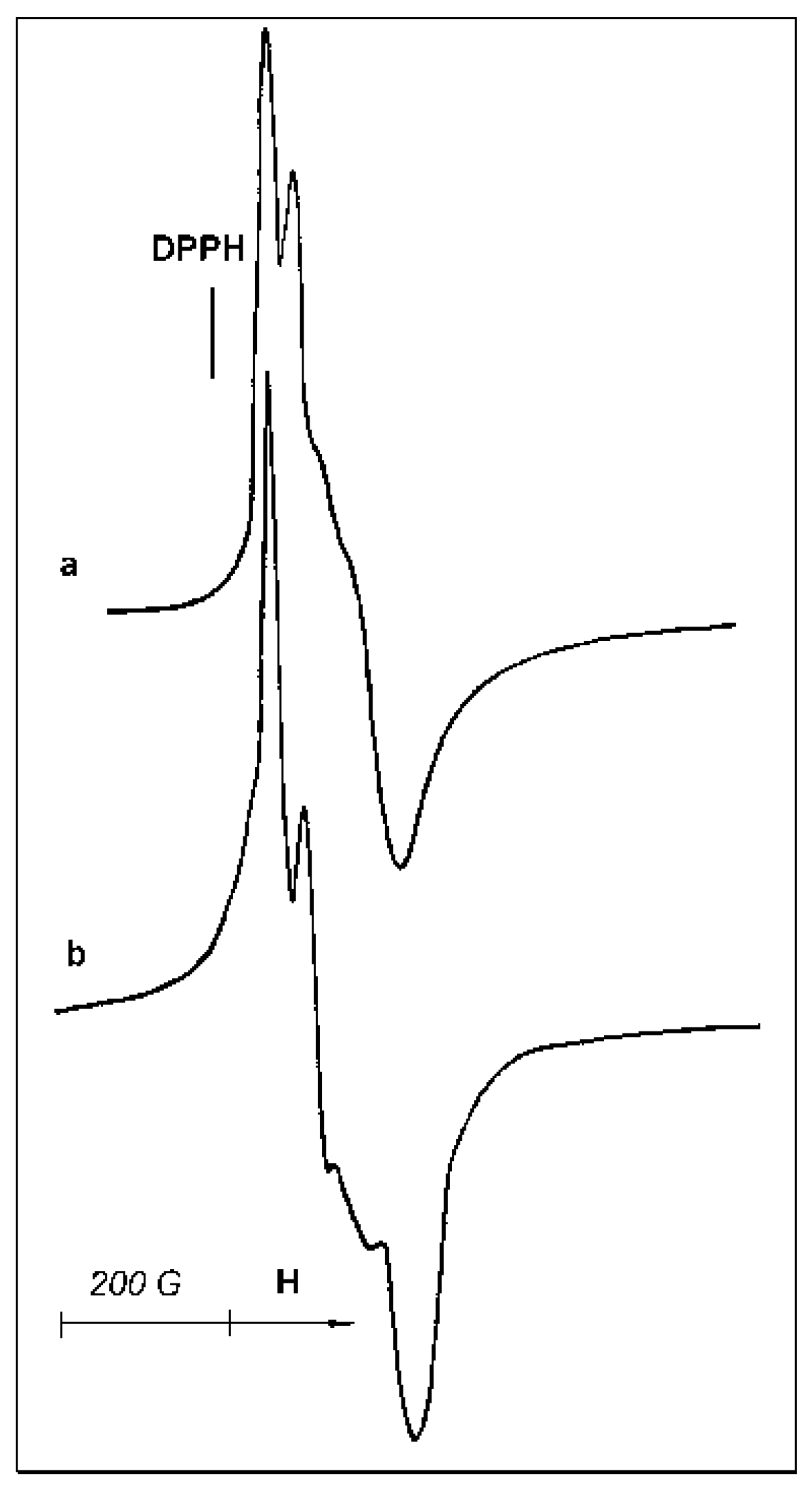
In ESR testing of samples, paramagnetic Ti
3+ ions located both on the surface and in the volume of catalyst particles are detected. However, peculiarities of sample reoxidation or interactions with ligand molecules allow us to draw some conclusions about the surface/bulk location of paramagnetic sites. The starting sample B demonstrates a rather strong Ti
3+ ESR signal at room temperature (
Figure 7a). CO adsorption causes only a minor change in the signal shape (
Figure 7b).
Acetonitrile adsorption on this catalyst is accompanied by more pronounced changes in the signal shape and asymmetry (
Figure 8a), demonstrating a measurable transformation of the paramagnetic site symmetry. Subsequent evacuation of the sample at 50 °C does not lead to the restoration of the parent ESR signal (
Figure 7a) but causes only minor changes of the signal shape (
Figure 8b), confirming that acetonitrile molecules are strongly bonded in the complex formed.
Interaction of sample B with air is accompanied by the appearance of a very weak narrow ESR signal at g = 2.003, indicative of the formation of a small amount of O
2− species. However, the low intensity of this signal casts into doubt the relationship between the newly formed O
2− species and the existing Ti
3+ paramagnetic sites. Moreover, the exposure of sample B to air does not cause an immediate disappearance of the ESR signal of paramagnetic Ti-sites; rather, it begins a slow decay of the signal (
Figure 7a) that lasts for hours. Therefore, a considerable portion of ESR-visible Ti
3+-ions seems to be located inside the bulk of the active phase.
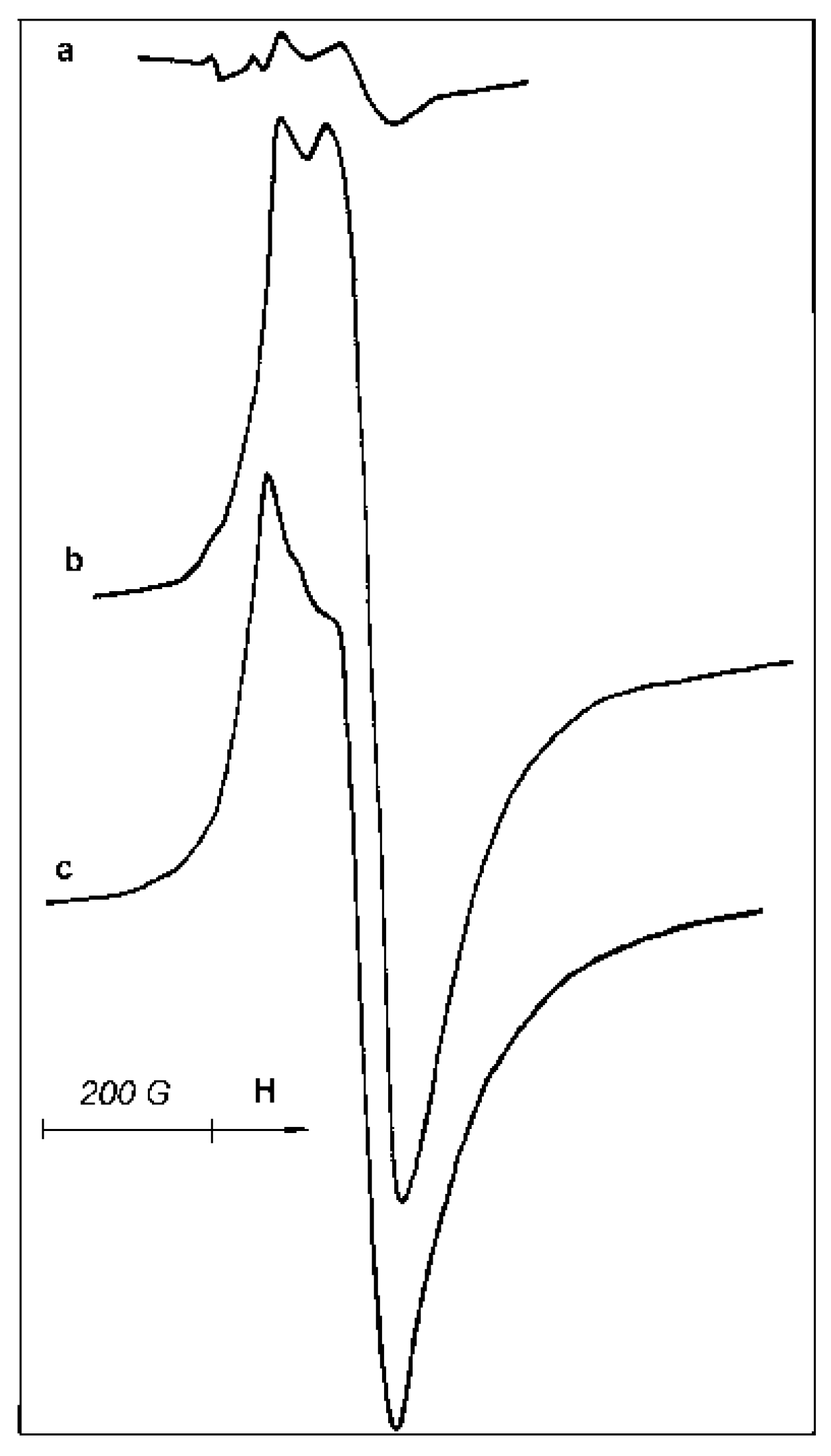
Two ESR signals in samples C and D are presented in
Figure 9a,b, respectively. Comparison of these signals, taken at identical magnifications from two identical probes, illustrates a sharp difference in the concentration of paramagnetic sites in these catalysts.
No changes in the ESR signal (
Figure 9b) take place as a result of CO adsorption on sample D. Rather insignificant irreversible transformation of the signal shape is observed after acetonitrile adsorption (
Figure 9c). Surprisingly, the catalyst is very resistant to air exposure at 20 °C: after 16 h of exposure to air, the ESR signal (
Figure 9b) of paramagnetic Ti-sites retains ~75% of the original intensity. Once more, a main part of paramagnetic Ti
3+-sites detected in sample D by ESR is located in hindered positions inside catalyst particles and takes no part in the interaction with gas phase molecules.
Two ESR signals, taken at −196 °C, for samples B and D are presented in
Figure 10a,b. It is clearly seen that the signals differ in their shape. Thus, the catalysts differ in the coordination/structure of the paramagnetic sites. Double integration of the two spectra permits us to conclude that the number of paramagnetic sites in sample D is maximal and exceeds the concentration of such centers in sample B by a factor of ~3.5.
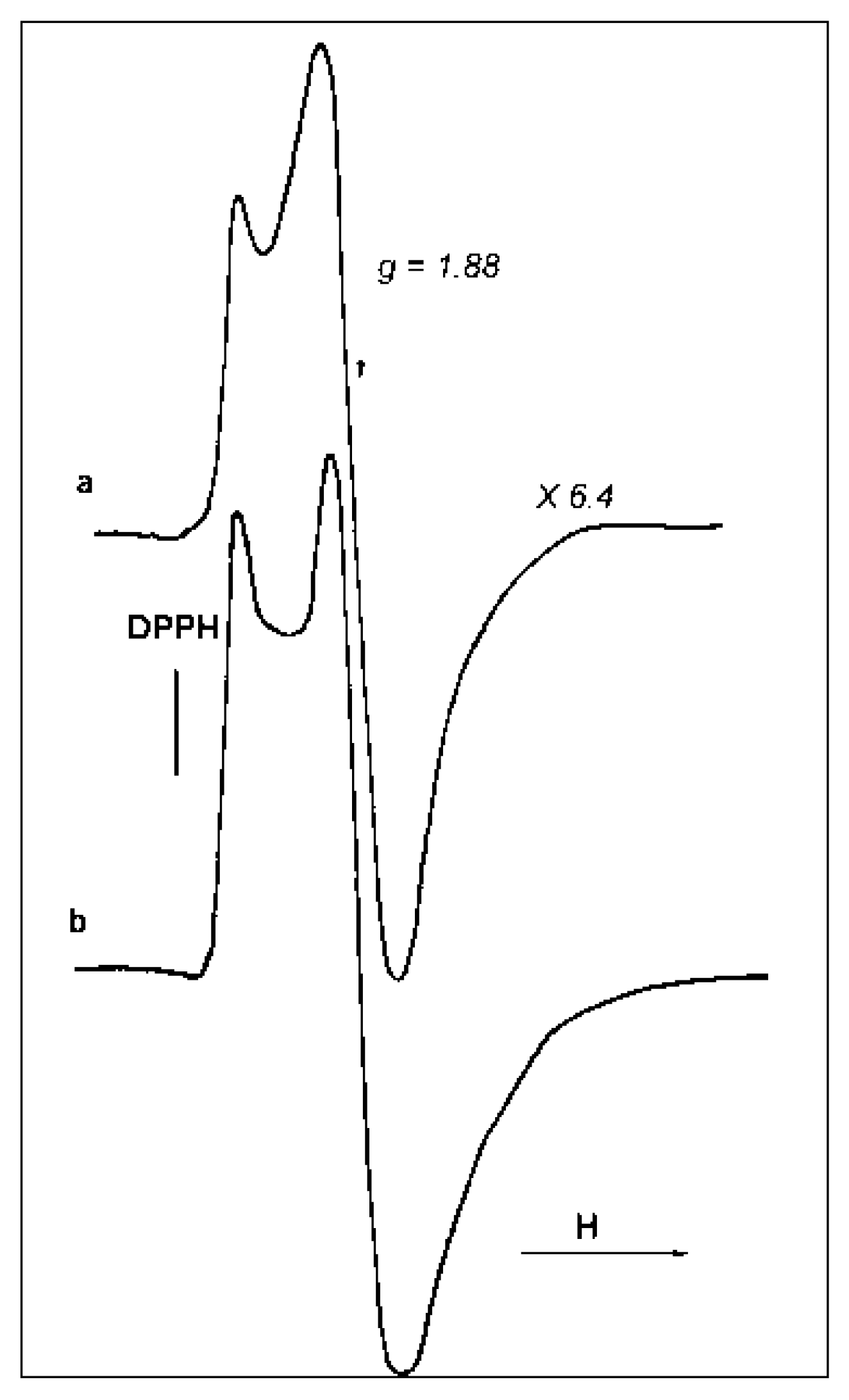
The ESR spectrum of sample D is compared with a standard (frozen TiCl
3 solution) taken as a reference for the evaluation of the number of “ESR-visible” Ti
3+-sites in the samples (
Figure 11). According to this evaluation, the number of paramagnetic centers in the catalyst D reaches 1–1.5% (wt % of Ti). Taking into account that the total Ti concentration in the samples averages 2–4 wt %, one can conclude that the considerable part (~50%) of Ti in D catalyst forms isolated paramagnetic sites. From the identity of the signal shapes, one can suppose that paramagnetic sites in this catalyst resemble isolated Ti
3+ species in the frozen TiCl
3 solution.
2.4. XAS
The Ti
K-edge normalized XANES spectra of the catalysts and the reference samples are depicted in
Figure 12. It is known that the pre-edge features of transition metals are related to the coordination number, oxidation state of adsorbing atom, and symmetry of the adsorbing atom site [
34,
35,
36]. The pre-edge features in the spectra of the Ziegler–Natta catalysts are similar to each other, but they differ from those of reference compounds: Ti-foil, TiO
2 rutile, TiCl
3, TiCl
4, and Ti(C
3H
7O)
4. There is one narrow peak in the spectra of the Ziegler–Natta polymerization catalysts, in contrast to three of them in the TiO
2 spectrum. There is one pre-edge peak in the spectrum of TiCl
4 as well, but the pre-peak normalized height of the samples under study is around 0.25, whereas the normalized height of the pre-peak for TiCl
4 is about 0.55 (
Figure S3, Supplementary Materials). Moreover, the pre-edge peak in the TiCl
4 spectrum is located at a lower energy (0.6 eV) compared with that in the spectra of the Ziegler–Natta catalysts under study. There are two pre-edge peaks in the spectrum of TiCl
3 and the position of the main peak is shifted to a higher energy (0.5 eV). There is one pre-edge peak in the spectrum of tetrabutoxy titanium located at the same energy as in the spectra of the Ziegler–Natta catalysts. Thus, the fingerprint approach indicates the absence of free TiCl
4 tetrahedral clusters in the Ziegler–Natta polymerization catalysts. It indicates that the Ti species in these catalysts are definitely not Ti
4+ ions surrounded by six oxygen atoms and not Ti
3+ ions surrounded by three chlorine atoms.
The position of the Ti K-edge in the spectra of the polymerization catalysts is lower than that in TiO2, TiCl3, and Ti(C3H7O)4 but similar to that in the spectrum of TiCl4. However, the shape of XANES and the height of the white line in the spectra of samples C and D differ from TiCl4. The spectrum of sample B differs from TiCl4 in the position and height of the pre-edge peak. This means that Ti in the studied Ziegler–Natta polymerization catalysts, perhaps, exists as a mixture of Ti4+, Ti3+, Ti2+, and/or Ti1+ electronic states. These results are in good agreement with the conclusions drawn from the XPS data (see above).
Furthermore, one can deduce by analyzing the position of the Ti K-edge and the intensity of the white line that the Ti species in samples C and D have, on average, a more electron-deficient character (surrounded by more electron-accepting neighbors), whereas sample B demonstrates a lower electron-deficient character (more electron-donating neighbors). The difference in the edge position can indicate the difference in the bond ionicity of Ti with nearest neighbors as well. In this case, the average bond ionicity is higher in the case of samples C and D and is lower in sample B.
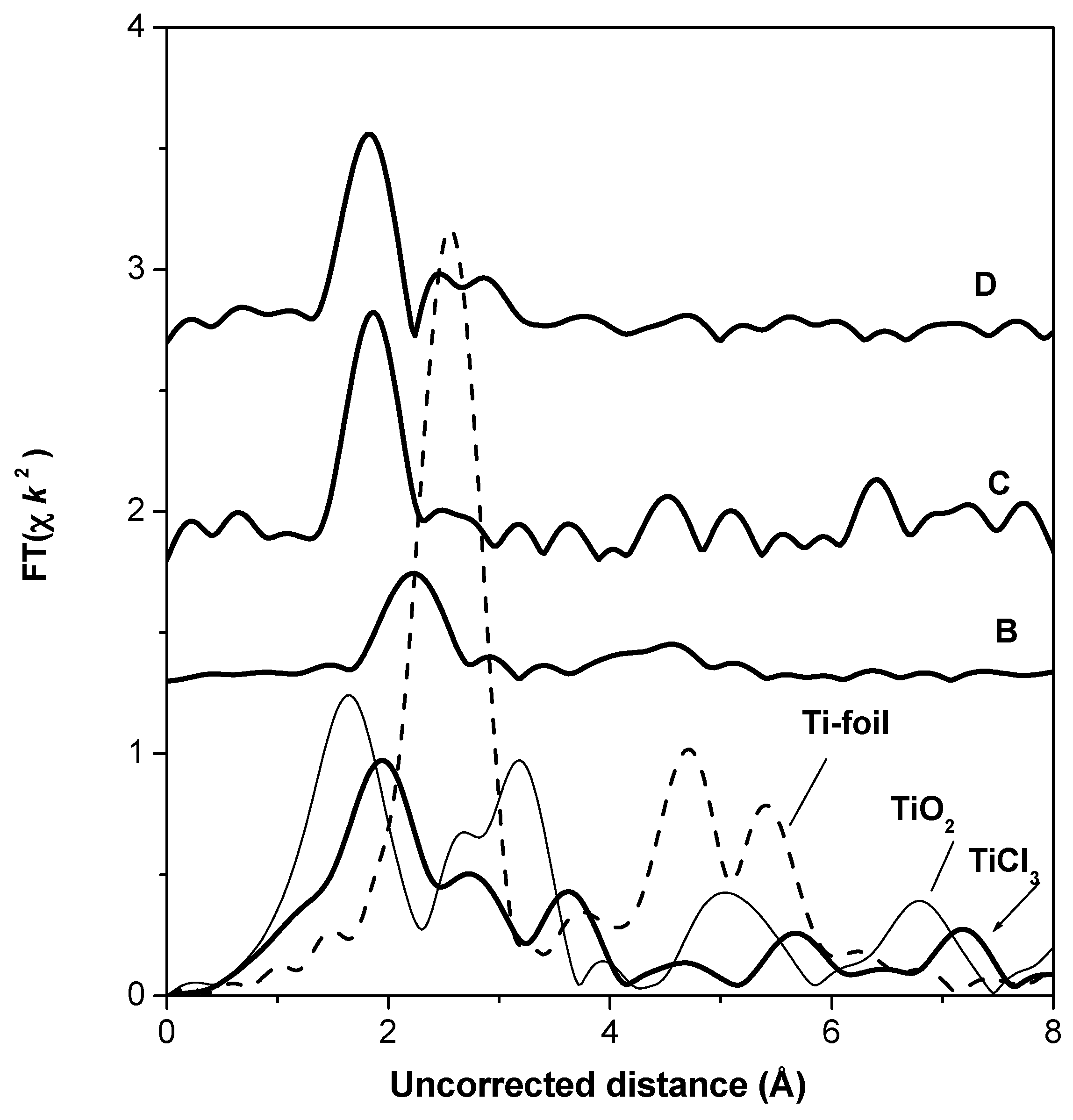
The Fourier transformations of XAS oscillations for the Ziegler–Natta polymerization catalysts and references are presented in
Figure 13. EXAFS oscillation of Ti(C
3H
7O)
4 reference compound is too noisy; therefore, FT is uncertain and is not shown. One peak is observed in the spectra of the B, C, and D catalysts. The position of this peak is near 2.2 Å uncorrected distance in the spectrum of sample B, whereas it is near 1.8 Å in the spectrum of the other two catalysts.
In the spectrum of TiO2, the first peak corresponds to the coordination shells containing O atoms at about 1.6 Å uncorrected distance, whereas in the spectrum of TiCl3 the first peak corresponds to the coordination shell containing Cl atoms.
The position of the first peak in the spectra of catalysts shows the possibility of the Ti–Cl atomic pair presence in the first coordination shell. The first peak of the Ti FT EXAFS spectra was fitted in both r- and k-spaces with a one-shell model—a chlorine shell around the central absorbing titanium atom (
Figure S4, Supplementary Materials). The results of the model fit are presented in
Table 3. It is seen that the first shell contains one Cl atom at the real distance 2.25 Å in sample C. The real distance in the Ti–Cl atomic pair in sample D is considerably shorter (2.23 Å), than in sample B (2.54 Å).
3. Materials and Methods
Four samples: A—MgCl
2 containing catalyst support; B—MgCl
2-based TiCl
4 catalyst of ethylene polymerization; С and D—MgCl
2-based TiCl
4 catalysts with the internal donor for propylene polymerization, were studied (
Table 4).
Diffuse-reflectance Fourier-transform infrared spectra of the polymerization catalysts were recorded at room temperature using a NICOLET “Protege” 460 spectrometer with a homemade diffuse-reflectance unit [
40]. To obtain a satisfactory signal-to-noise ratio, 200 scans were collected per spectrum. The spectra were measured from 400 to 4000 cm
−1 with a resolution of 4 cm
−1. The powdered samples of the catalysts were placed in the ampoules using a glove box under a dried Ar atmosphere. The same quartz ampoules were used for ESR measurements. The probe molecules CO (1.3 kPa), CD
3CN (12 kPa), and CCl
3D (18.7 kPa), were adsorbed at room temperature. Before adsorption, non-activated samples were treated in a vacuum (0.13 Pa) for 20 min at ambient temperature.
Carbon monoxide and d
3-acetonitrile were used as probe molecules for Lewis acid sites because they differ in their basicity and can differentiate between “exposed” Lewis acid sites, i.e., the sites with a low-coordinated cation exposing its electron vacancy for the interaction (like trigonal sites), and “shielded” Lewis acid sites, i.e., sites with a pseudo-saturated coordination sphere. In the latter case, a probe-molecule like CD
3CN is capable of withdrawing the cation from its coordination sphere and increasing its coordination number. For instance, when we have a MO
4 tetrahedron in which the metal ion is shielded by four oxygen atoms, adsorption of a strong ligand (probe molecule) may result in the insertion of the fifth ligand into the coordination sphere of the metal [
41]. In addition to probe molecules for acid sites and low-coordinated ions, we also used a probe molecule for basic sites, i.e., chlorine atoms of the metal chlorides forming the support and the catalyst, as well as oxygen atoms in the composition of the organic electron-donor modifiers used to activate the catalyst. For this purpose, the molecule of deuterated chloroform, as a weak acid, was chosen because of the presence of intense CH bonds on the initial catalyst surface before the adsorption of the probe molecule and thus the CH-containing probes cannot be used for this purpose. Although the background in the region of C–D vibrations was not good for reliable spectra measurement, we succeeded in collecting some data about the basic sites at the surface of the polymerization catalysts. Up to now we studied only the adsorption of CHCl
3 on various solid carriers and catalysts and some representative shifts reflecting the measure of basicity due to the surface
…O
…H–CCl
3 complexes are collected in
Table 5. Obviously, the stronger the O
…H interaction in this complex, the larger the red shift of the C–H stretching vibration, and vice versa. The corresponding shifts in the C–D region are approximately equal to the value of the C–H shift divided by √2 [
42]. For sample D the spectrum was also taken after heating in СО at 85 °С for 10 min. For the same sample, adsorption of CDCl
3 at room temperature was carried out.
XPS spectra were recorded using a XSAM 800 spectrometer with Mg Kα X-ray (1253.6 eV) source. The base pressure in the XPS chamber was about 1.3 × 10
−5 Pa. The spectrometer was calibrated using the binding energy (E
b) of the Au 4f
7/2 level = 84.0 eV and Ni 2p
3/2 = 852.7 eV. Survey spectra were collected between 20 and 1130 eV. Detailed spectra were recorded for the region of C 1s, O 1s, Mg 2p, Cl 2p, Al 2p, and Ti 2p with a 0.1 eV step. The C 1s line at 285.0 eV was used for energy calibration. The Mg 2p line (51.6 eV) was used as an internal standard for the carrier. The surface atomic composition and atomic ratio were calculated after a Shirley type background subtraction [
43] by dividing the peak area by the photo-ionization cross-sections [
44]. The experimental Ti 2p spectra have been deconvoluted in a series of mixed Gaussian–Lorentzian shape contributions.
ESR spectra were taken in the X-band (λ ≅ 3.2 cm) at 20 °C and −196 °C using a reflecting-type ESR spectrometer equipped with a 4104OR cavity and quartz Dewar vessel. The ESR signals were registered in the absence of saturation in the field range of 2000–4000 G. The Excel program was used for spectra processing (baseline correction, double integration). DPPH and a frozen solution of 8.5 wt % TiCl3 in concentrated HCl were used as standards for g-factor calculation and quantitative Ti3+ ESR analysis, respectively.
The samples of catalysts were inserted into identical quartz ampoules in a glove box for combined use in ESR study and DRIFT spectroscopy. The amount of the sample in the cylindrical ESR part of the ampoule (3.5 mm in diameter) exceeded the volume of the sample in the resonator (>30 mm height). ESR spectra were registered at 20 °C and −196 °C immediately after the DRIFT measurements. Next, closed ampoules were connected to the adsorption setup and the samples were treated with CO or d3-acetonitrile. Then DRIFT and ESR measurements at 20 °C and −196 °C were repeated. The solution of 8.5 wt % TiCl3 in (30% HCl + H2O) was used as a standard for the comparative evaluation of the “ESR-visible” fraction of Ti3+ in the samples. Double integration of the ESR spectrum of this standard provided the absolute value for comparison with double integrals of the signals under study. The shape of the ESR lines does not play any role, and the spectra simulation was not required. For the sake of accuracy, the series of the samples were measured consecutively, with ampoules placed in the same position inside the ESR resonator one after another.
X-ray absorption spectra (Ti K-edge at 4966 eV) were measured at HASYLAB E4 station (DESY, Hamburg, Germany). The X-ray beam was monochromatized with Si(111) double crystal detuned to 50% of the maximum intensity to avoid higher harmonics contamination of the monochromatized beam. The spectra μ(E) were measured at −196 °C in a transmission mode using ionization chambers. The spectrum of Ti metal foil (between the second and third ionization chambers) was recorded at the same time for energy calibration purposes.
Special care was taken to avoid air contamination. For sample preparation, a mixture of about 10 mg of the catalyst (the optimum weight to maximize the signal-to-noise ratio) and 20 mg of polyethylene powder (spectrophotometric grade, Aldrich) as a binding agent was pressed into a 13-mm diameter pellet in a glove box (a dried Ar atmosphere) and completely wrapped in Kapton polyimide tape with silicone adhesive. The aim of dilution was to provide transmission of approximately 30% of the beam. However, a good signal to noise ratio was not always achieved, because a significant portion of the beam absorption came from other atoms, such as Mg, Al, Si, and Cl. Reference spectra for Ti0 and Ti4+ oxidation states were recorded using standard reference samples (Ti-foil, TiO2 (rutile), TiCl3, TiCl4, and Ti(C3H7O)4).
Data treatment was carried out with the software package VIPER [
45]. In the spectra of the absorption coefficient μ, a Victorian polynomial was fitted to the pre-edge region for background subtraction. A smooth atomic background, μ
0(
k), was evaluated using smoothed cubic splines. The radial distribution function FT[
k2χ(
k)] was obtained by Fourier transformation of
k2-weighted experimental function χ = (μ(
k) − μ
0(
k))/μ
0(
k) performed with a Bassel window. For the determination of structural parameters, theoretical references calculated by the FEFF8.10 code were used [
46]. Duplicate spectra were recorded to ensure data reproducibility.

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}