Synthesis of Magnesium Nickel Boride Aggregates via Borohydride Autogenous Pressure




Abstract
:
1. Introduction
2. Materials and Methods
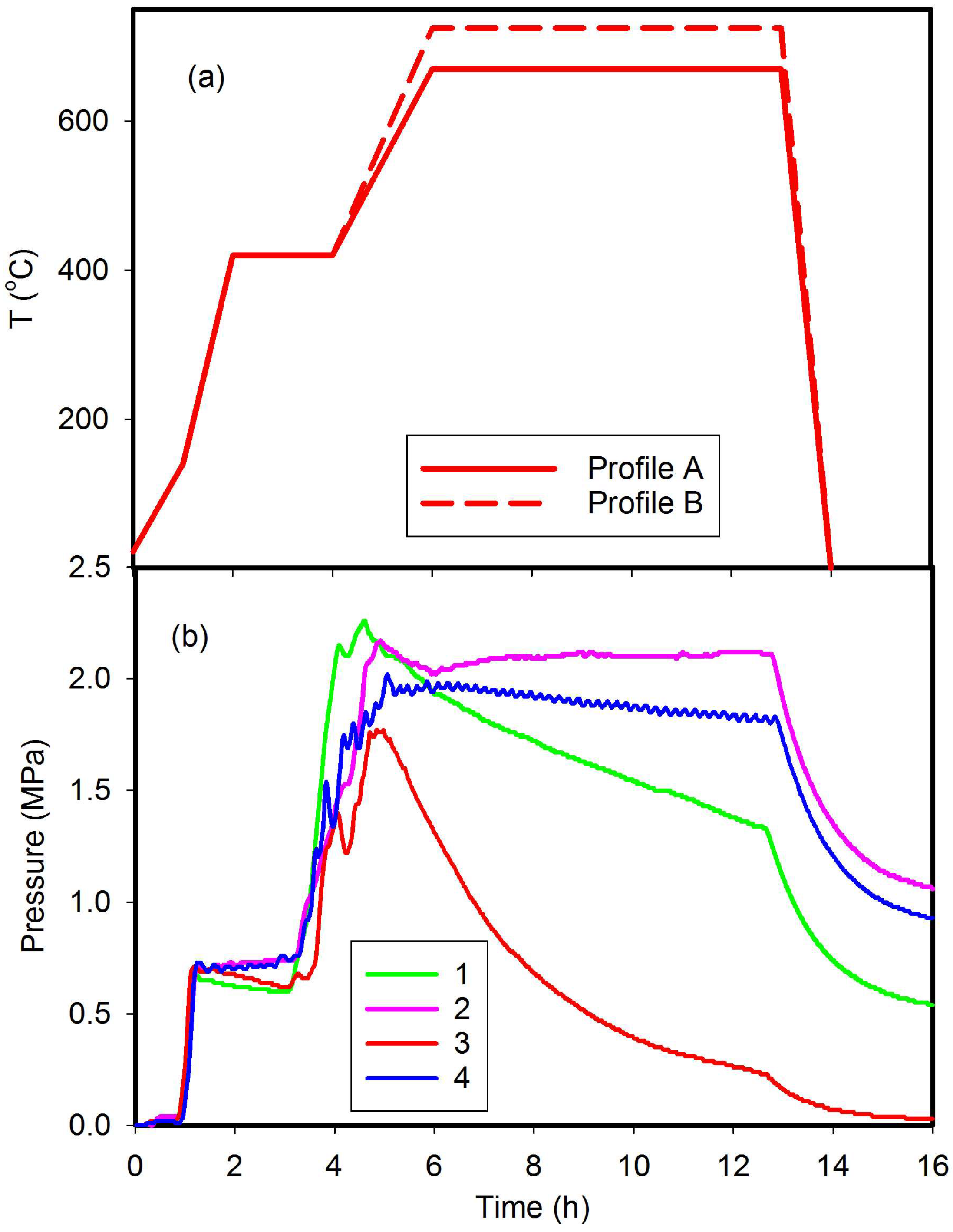
2.1. Boride Synthesis
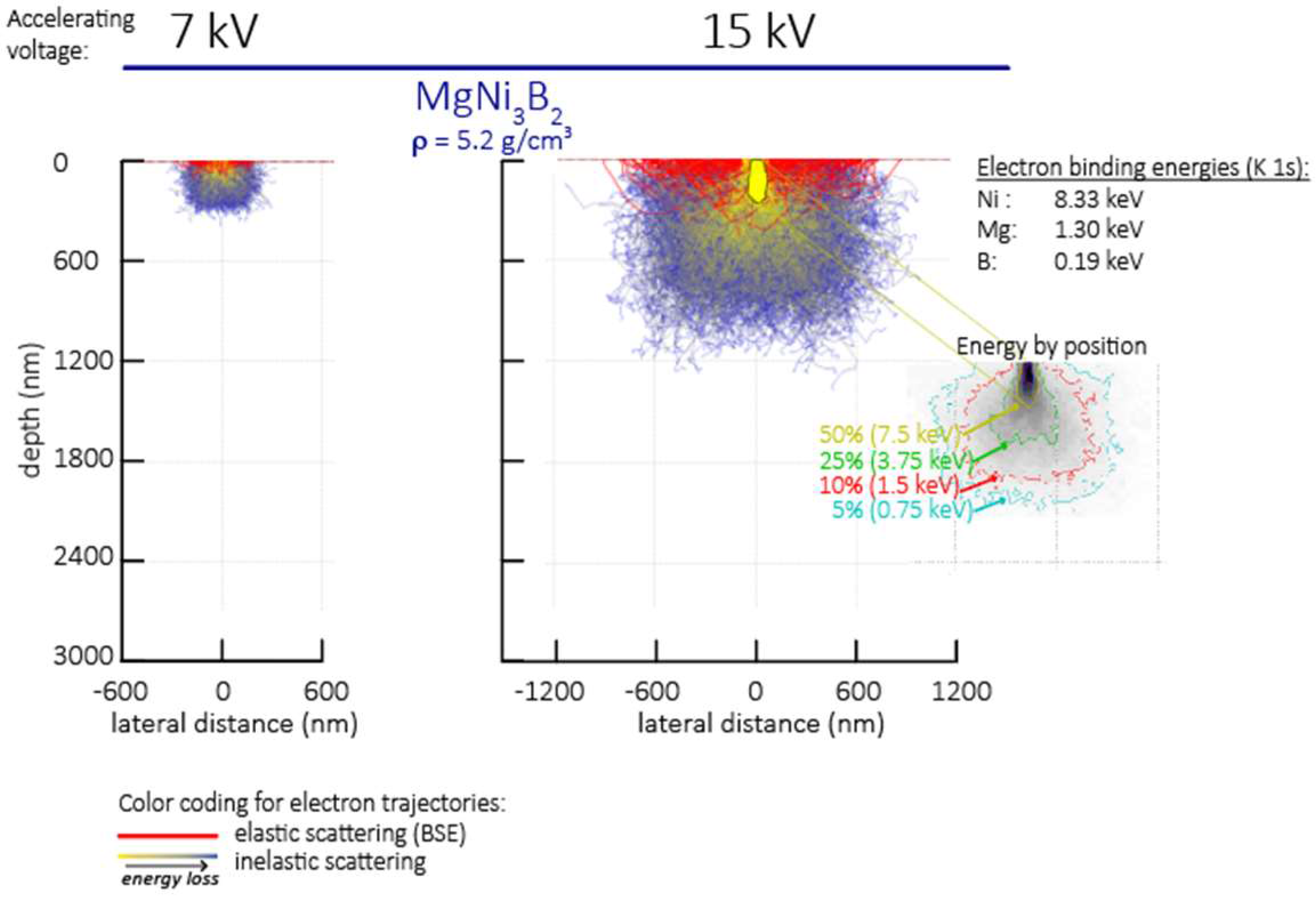
2.2. Characterisation
3. Results
3.1. Synthesis
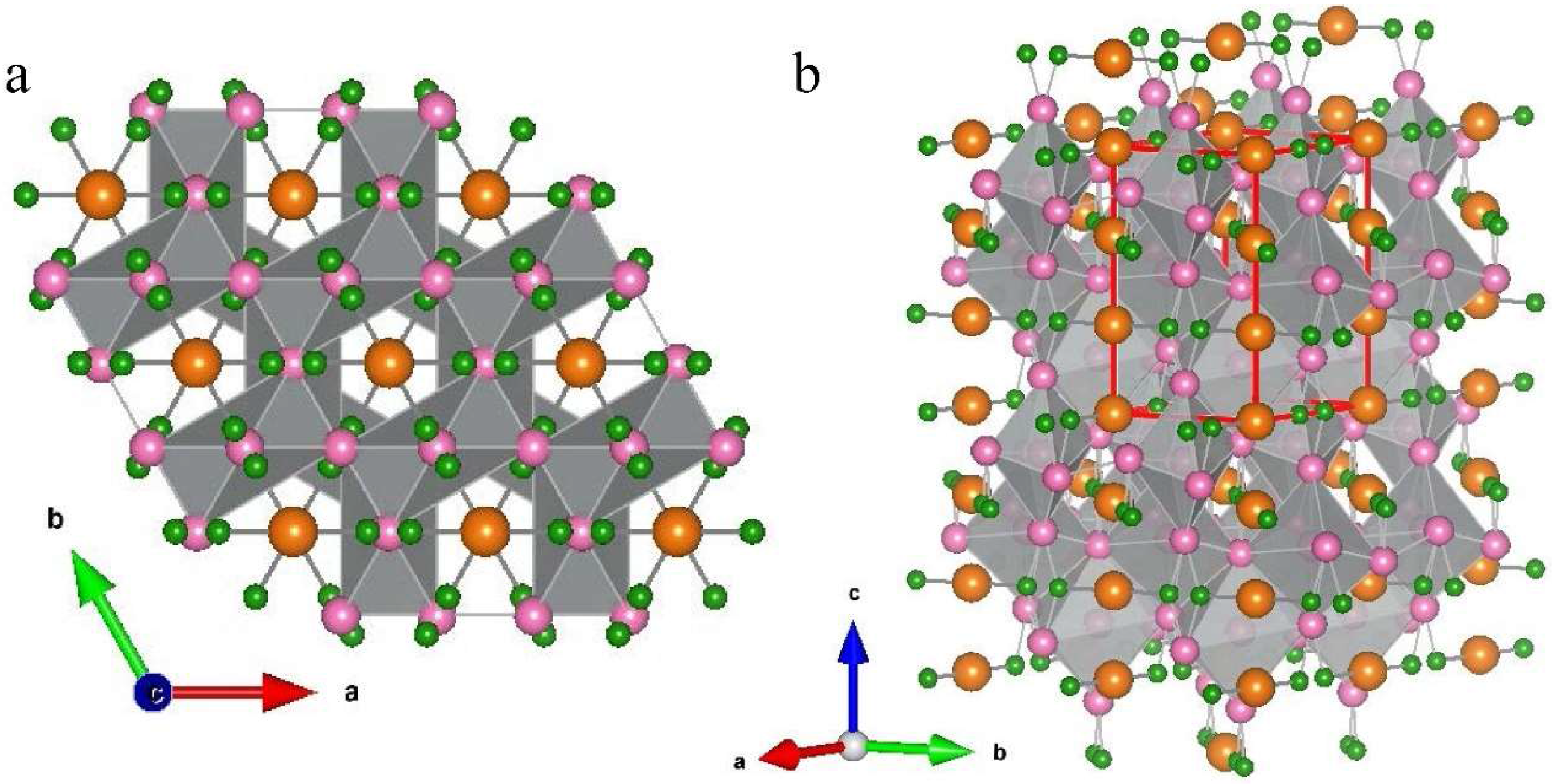
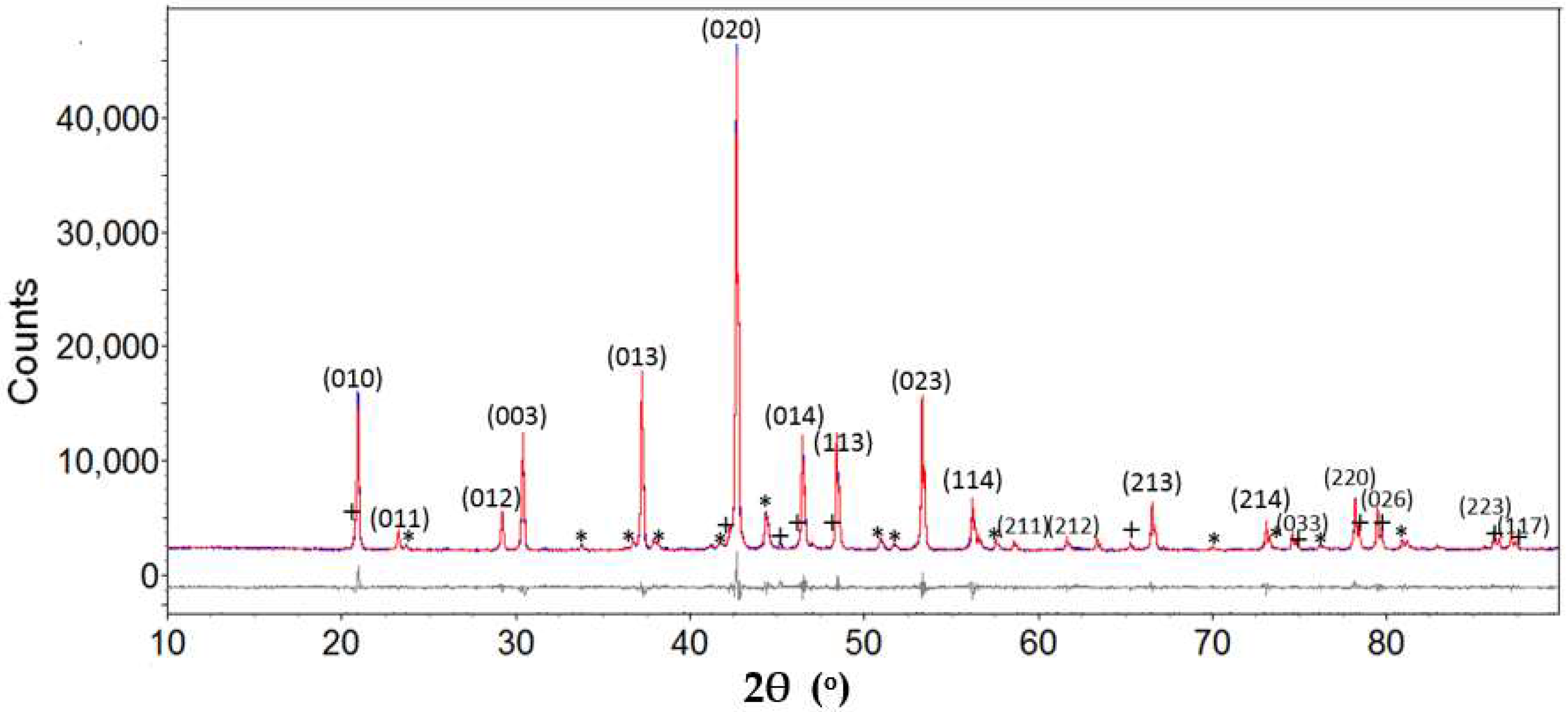
3.2. Structure Determination
3.3. Morphology and Microstructure
4. Discussion
4.1. Synthesis of MgNi3B2
4.2. Microstructures
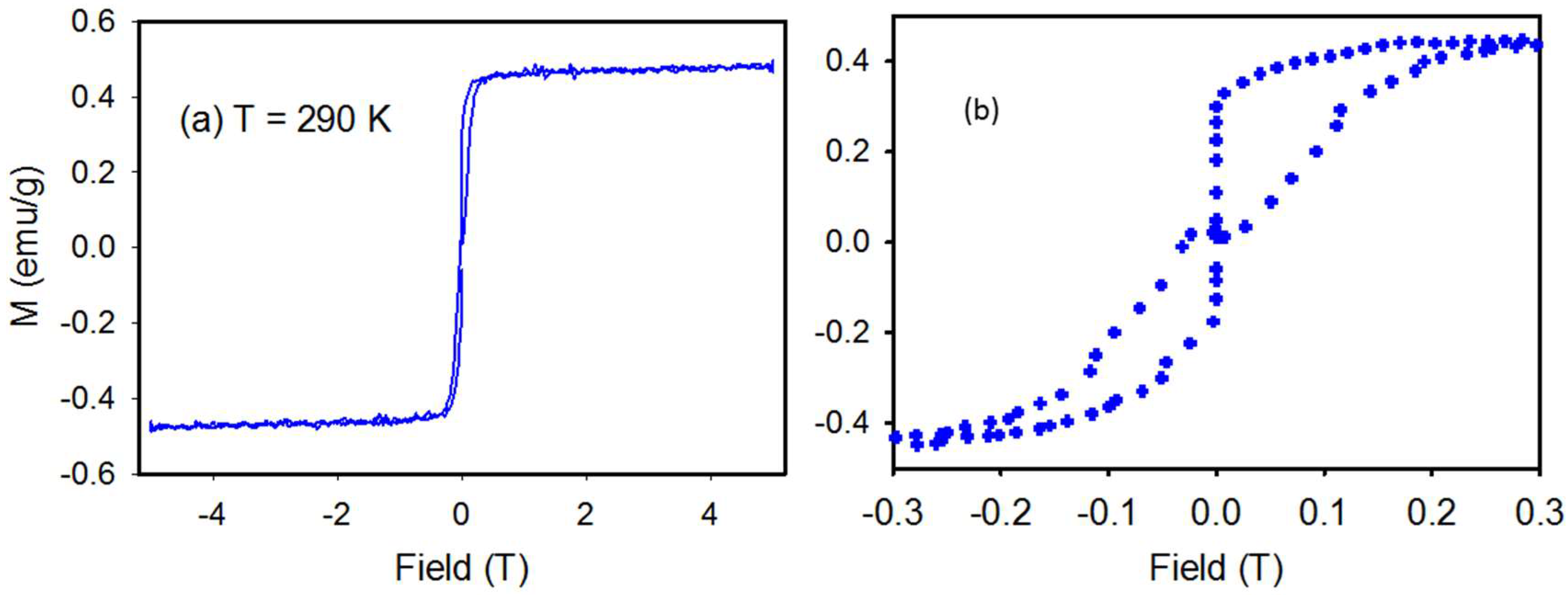
4.3. Magnetic Properties
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Zhang, J.; Zhu, Y.; Wang, Y.; Pu, Z.; Li, L. Electrochemical hydrogen storage properties of Mg2−xAlx Ni (x = 0, 0.3, 0.5, 0.7) prepared by hydriding combustion synthesis and mechanical milling. International. Int. J. Hydrog. Energy 2012, 37, 18140–18147. [Google Scholar] [CrossRef]
- Cova, F.; Larochette, P.A.; Gennari, F. Hydrogen sorption in MgH2-based composites: The role of Ni and LiBH4 additives. Int. J. Hydrog. Energy 2012, 37, 15210–15219. [Google Scholar] [CrossRef]
- Shao, H.; Asano, K.; Enoki, H.; Akiba, E. Fabrication and hydrogen storage property study of nanostructured Mg-Ni-B ternary alloys. J. Alloys Compd. 2009, 479, 409–413. [Google Scholar] [CrossRef]
- Liao, C.Z.; Dong, C.; Zeng, L.M.; He, B.; Cao, W.H.; Yang, L.H. Crystal structure and physical properties of the new ternary compound MgNi7B3. J. Alloys Compd. 2010, 493, 31–34. [Google Scholar] [CrossRef]
- Manfrinetti, P.; Pani, M.; Dhar, S.K.; Kulkarni, R. Structure, transport and magnetic properties of MgNi3B2. J. Alloys Compd. 2007, 428, 94–98. [Google Scholar] [CrossRef]
- Pedneault, S.; Huot, J.; Rouéa, L. Nanostructured Mg2Ni materials prepared by cold rolling and used as negative electrode for Ni–MH batteries. J. Power Sources 2008, 185, 566–569. [Google Scholar] [CrossRef]
- Aswal, D.K.; Shashwati, S.; Singh, A.; Chandrasekhar Rao, T.V.; Vyas, J.C.; Gupta, L.C.; Gupta, S.K.; Sahni, V.C. Synthesis and characterization of MgB2 superconductor. Phys. C Supercond. 2001, 363, 149–154. [Google Scholar] [CrossRef]
- Takano, Y.; Oguro, N.; Kaieda, Y.; Togano, K. Superconducting properties of combustion synthesized MgB2. Phys. C Supercond. 2004, 412, 125–129. [Google Scholar] [CrossRef]
- Portehault, D.; Devi, S.; Beaunier, P.; Gervais, C.; Giordano, C.; Sanchez, C.; Antonietti, M. A general solution route toward metal boride nanocrystals. Angew. Chem. Int. Ed. 2011, 50, 3262–3265. [Google Scholar] [CrossRef] [PubMed]
- Carenco, S.; Portehault, D.; Boissière, C.; Mézailles, N.; Sanchez, C. Nanoscaled metal borides and phosphides: Recent developments and perspectives. Chem. Rev. 2013, 113, 7981–8065. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Vajo, J.J.; Cumberland, R.W.; Liu, P.; Hwang, S.J.; Kim, C.; Bowman, R.C. Hydrogenation of magnesium nickel boride for reversible hydrogen storage. Phys. Chem. Lett. 2010, 1, 69. [Google Scholar] [CrossRef]
- Grasso, G.; Malagoli, A.; Modica, M.; Tumino, A.; Ferdeghini, C.; Siri, A.S.; Vignola, C.; Martini, L.; Previtali, V.; Volpini, G. Fabrication and properties of monofilamentary MgB2 superconducting tapes. Supercond. Sci. Technol. 2003, 16, 271. [Google Scholar] [CrossRef]
- Bellingeri, E.; Malagoli, A.; Modica, M.; Braccini, V.; Siri, A.S.; Grasso, G. Neutron scattering studies of superconducting MgB2 tapes. Supercond. Sci. Technol. 2003, 16, 276–280. [Google Scholar] [CrossRef]
- Gross, K.J.; Zuttel, A.; Schlapbach, L. On the possibility of metal hydride formation Part I. The synthesis of MgNi B by mechanical milling and sintering. J. Alloys Compd. 1998, 274, 234. [Google Scholar] [CrossRef]
- Jung, W. Preparation and crystal structure of MgNi3B2 and Li1.2Ni2.5B2. Z. Naturforsch. 1977, 32, 1371. [Google Scholar] [CrossRef]
- Vajeeston, P.; Ravindran, P.; Ravi, C.; Asokamani, R. Electronic structure, bonding, and ground-state properties of AlB2-type transition-metal diborides. Phys. Rev. B 2001, 63, 045115. [Google Scholar] [CrossRef]
- Mackinnon, I.D.R.; Shahbazi, M.; Alarco, J.A.; Talbot, P.C. Low temperature decomposition of metal borohydride drives autogenous synthesis of MgB2. Supercond. Sci. Technol. 2017, 30, 055004. [Google Scholar] [CrossRef]
- Mackinnon, I.D.R.; Winnett, A.; Alarco, J.A.; Talbot, P.C. Synthesis of MgB2 at low temperature and autogenous pressure. Materials 2014, 7, 3901–3918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, R.P.; Pease, R.N. A preliminary study of the kinetics of pyrolysis of Diborane. J. Am. Chem. Soc. 1951, 73, 2132–2134. [Google Scholar] [CrossRef]
- McCusker, L.B.; Von Dreele, R.B.; Cox, D.E.; Louer, D.; Scardi, P. Rietveld refinement guidelines. J. Appl. Crystallogr. 1999, 32, 36–50. [Google Scholar] [CrossRef]
- Momma, K.; Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]
- Bastin, G.F.; Heijligers, H.J.M. Quantitative electron probe microanalysis of Boron. J. Solid State Chem. 2000, 154, 177–187. [Google Scholar] [CrossRef]
- Bastin, G.F.; Dijkstra, J.M.; Heijligers, H.J.M. PROZA96: An improved matrix correction program for electron probe microanalysis, based on a double gaussian phi(rho z) approach. X-Ray Spectrom. 1998, 27, 3–10. [Google Scholar] [CrossRef]
- Drouin, D.; Couture, A.R.; Joly, D.; Tastet, X.; Aimez, V.; Gauvin, R. CASINO V2. 42—A fast and and easy-to-use modeling tool for scanning electron microscopy and microanalysis users. Scanning 2007, 29, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, J. Scanning Electron Microscopy and X-ray Microanalysis, Kluwer Academic, Plenum Publishers; Springer: New York, NY, USA, 2003. [Google Scholar]
- Rongeat, C.; Grosjean, M.-H.; Ruggeri, S.; Dehmas, M.; Bourlot, S.; Marcotte, S.; Rou’e, L. Evaluation of different approaches for improving the cycle life of MgNi-based electrodes for Ni-MH batteries. J. Power Sources 2006, 158, 747–753. [Google Scholar] [CrossRef]
- Varin, R.A.; Czujko, T.; Wronski, Z.S. Nanomaterials for Solid State Hydrogen Storage; Springer: New York, NY, USA, 2009. [Google Scholar]
- Shao, J.; Xiao, X.; Chen, L.; Fan, X.; Li, S.; Ge, H.; Wang, Q. Enhanced hydriding—Dehydriding performance of 2LiBH4–MgH2 composite by the catalytic effects of transition metal chlorides. J. Mater. Chem. 2012, 22, 20764–20772. [Google Scholar] [CrossRef]
- Shao, J.; Xiao, X.; Chen, L.; Fan, X.; Han, L.; Li, S.; Ge, H.; Wang, Q. Enhanced hydriding—Dehydriding performance of a 2LiH–MgB2 composite by the catalytic effects of Ni–B nanoparticles. J. Mater. Chem. A 2013, 1, 10184–10192. [Google Scholar] [CrossRef]
- Puszkiel, J.A.; Gennari, F.C. Reversible hydrogen storage in metal-doped Mg–LiBH4 composites. Scripta Mater. 2009, 60, 667–670. [Google Scholar] [CrossRef]
- Liao, C.-Z.; Dong, C.; Shih, K.; Zeng, L.; He, B.; Cao, W.; Yang, L. Preparation and properties of a new ternary phase Mg3+xNi7-xB2 (0.17–0.66) and its Cu-doping effect. J. Solid State Chem. 2015, 226, 24–28. [Google Scholar] [CrossRef]
- Bragg, J.K.; McCarthy, L.V.; Norton, F.J. Kinetics of Pyrolysis of Diborane. J. Am. Chem. Soc. 1951, 73, 2134–2140. [Google Scholar] [CrossRef]
- Borer, K.; Littlewood, A.B.; Phillips, C.S.G. A gas-chromatographic study of the diborane pyrolysis. J. Inorg. Nucl. Chem. 1960, 15, 316–319. [Google Scholar] [CrossRef]
- Zhu, C.; Hayashi, H.; Saita, I.; Akiyama, T. Direct synthesis of MgH2 nanofibers at different hydrogen pressures. Int. J. Hydrog. Energy 2009, 34, 7283–7290. [Google Scholar] [CrossRef]
- Sastri, M.V.; Viswanathan, B.; Murthy, S.S. Metal Hydrides-Fundamentals and Applications; Narosa Publishing House: New Delhi, India, 1998. [Google Scholar]
- Varin, R.A.; Czujko, T.; Wasmund, E.B.; Wronski, Z.S. Catalytic effects of various forms of nickel on the synthesis rate and hydrogen desorption properties of nanocrystalline magnesium hydride (MgH2) synthesized by controlled reactive mechanical milling (CRMM). J. Alloys Compd. 2007, 432, 217–231. [Google Scholar] [CrossRef]
- Polanski, M.; Nielsen, T.K.; Kunce, I.; Norek, M.; Płocinski, T.; Jaroszewicz, L.R.; Gundlach, C.; Jensen, T.R.; Bystrzycki, J. Mg2NiH4 synthesis and decomposition reactions. Intnl. J. Hydrog. Energy 2013, 38, 4003–4010. [Google Scholar] [CrossRef]
- Czujko, T.; Varin, R.A.; Wronski, Z.; Zaranski, Z.; Durejko, T. Synthesis and hydrogen desorption properties of nanocomposite magnesium hydride with sodium borohydride (MgH2 + NaBH4). J. Alloys Compd. 2007, 427, 291–299. [Google Scholar] [CrossRef]
- Prikhna, T.A.; Gawalek, W.; Savchuk, Y.M.; Moshchil, V.E.; Sergienko, N.V.; Surzhenko, A.B.; Wendt, M.; Dub, S.N.; Melnikov, V.S.; Schmidt, C.; et al. High-pressure synthesis of a bulk superconductive MgB2-based material. Phys. C Supercond. 2003, 386, 565–568. [Google Scholar] [CrossRef]
- Baranowski, B.; Filipek, S.M. 45 Years of nickel hydride—History and perspectives. J. Alloys Compd. 2005, 404, 2–6. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Run No. | NaBH4 (g) | Tmax £ (°C) | tTm (h) | Pmax MPa | % MgNi3B2 | Other Phases ** |
|---|---|---|---|---|---|---|
| 1 | 0.76 | 725 | 8 | 2.2 | 88% | MgNi6.7B2 MgNi7B3 |
| 2 | 0.76 | 670 | 8 | 2.1 | 70% | MgNi6.7B2 MgNi7B3 Ni2B Ni3B |
| 3 | 0.72 | 670 | 8 | 1.7 | 76% | MgNi6.7B2 MgNi7B3 Ni2B Ni3B |
| 4 | 0.72 | 670 | 8 | 2.0 | 90% | MgNi6.7B2 Ni2B MgNi7B3 |
| 5 | 0.72 | 670 | 40 | 2.0 | 85% | MgNi6.7B2 MgNi7B3 Ni3B |
| 6 | 0.72 | 725 | 30 | 2.5 | 76% | Ni2B (12%) MgNi7B3 (11%) |
| Parameter | MgNi3B2 (This Work) | MgNi3B2 [5] (Single Crystal) |
|---|---|---|
| Space group | P6422 | P6422 |
| a (Å) | 4.8799 (1) | 4.8800 (1) |
| c (Å) | 8.7884 (2) | 8.7870 (1) |
| Cell volume (Å3) | 181.2 (1) | 181.22 |
| Rwp | 3.8% | 4.0% |
| Rp | 2.7% | 1.9% |
| GoF | 1.98 | 0.93 |
| Atom: Ni2 (z; 6f) | 0.2062 (1) | 0.2083 (1) |
| Atom: B (x; 6i) | 0.6102 (7) | 0.6110 (8) |
| Element (wt %) | Stoichiometry | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compound | n + | B | Mg | Ni | Total | B | Mg | Ni | Total |
| Run 3 | |||||||||
| Ni2B (core 1) | 4 | 8.36 (0.47) | 0.04 (0.01) | 89.8 (0.9) | 98.2 (1.3) | 1 | 0.00 (0.00) | 1.98 (0.10) | 2.98 (0.10) |
| Ni3B (core 2) | 5 | 5.58 (0.32) | 0.03 (0.01) | 93.5 (1.3) | 99.1 (1.5) | 1 | 0.00 (0.00) | 3.09 (0.17) | 4.09 (0.17) |
| Ni4B3 (mid-core) | 5 | 12.0 (0.1) | 0.04 (0.02) | 87.9 (0.5) | 99.9 (0.4) | 3 | 0.00 (0.00) | 4.06 (0.06) | 7.06 (0.06) |
| MgNi3B2 (shell) | 5 | 9.01 (0.34) | 11.5 (0.1) | 79.2 (1.4) | 99.6 (1.3) | 2 | 1.13 (0.05) | 3.24 (0.16) | 6.37 (0.21) |
| Run 5 | |||||||||
| MgNi10.5B3 (core1) | 3 | 4.76 (0.11) | 3.65 (0.37) | 90.2 (0.4) | 98.6 (0.1) | 3 | 1.02 (0.04) | 10.50 (0.09) | 14.52 (0.10) |
| Ni3B (core2) | 2 | 5.53 | bdl | 93.2 | 98.7 | 1 | bdl ** | 3.11 | 4.11 |
| Ni4B3 (mid-core) | 1 | 12.2 | bdl | 87.2 | 99.5 | 3 | bdl ** | 3.94 | 6.94 |
| MgNi3B2 (shell) | 8 | 9.27 (0.14) | 11.7 (0.1) | 78.1 (1.0) | 99.1 (0.9) | 2 | 1.12 (0.02) | 3.10 (0.07) | 6.22 (0.09) |
| MgNi4B2 | 1 | 7.83 | 8.73 | 84.1 | 100.7 | 2 | 0.99 | 3.96 | 6.95 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shahbazi, M.; Cathey, H.E.; Mackinnon, I.D.R. Synthesis of Magnesium Nickel Boride Aggregates via Borohydride Autogenous Pressure. Materials 2018, 11, 480. https://doi.org/10.3390/ma11040480
Shahbazi M, Cathey HE, Mackinnon IDR. Synthesis of Magnesium Nickel Boride Aggregates via Borohydride Autogenous Pressure. Materials. 2018; 11(4):480. https://doi.org/10.3390/ma11040480
Chicago/Turabian StyleShahbazi, Mahboobeh, Henrietta E. Cathey, and Ian D. R. Mackinnon. 2018. "Synthesis of Magnesium Nickel Boride Aggregates via Borohydride Autogenous Pressure" Materials 11, no. 4: 480. https://doi.org/10.3390/ma11040480
APA StyleShahbazi, M., Cathey, H. E., & Mackinnon, I. D. R. (2018). Synthesis of Magnesium Nickel Boride Aggregates via Borohydride Autogenous Pressure. Materials, 11(4), 480. https://doi.org/10.3390/ma11040480