Fe-Based Nano-Materials in Catalysis

 ,
,  , and
, and
Abstract
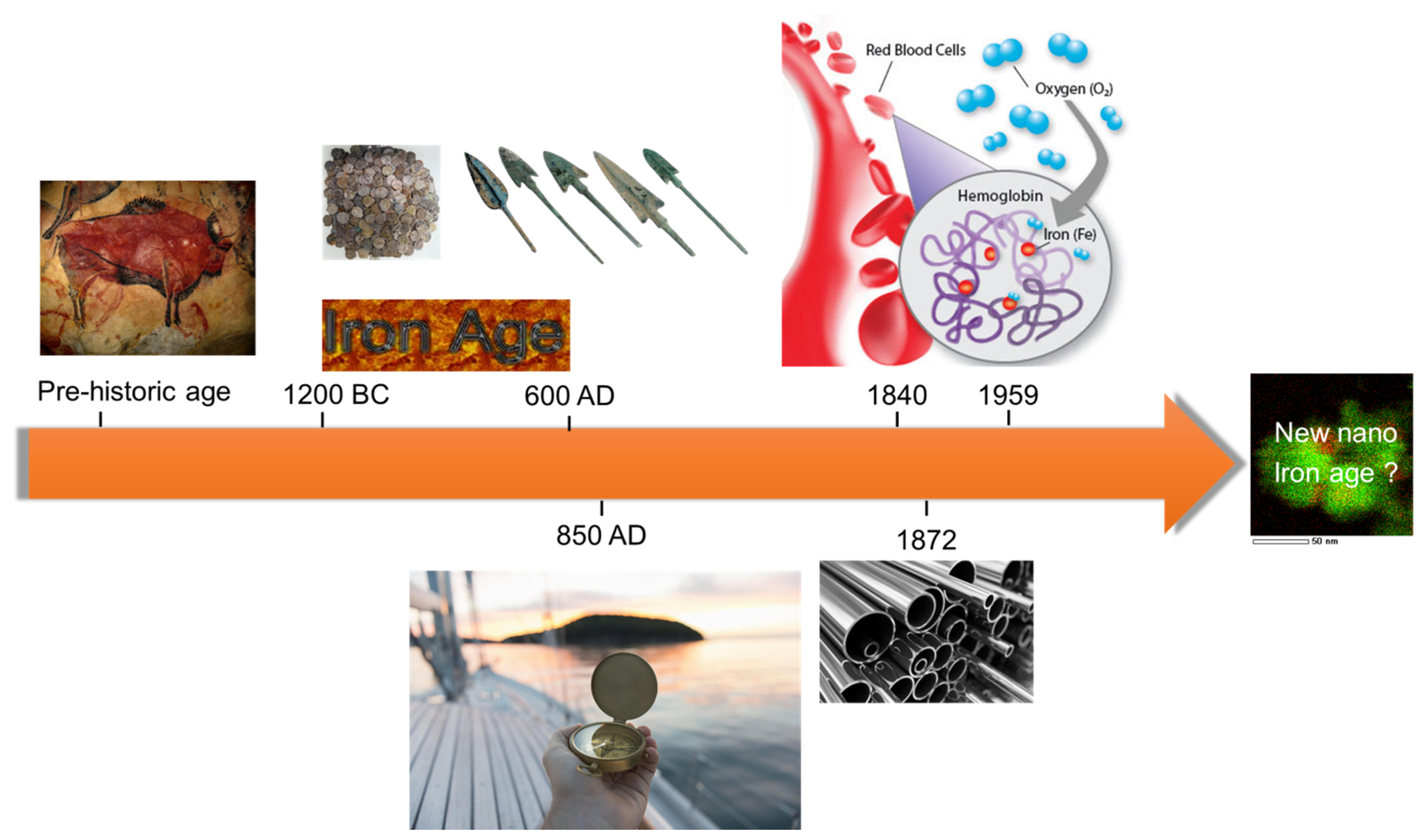
:1. Introduction and Motivation
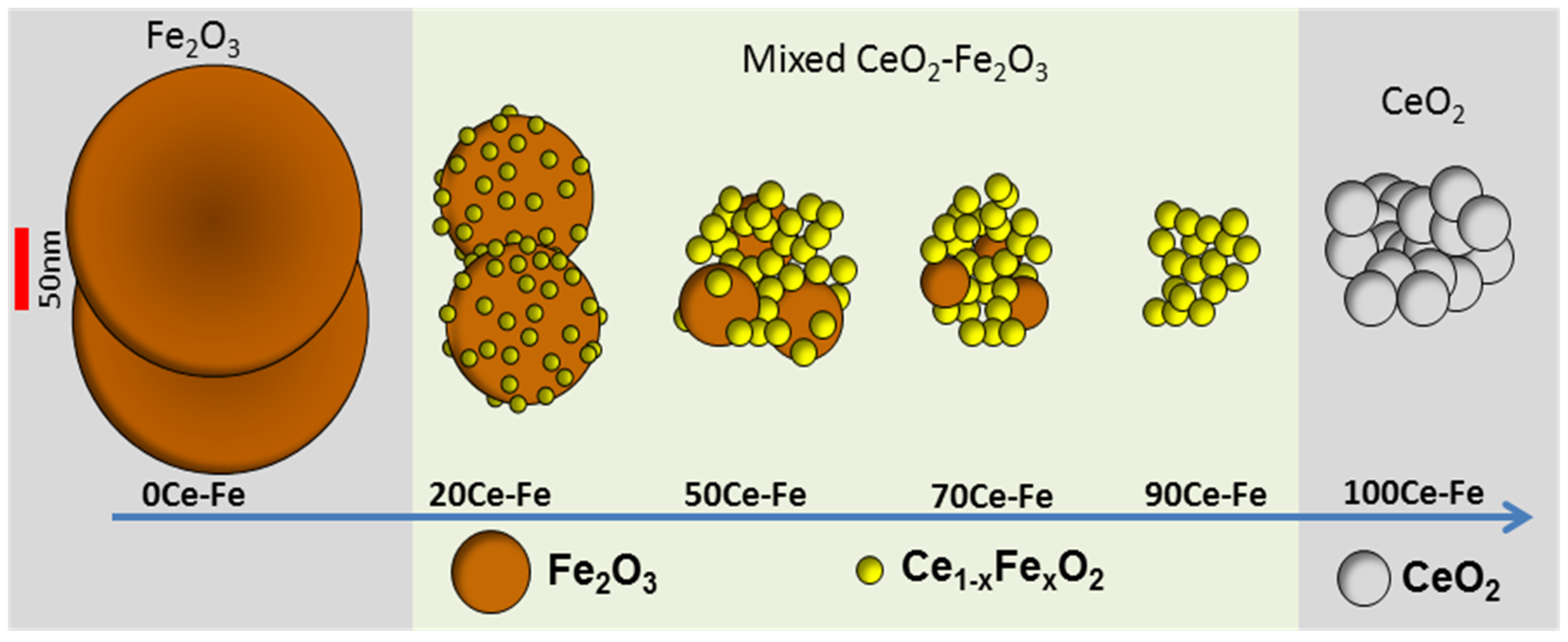
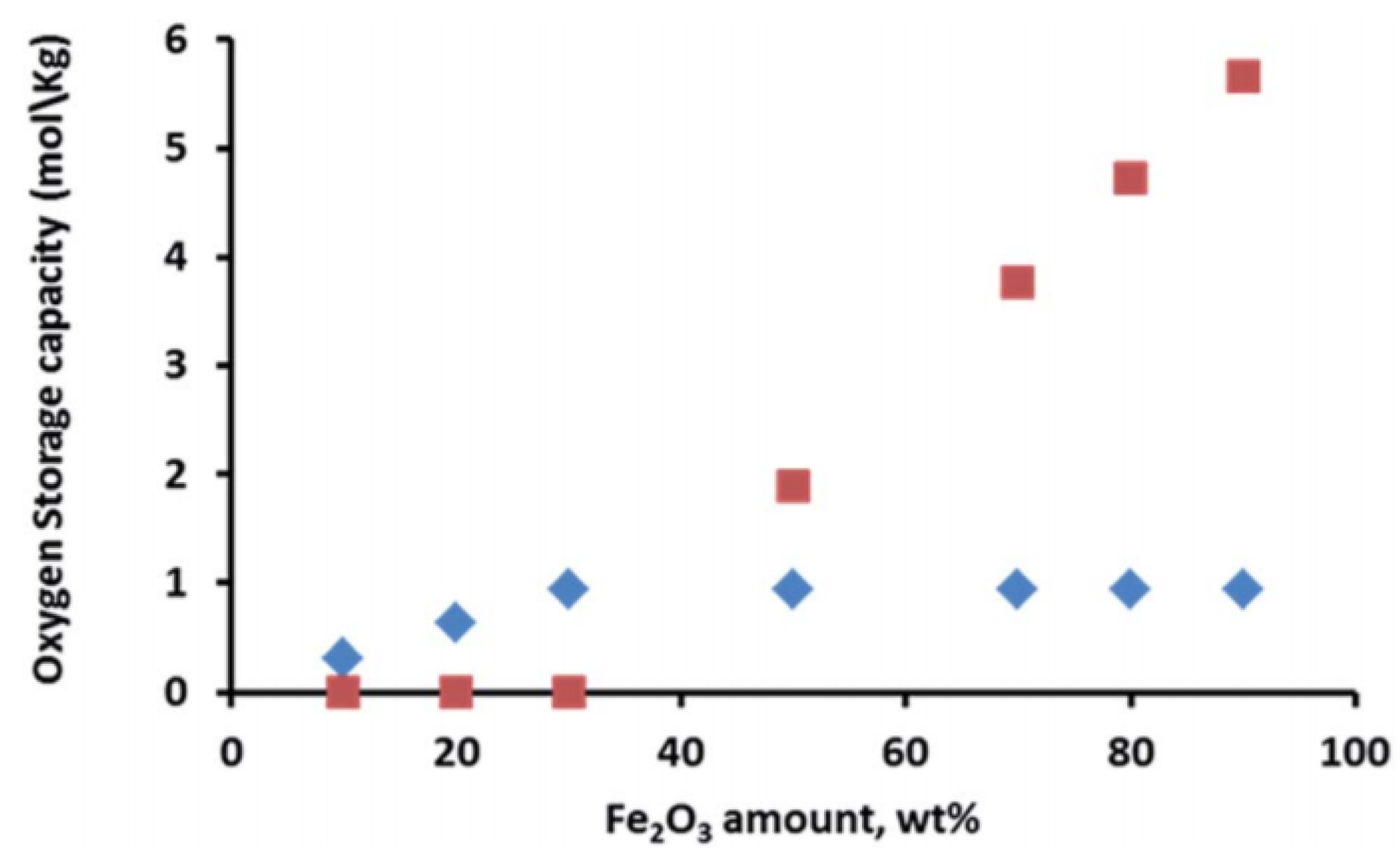
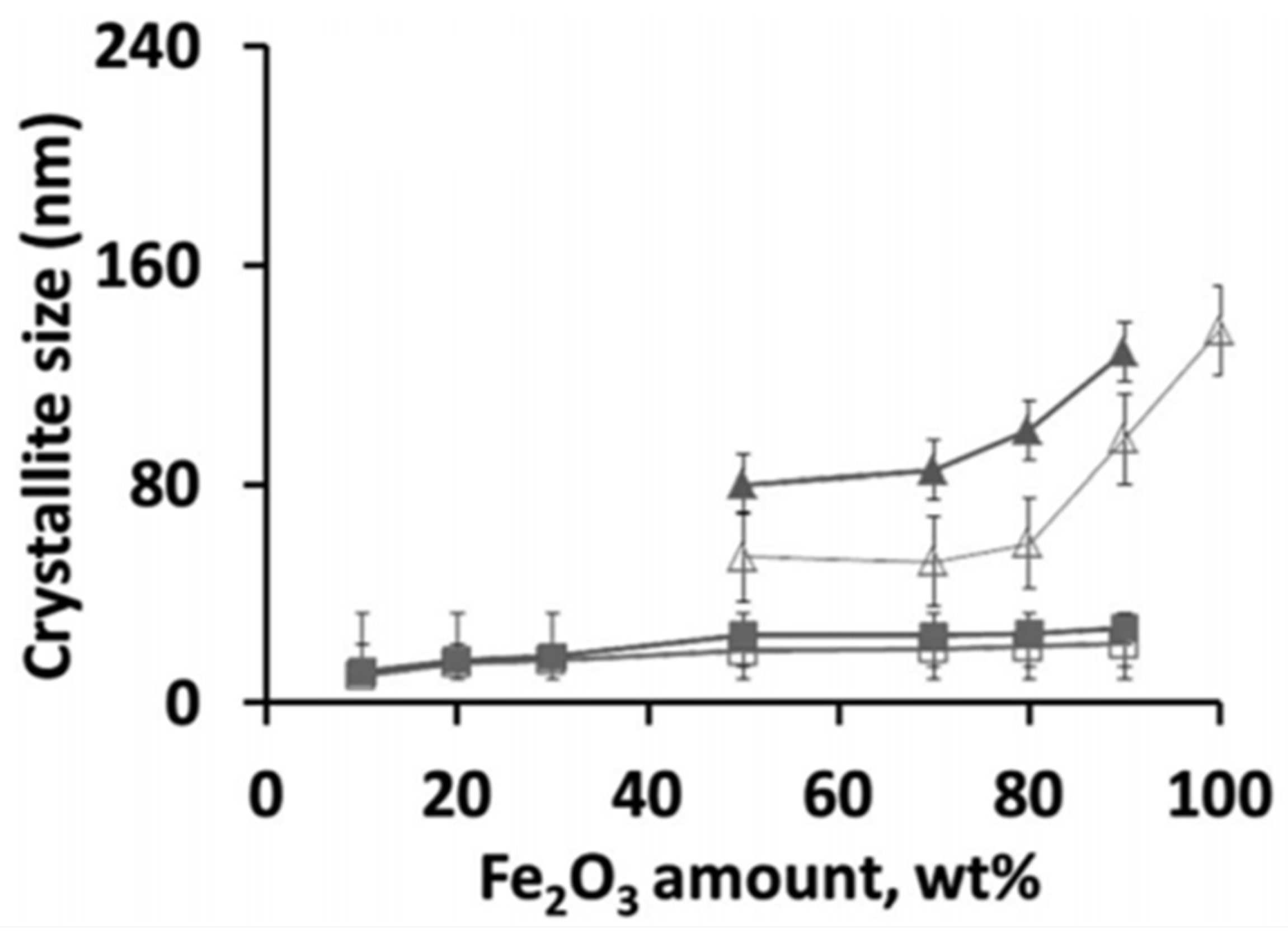
2. Fe in CeO2 for Chemical Looping
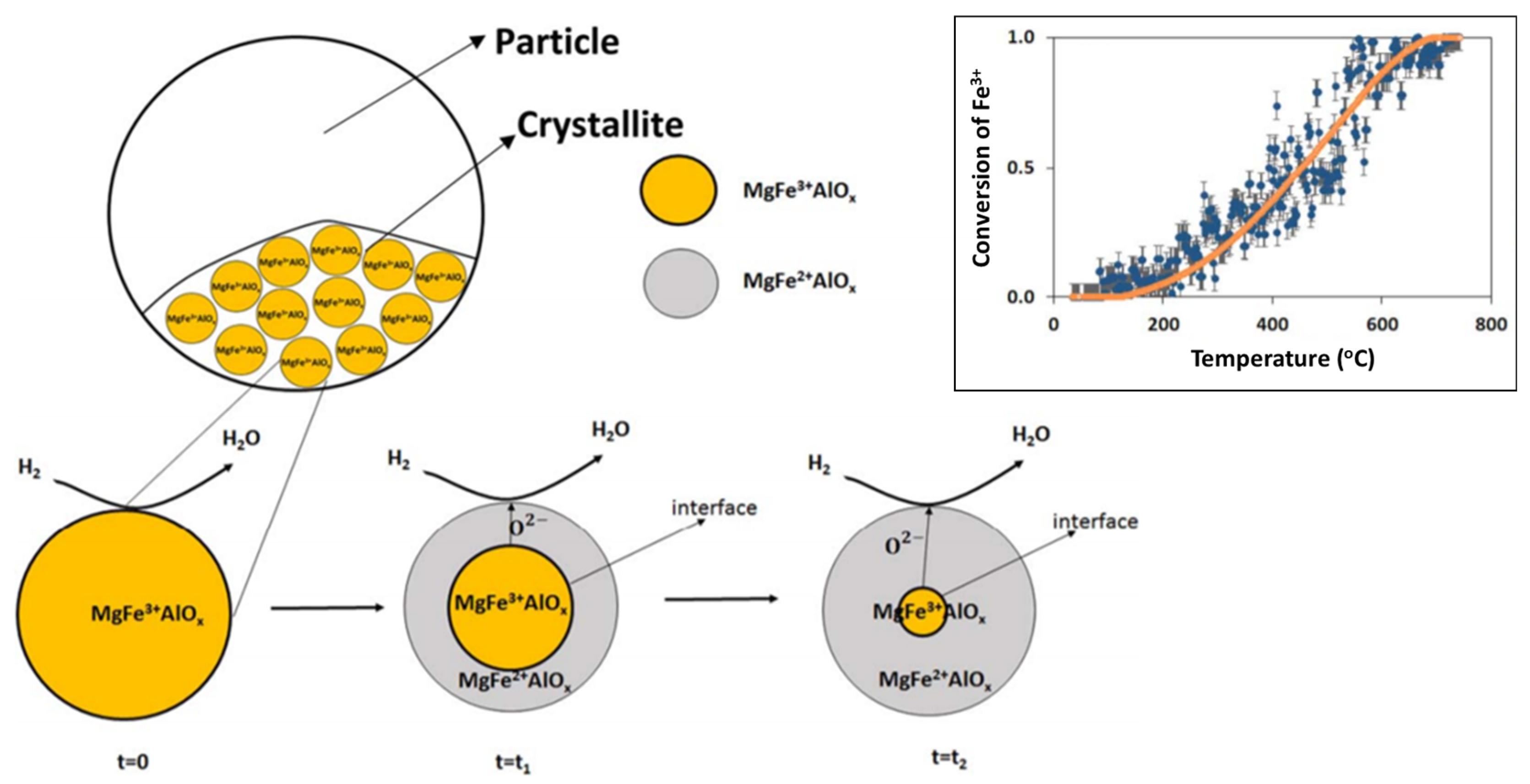
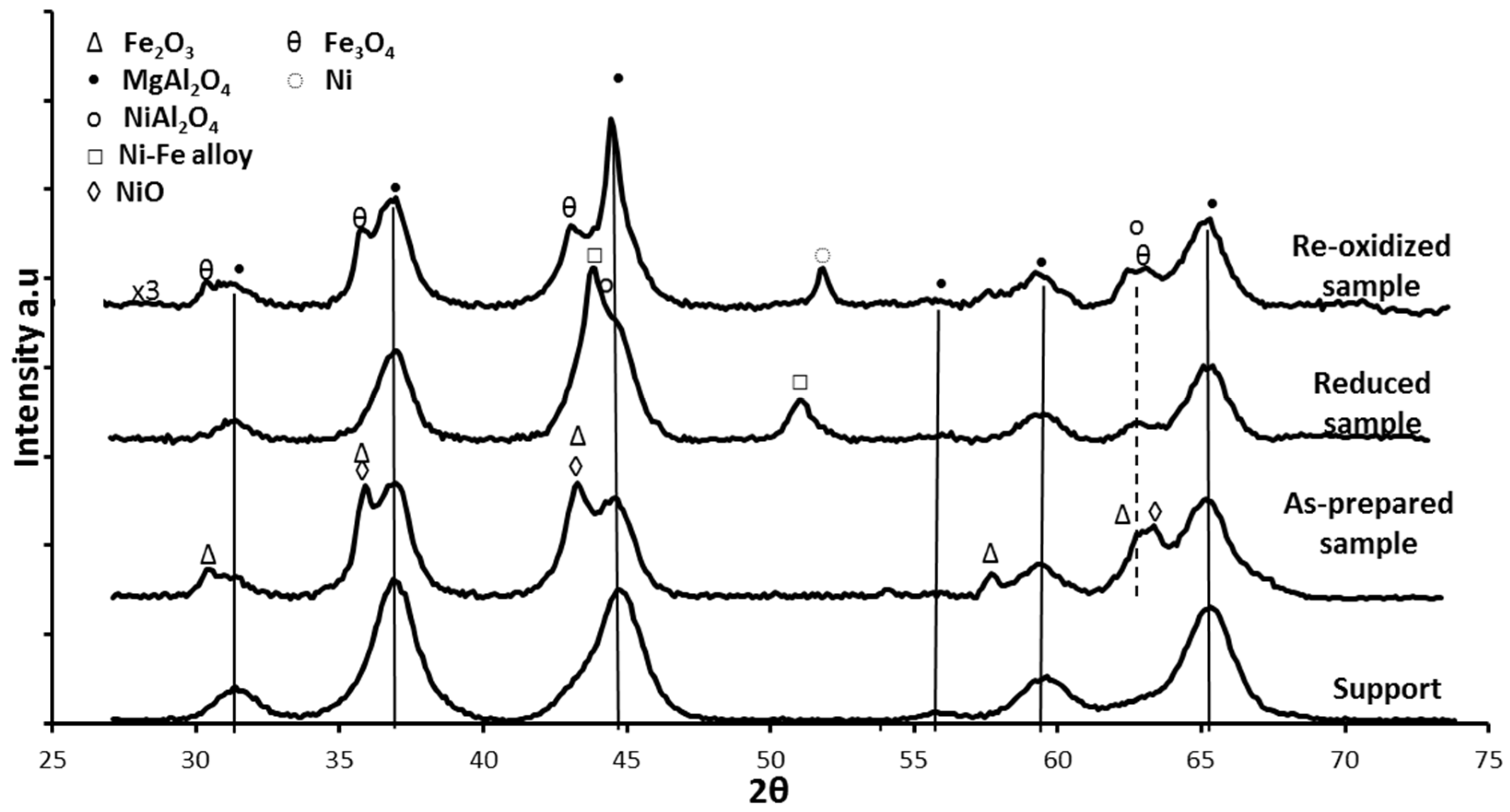
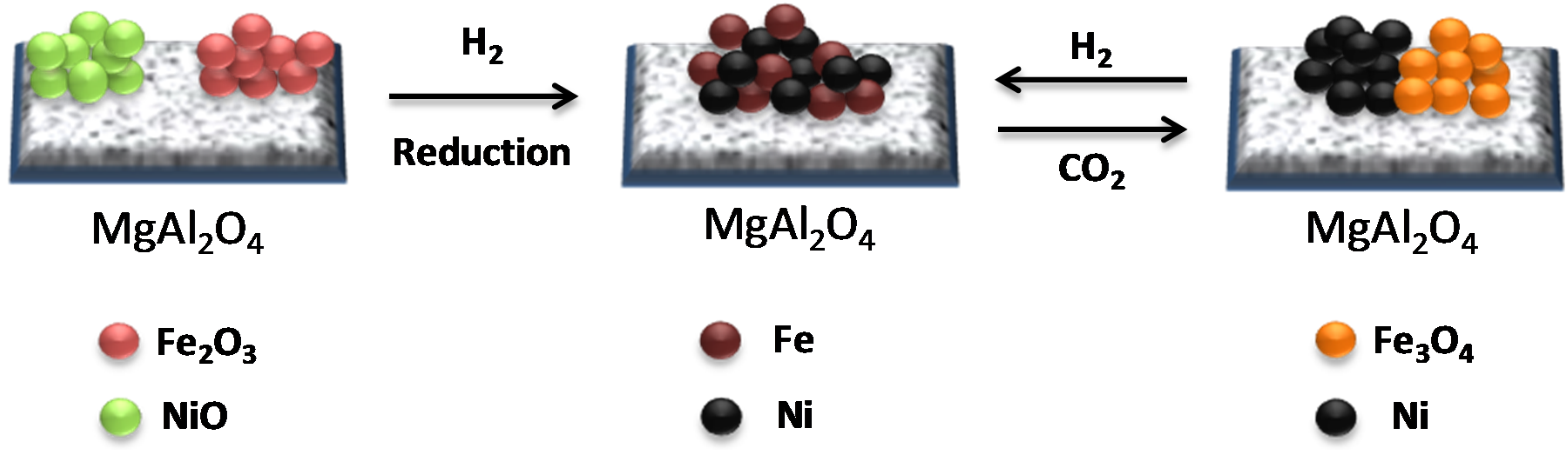
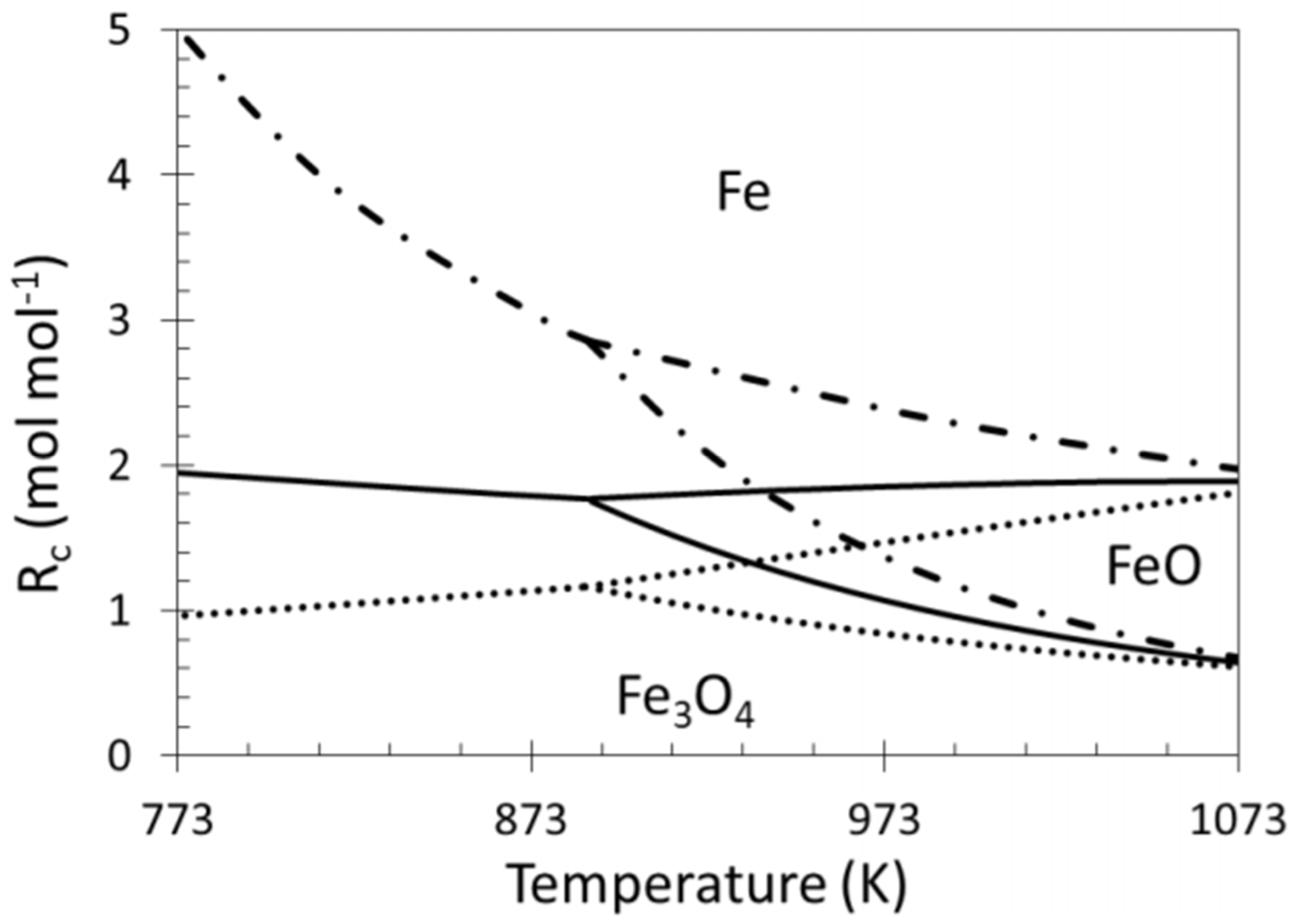
3. Fe in Spinels for Chemical Looping
4. Fe in Nano-Alloys for Catalysis
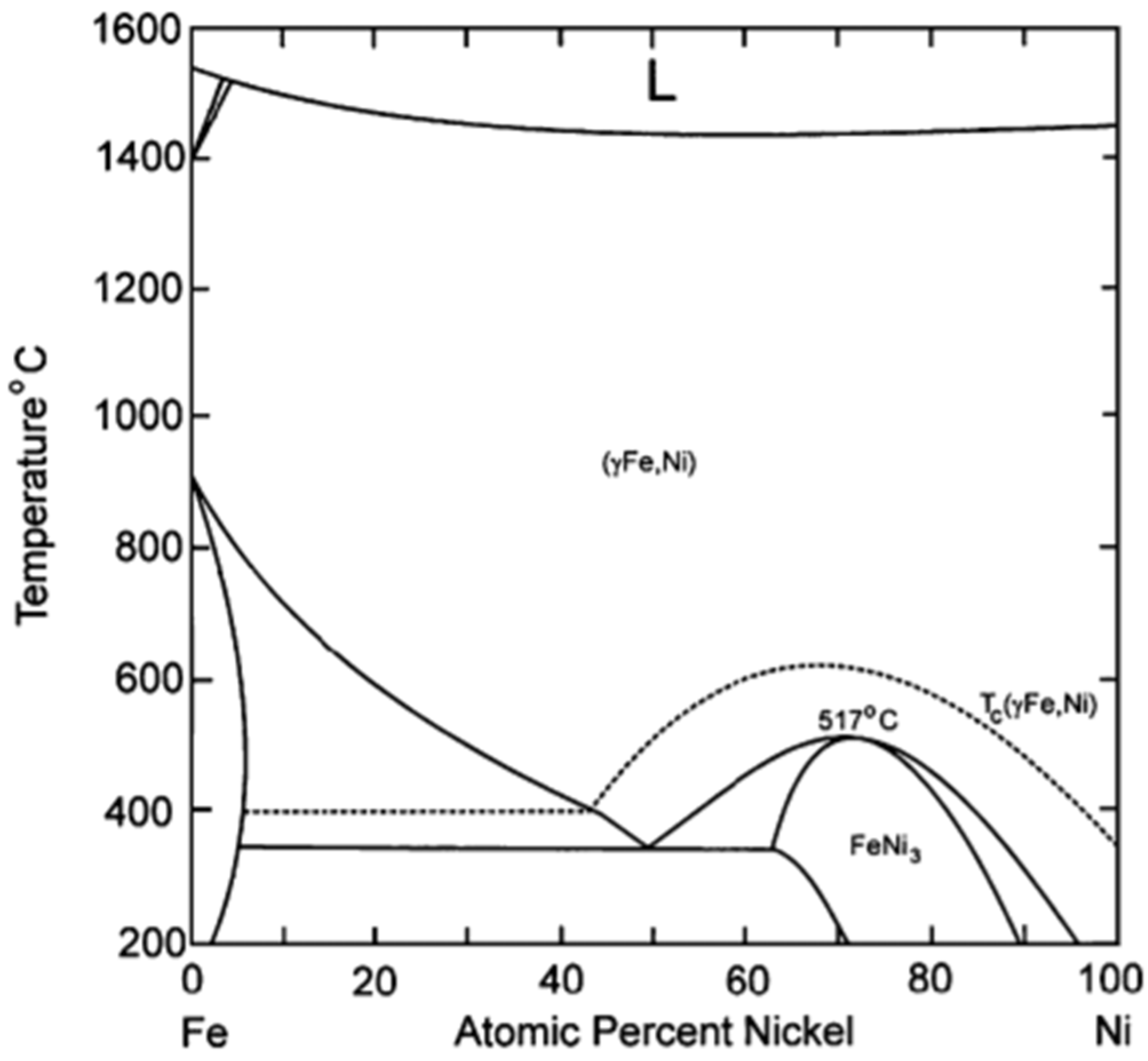
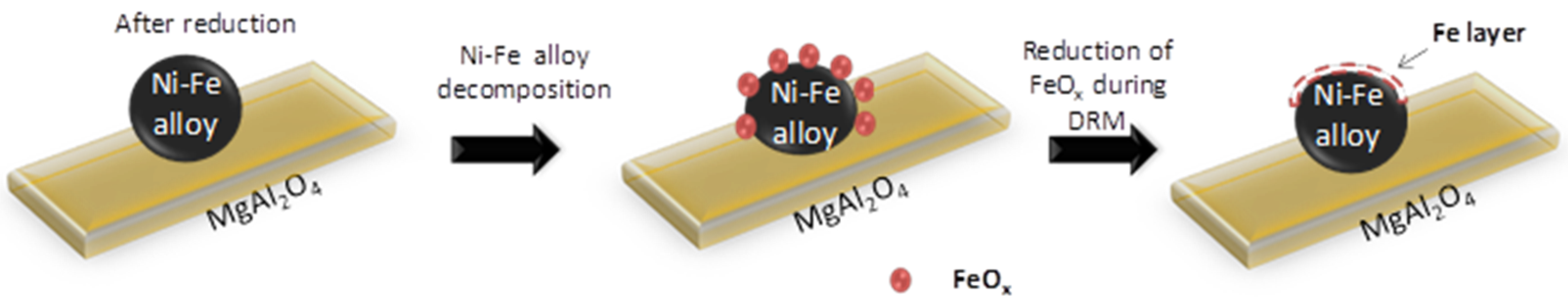
4.1. Fe–Ni Nano-Alloy
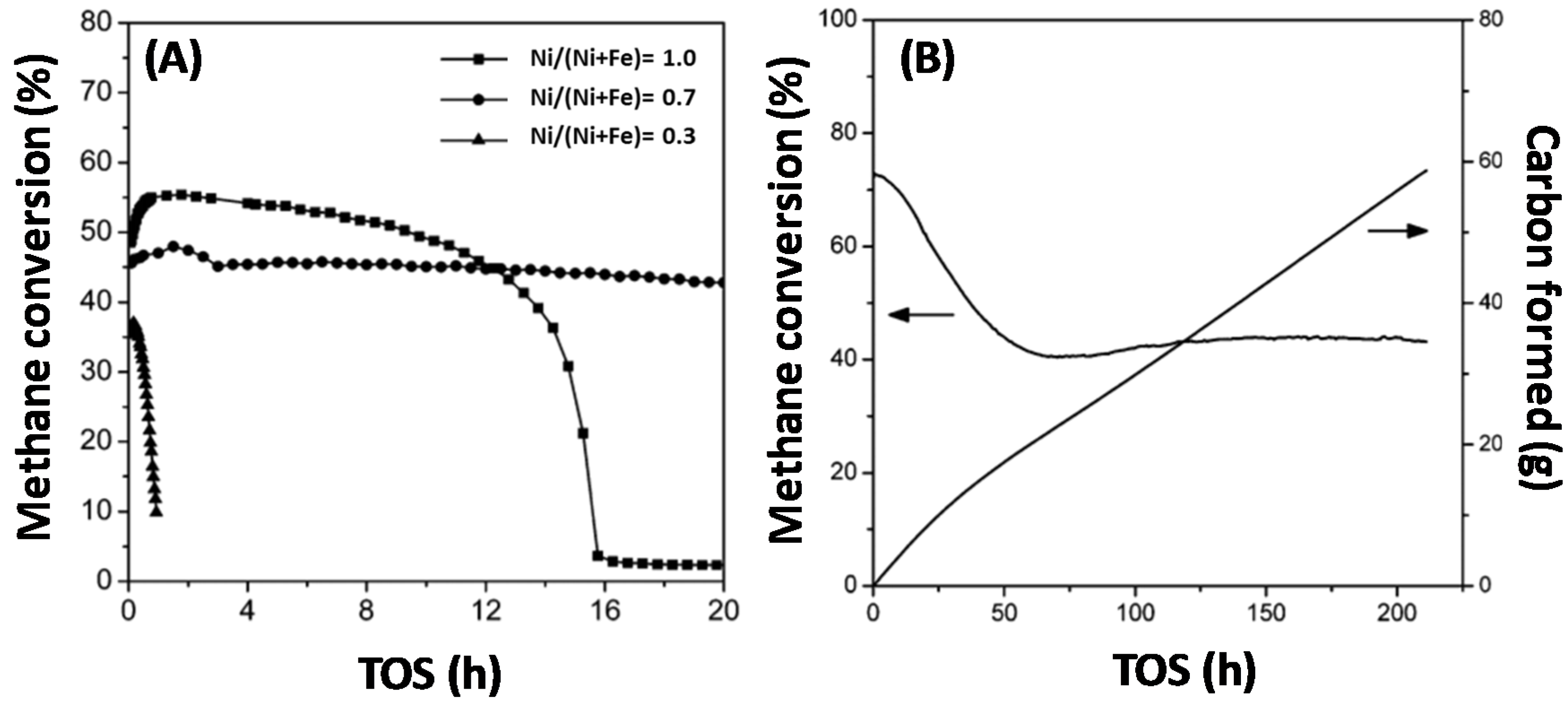
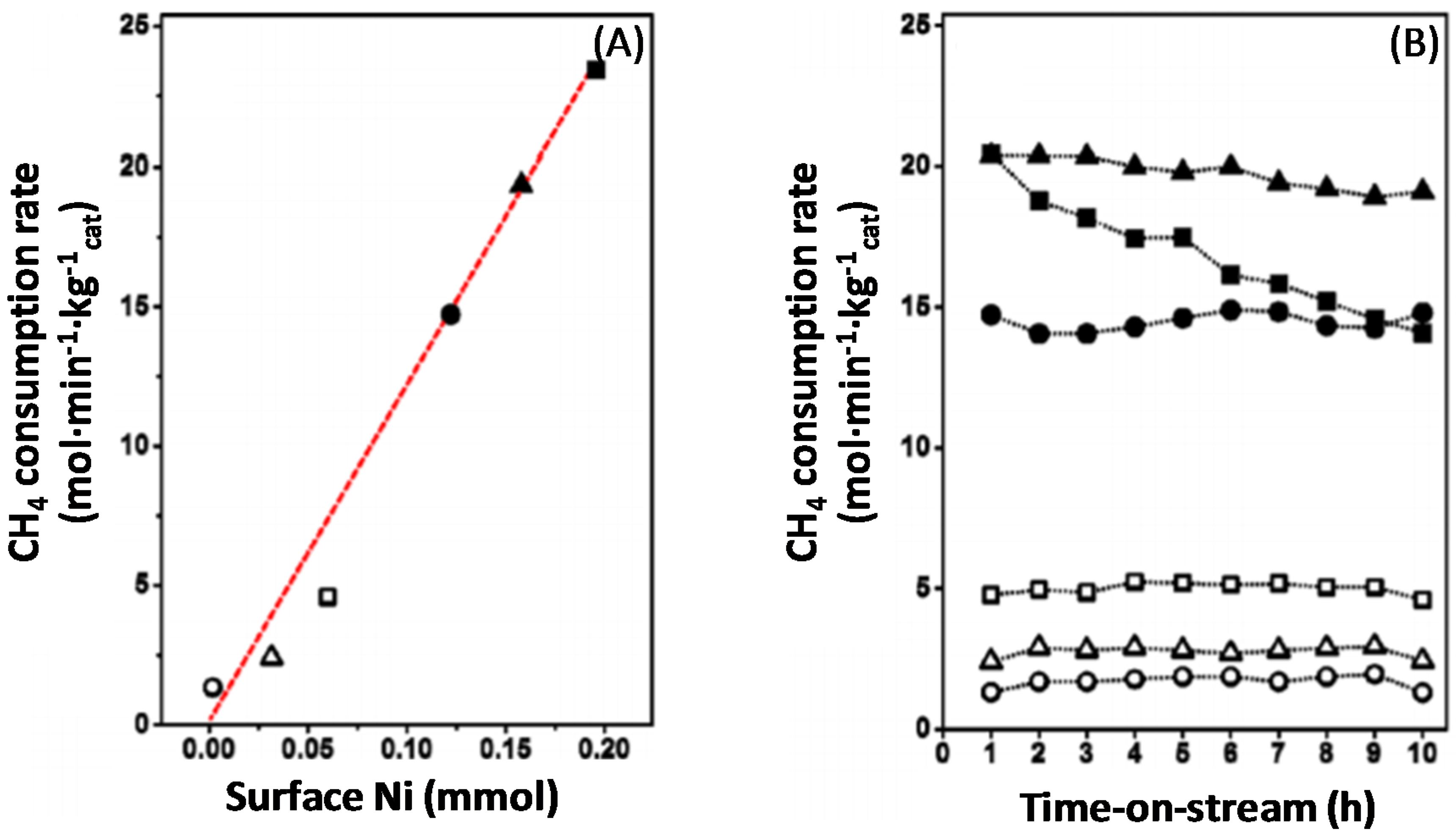
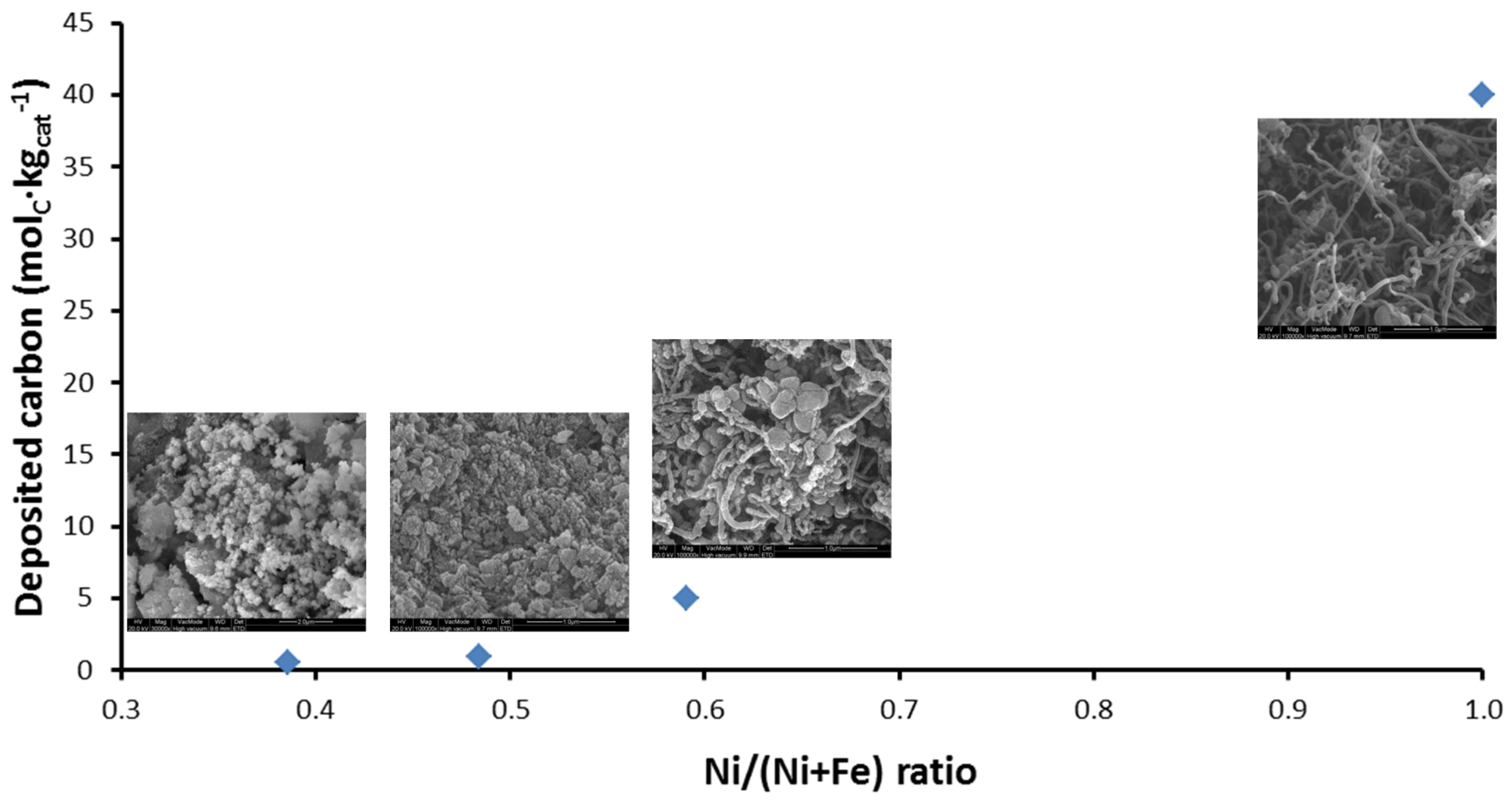
4.1.1. Activity during Methane Decomposition
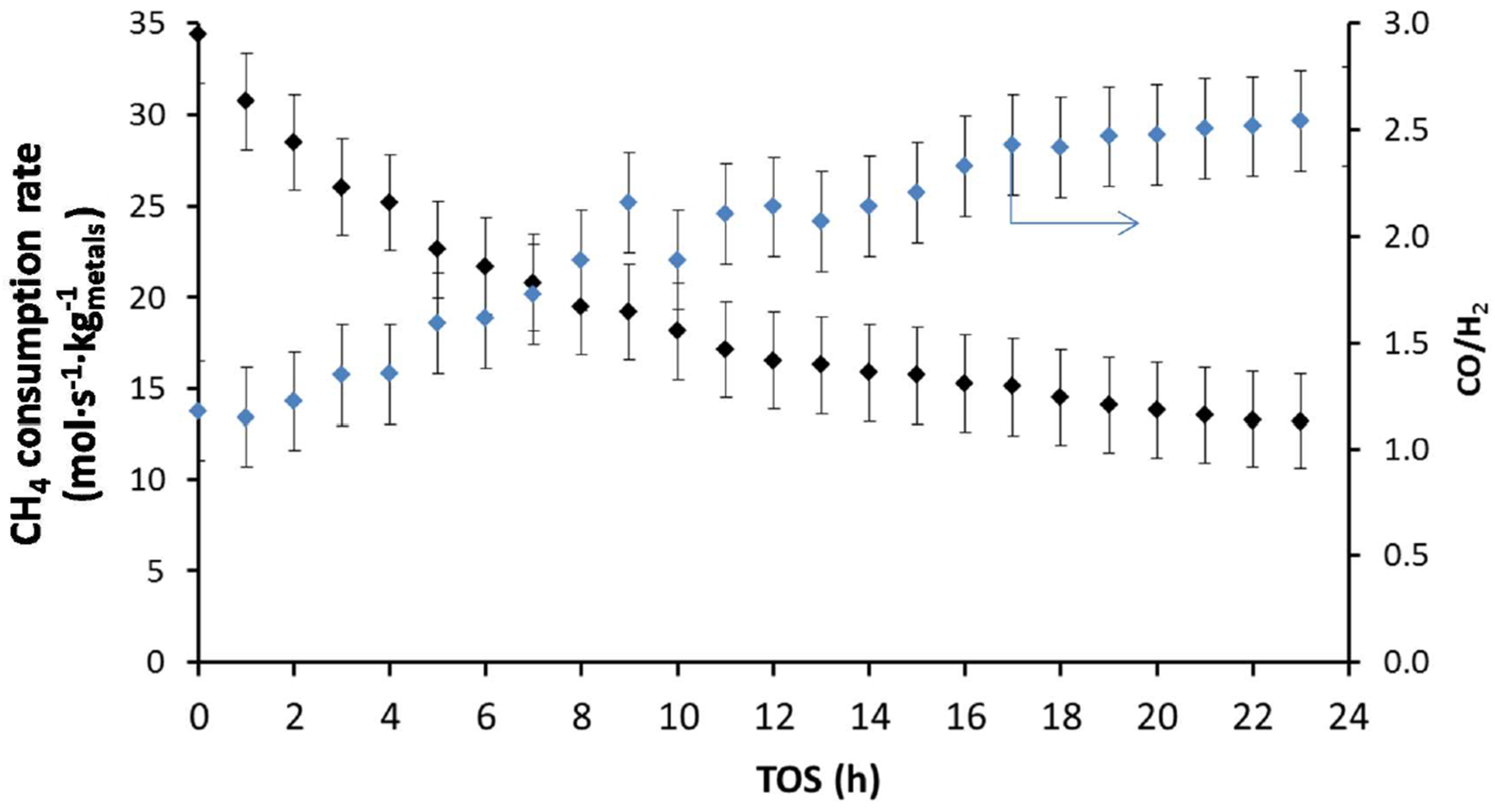
4.1.2. Activity during Syngas Production
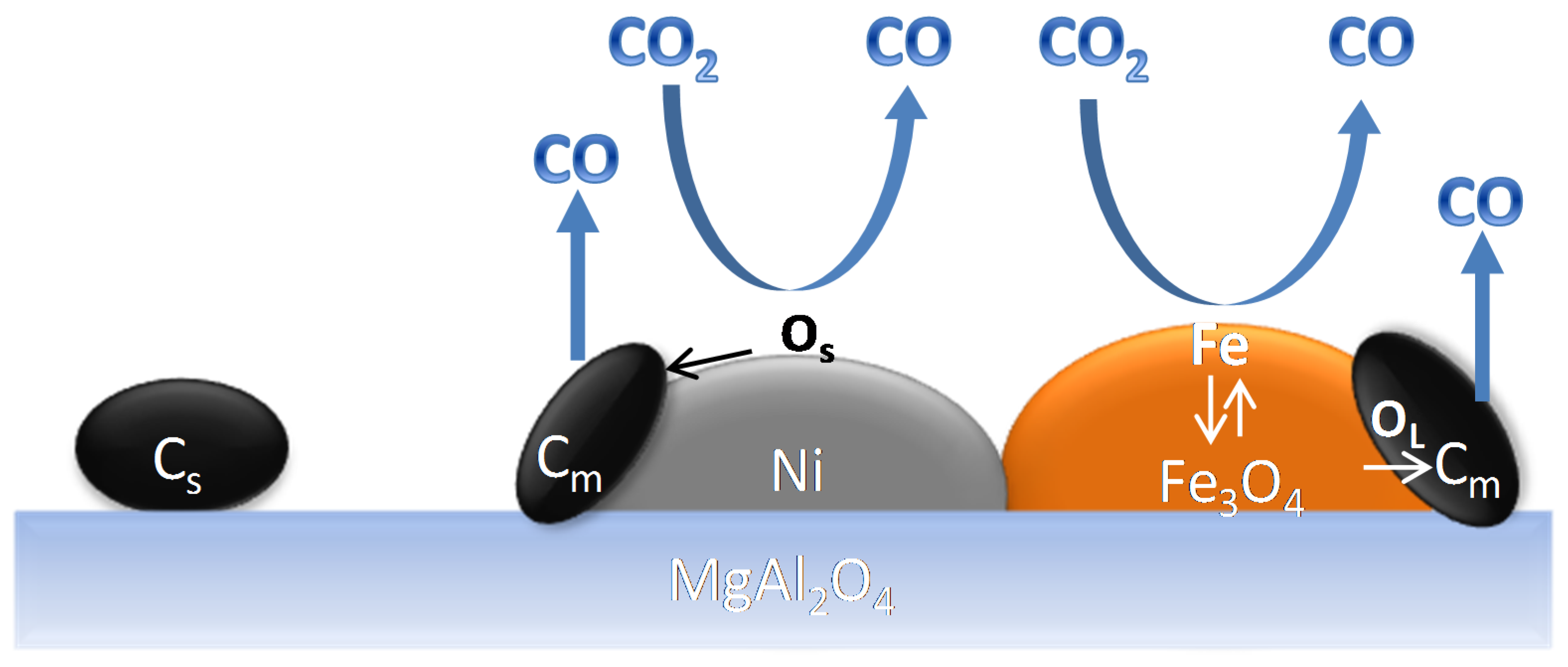
4.1.3. Catalyst Regeneration: Carbon Removal by CO2
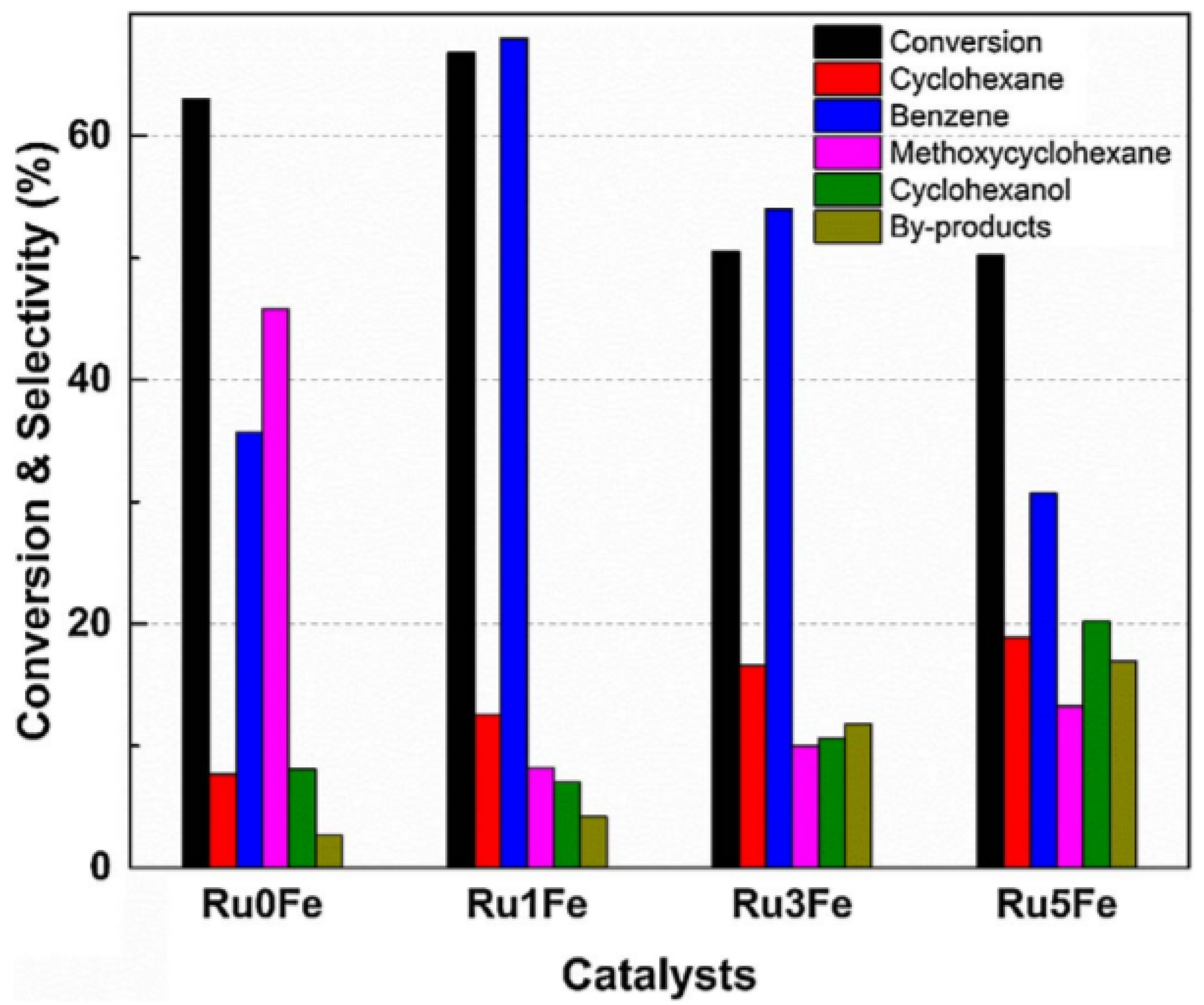
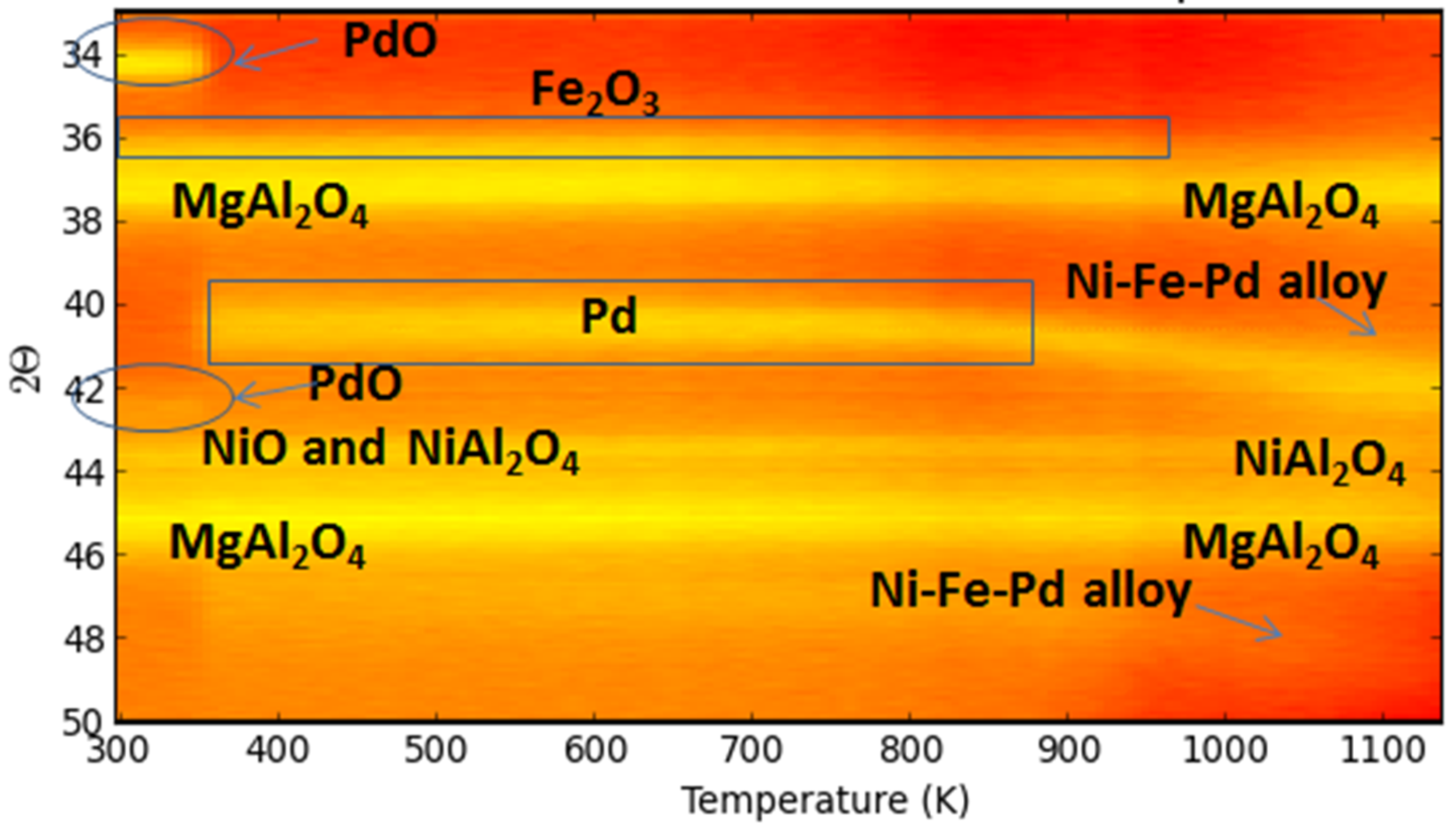
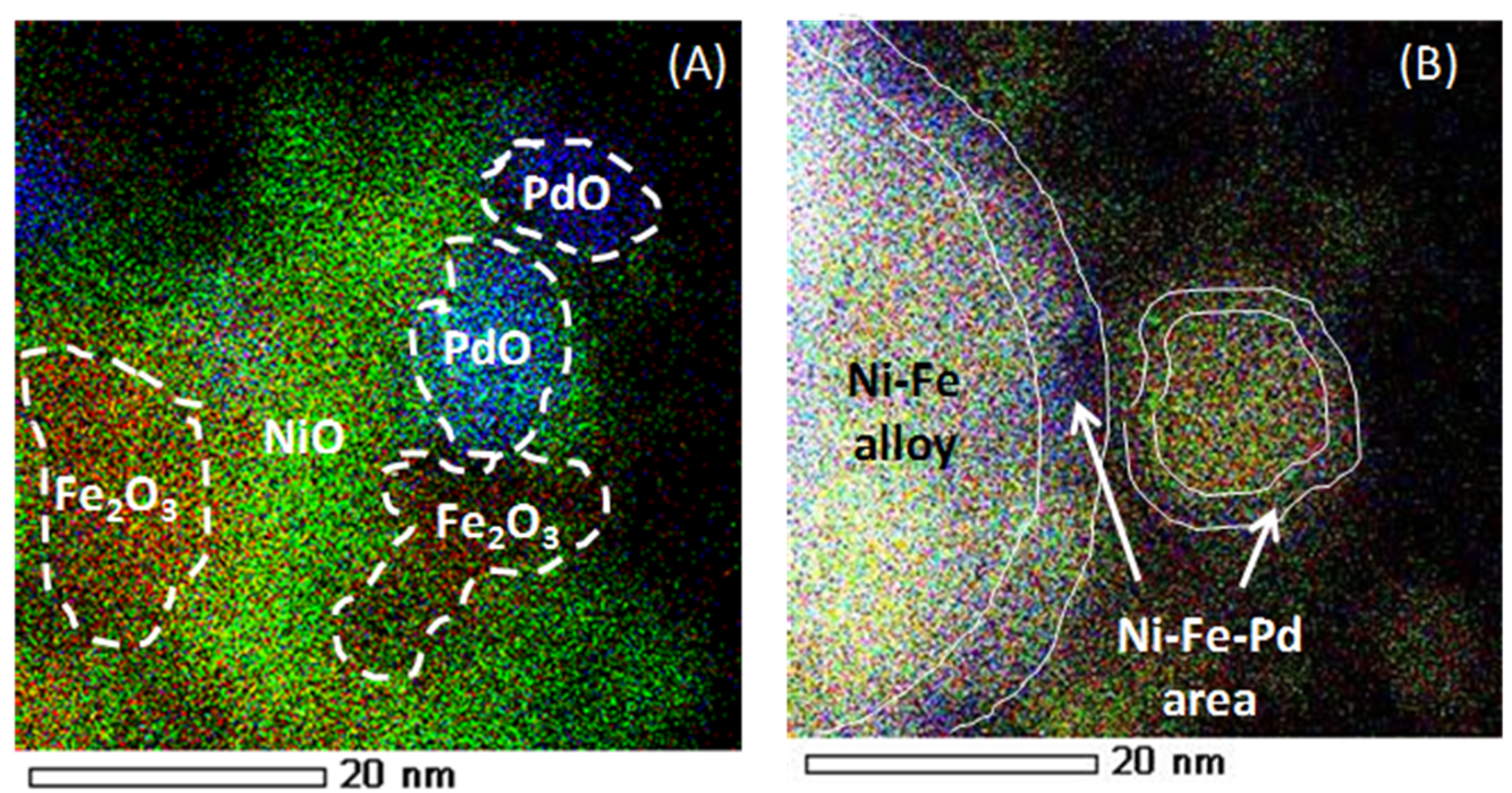
4.2. Trimetallic Fe-Containing Alloys for Hydrocarbon Conversion
5. Summary and Outlook: The Role of Fe
- (1)
- The addition of Fe, either in bimetallic catalysts or incorporated into the support lattice, can provide redox functionality to the catalyst, which helps to suppress carbon formation.
- (2)
- The mechanism of carbon species removal by CO2 over bimetallic Fe–Ni is different from that over a monometallic Ni catalyst.
- (3)
- The redox properties of Fe can be exploited in different processes.
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Parkinson, G.S. Iron oxide surfaces. Surf. Sci. Rep. 2016, 71, 272–365. [Google Scholar] [CrossRef]
- Bragg, W.H. The structure of magnetite and the spinels. Nature 1915, 95, 561. [Google Scholar] [CrossRef]
- Coltrain, B.K.; Herron, N.; Busch, D.H. Oxygen Activation by Transition Metal Complexes of Macrobicyclic Cyclidene Ligands. In The Activation of Dioxygen and Homogeneous Catalytic Oxidation; Barton, D.H.R., Martell, A.E., Sawyer, D.T., Eds.; Springer: Boston, MA, USA, 1993; pp. 359–380. [Google Scholar]
- Dlouhy, A.C.; Outten, C.E. The Iron Metallome in Eukaryotic Organisms. Met. Ions Life Sci. 2013, 12, 241–278. [Google Scholar] [PubMed]
- Yan, L.; Zhang, S.; Chen, P.; Liu, H.; Yin, H.; Li, H. Magnetotactic bacteria, magnetosomes and their application. Microbiol. Res. 2012, 167, 507–519. [Google Scholar] [CrossRef] [PubMed]
- Encyclopedia, A. Prehistoric Colour Palette. Available online: http://www.visual-arts-cork.com/index.htm (accessed on 14 May 2018).
- Bellis, M. The Compass and Other Magnetic Innovations. Available online: https://www.thoughtco.com/ (accessed on 14 May 2018).
- Davis, C.P. Hemoglobin (Low and High Range Causes). Available online: https://www.medicinenet.com/script/main/hp.asp (accessed on 14 May 2018).
- Xu, P.; Zeng, G.M.; Huang, D.L.; Feng, C.L.; Hu, S.; Zhao, M.H.; Lai, C.; Wei, Z.; Huang, C.; Xie, G.X.; et al. Use of iron oxide nanomaterials in wastewater treatment: A review. Sci. Total Environ. 2012, 424, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Jagadeesh, R.V.; Surkus, A.-E.; Junge, H.; Pohl, M.-M.; Radnik, J.; Rabeah, J.; Huan, H.; Schünemann, V.; Brückner, A.; Beller, M. Nanoscale Fe2O3-Based Catalysts for Selective Hydrogenation of Nitroarenes to Anilines. Science 2013, 342, 1073–1076. [Google Scholar] [CrossRef] [PubMed]
- Bagheri, S.; Julkapli, N.M. Modified iron oxide nanomaterials: Functionalization and application. J. Magn. Magn. Mater. 2016, 416, 117–133. [Google Scholar] [CrossRef]
- Bykova, E.; Dubrovinsky, L.; Dubrovinskaia, N.; Bykov, M.; McCammon, C.; Ovsyannikov, S.V.; Liermann, H.P.; Kupenko, I.; Chumakov, A.I.; Ruffer, R.; et al. Structural complexity of simple Fe2O3 at high pressures and temperatures. Nat. Commun. 2016, 7, 10661. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Zhao, Y.; Fu, G.; Duchesne, P.N.; Gu, L.; Zheng, Y.; Weng, X.; Chen, M.; Zhang, P.; Pao, C.-W.; et al. Interfacial Effects in Iron-Nickel Hydroxide–Platinum Nanoparticles Enhance Catalytic Oxidation. Science 2014, 344, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Handa, S.; Wang, Y.; Gallou, F.; Lipshutz, B.H. Sustainable Fe–ppm Pd nanoparticle catalysis of Suzuki-Miyaura cross-couplings in water. Science 2015, 349, 1087–1091. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Yuan, K.; Wang, Y.; Li, G.; Guo, J.; Gu, L.; Hu, W.; Zhao, H.; Tang, Z. Metal–organic frameworks as selectivity regulators for hydrogenation reactions. Nature 2016, 539, 76–80. [Google Scholar] [CrossRef] [PubMed]
- Tartaj, P.; Morales, M.P.; Gonzalez-Carreño, T.; Veintemillas-Verdaguer, S.; Serna, C.J. The Iron Oxides Strike Back: From Biomedical Applications to Energy Storage Devices and Photoelectrochemical Water Splitting. Adv. Mater. 2011, 23, 5243–5249. [Google Scholar] [CrossRef] [PubMed]
- Theofanidis, S.A.; Galvita, V.V.; Poelman, H.; Marin, G.B. Enhanced Carbon-Resistant Dry Reforming Fe-Ni Catalyst: Role of Fe. ACS Catal. 2015, 5, 3028–3039. [Google Scholar] [CrossRef]
- Ashok, J.; Kawi, S. Nickel–Iron Alloy Supported over Iron–Alumina Catalysts for Steam Reforming of Biomass Tar Model Compound. ACS Catal. 2014, 4, 289–301. [Google Scholar] [CrossRef]
- Li, D.; Koike, M.; Wang, L.; Nakagawa, Y.; Xu, Y.; Tomishige, K. Regenerability of Hydrotalcite-Derived Nickel–Iron Alloy Nanoparticles for Syngas Production from Biomass Tar. ChemSusChem 2014, 7, 510–522. [Google Scholar] [CrossRef] [PubMed]
- Koike, M.; Li, D.; Nakagawa, Y.; Tomishige, K. A Highly Active and Coke-Resistant Steam Reforming Catalyst Comprising Uniform Nickel–Iron Alloy Nanoparticles. ChemSusChem 2012, 5, 2312–2314. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, D.; Koike, M.; Koso, S.; Nakagawa, Y.; Xu, Y.; Tomishige, K. Catalytic performance and characterization of Ni-Fe catalysts for the steam reforming of tar from biomass pyrolysis to synthesis gas. Appl. Catal. A 2011, 392, 248–255. [Google Scholar] [CrossRef]
- Hu, J.; Buelens, L.; Theofanidis, S.-A.; Galvita, V.V.; Poelman, H.; Marin, G.B. CO2 conversion to CO by auto-thermal catalyst-assisted chemical looping. J. CO2 Util. 2016, 16, 8–16. [Google Scholar] [CrossRef]
- Haider, M.A.; Gogate, M.R.; Davis, R.J. Fe-promotion of supported Rh catalysts for direct conversion of syngas to ethanol. J. Catal. 2009, 261, 9–16. [Google Scholar] [CrossRef]
- Burch, R.; Petch, M.I. Investigation of the synthesis of oxygenates from carbon monoxide/hydrogen mixtures on supported rhodium catalysts. Appl. Catal. A 1992, 88, 39–60. [Google Scholar] [CrossRef]
- Liu, Y.; Tuysuz, H.; Jia, C.-J.; Schwickardi, M.; Rinaldi, R.; Lu, A.-H.; Schmidt, W.; Schuth, F. From glycerol to allyl alcohol: Iron oxide catalyzed dehydration and consecutive hydrogen transfer. Chem. Commun. 2010, 46, 1238–1240. [Google Scholar] [CrossRef] [PubMed]
- Konaka, A.; Tago, T.; Yoshikawa, T.; Nakamura, A.; Masuda, T. Conversion of glycerol into allyl alcohol over potassium-supported zirconia–iron oxide catalyst. Appl. Catal. B 2014, 146, 267–273. [Google Scholar] [CrossRef]
- Grossale, A.; Nova, I.; Tronconi, E. Study of a Fe–zeolite-based system as NH3-SCR catalyst for diesel exhaust aftertreatment. Catal. Today 2008, 136, 18–27. [Google Scholar] [CrossRef]
- Colombo, M.; Nova, I.; Tronconi, E.; Schmeißer, V.; Bandl-Konrad, B.; Zimmermann, L. NO/NO2/N2O–NH3 SCR reactions over a commercial Fe-zeolite catalyst for diesel exhaust aftertreatment: Intrinsic kinetics and monolith converter modelling. Appl. Catal. B 2012, 111–112, 106–118. [Google Scholar] [CrossRef]
- Kandel, K.; Anderegg, J.W.; Nelson, N.C.; Chaudhary, U.; Slowing, I.I. Supported iron nanoparticles for the hydrodeoxygenation of microalgal oil to green diesel. J. Catal. 2014, 314, 142–148. [Google Scholar] [CrossRef]
- Li, X.; Zhai, Z.; Tang, C.; Sun, L.; Zhang, Y.; Bai, W. Production of propionic acid via hydrodeoxygenation of lactic acid over FexOy catalysts. RSC Adv. 2016, 6, 62252–62262. [Google Scholar] [CrossRef]
- Luska, K.L.; Bordet, A.; Tricard, S.; Sinev, I.; Grünert, W.; Chaudret, B.; Leitner, W. Enhancing the Catalytic Properties of Ruthenium Nanoparticle-SILP Catalysts by Dilution with Iron. ACS Catal. 2016, 6, 3719–3726. [Google Scholar] [CrossRef]
- Tamura, M.; Yonezawa, D.; Oshino, T.; Nakagawa, Y.; Tomishige, K. In Situ Formed Fe Cation Modified Ir/MgO Catalyst for Selective Hydrogenation of Unsaturated Carbonyl Compounds. ACS Catal. 2017, 7, 5103–5111. [Google Scholar] [CrossRef]
- Zeng, D.; Liu, S.; Gong, W.; Wang, G.; Qiu, J.; Chen, H. Effect of Surface Properties of Iron Oxide Sorbents on Hydrogen Sulfide Removal from Odor. CLEAN 2015, 43, 975–979. [Google Scholar] [CrossRef]
- Pérez-Rodríguez, S.; Pastor, E.; Lázaro, M.J. Noble metal-free catalysts supported on carbon for CO2 electrochemical reduction. J. CO2 Util. 2017, 18, 41–52. [Google Scholar] [CrossRef]
- Zhang, L.; Wu, H.B.; Lou, X.W. Iron-Oxide-Based Advanced Anode Materials for Lithium-Ion Batteries. Adv. Energy Mater. 2014, 4, 1300958. [Google Scholar] [CrossRef]
- Galvita, V.V.; Filez, M.; Poelman, H.; Bliznuk, V.; Marin, G.B. The Role of Different Types of CuO in CuO-CeO2/Al2O3 for Total Oxidation. Catal. Lett. 2014, 144, 32–43. [Google Scholar] [CrossRef]
- Meledina, M.; Turner, S.; Galvita, V.V.; Poelman, H.; Marin, G.B.; Van Tendeloo, G. Local environment of Fe dopants in nanoscale Fe: CeO2−x oxygen storage material. Nanoscale 2015, 7, 3196–3204. [Google Scholar] [CrossRef] [PubMed]
- Najera, M.; Solunke, R.; Gardner, T.; Veser, G. Carbon capture and utilization via chemical looping dry reforming. Chem. Eng. Res. Des. 2011, 89, 1533–1543. [Google Scholar] [CrossRef]
- Cho, W.C.; Kim, C.G.; Jeong, S.U.; Park, C.S.; Kang, K.S.; Lee, D.Y.; Kim, S.D. Activation and Reactivity of Iron Oxides as Oxygen Carriers for Hydrogen Production by Chemical Looping. Ind. Eng. Chem. Res. 2015, 54, 3091–3100. [Google Scholar] [CrossRef]
- Abad, A.; Mattisson, T.; Lyngfelt, A.; Johansson, M. The use of iron oxide as oxygen carrier in a chemical-looping reactor. Fuel 2007, 86, 1021–1035. [Google Scholar] [CrossRef]
- Zhu, M.; Wachs, I.E. Iron-Based Catalysts for the High-Temperature Water–Gas Shift (HT-WGS) Reaction: A Review. ACS Catal. 2016, 6, 722–732. [Google Scholar] [CrossRef]
- Meshkani, F.; Rezaei, M. A highly active and stable chromium free iron based catalyst for H2 purification in high temperature water gas shift reaction. Int. J. Hydrog. Energy 2014, 39, 18302–18311. [Google Scholar] [CrossRef]
- Galvita, V.; Sundmacher, K. Cyclic water gas shift reactor (CWGS) for carbon monoxide removal from hydrogen feed gas for PEM fuel cells. Chem. Eng. J. 2007, 134, 168–174. [Google Scholar] [CrossRef]
- Galvita, V.; Hempel, T.; Lorenz, H.; Rihko-Struckmann, L.K.; Sundmacher, K. Deactivation of Modified Iron Oxide Materials in the Cyclic Water Gas Shift Process for CO-Free Hydrogen Production. Ind. Eng. Chem. Res. 2008, 47, 303–310. [Google Scholar] [CrossRef]
- Galvita, V.; Schröder, T.; Munder, B.; Sundmacher, K. Production of hydrogen with low COx-content for PEM fuel cells by cyclic water gas shift reactor. Int. J. Hydrog. Energy 2008, 33, 1354–1360. [Google Scholar] [CrossRef]
- Sebastián, D.; Serov, A.; Artyushkova, K.; Gordon, J.; Atanassov, P.; Aricò, A.S.; Baglio, V. High Performance and Cost-Effective Direct Methanol Fuel Cells: Fe-N-C Methanol-Tolerant Oxygen Reduction Reaction Catalysts. ChemSusChem 2016, 9, 1986–1995. [Google Scholar] [CrossRef] [PubMed]
- Galvita, V.V.; Poelman, H.; Marin, G.B. Combined Chemical Looping: New Possibilities for Energy Storage and Conversion. Energy Fuels 2017, 31, 11509–11514. [Google Scholar] [CrossRef]
- Galvita, V.V.; Poelman, H.; Fornero, E.; Saeys, M.; Marin, G.B. Development and Performance of Iron Based Oxygen Carriers for Chemical Looping; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2017. [Google Scholar]
- Buelens, L.C.; Galvita, V.V.; Poelman, H.; Detavernier, C.; Marin, G.B. Super-dry reforming of methane intensifies CO2 utilization via Le Chatelier’s principle. Science 2016, 354, 449–452. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.D.; Siriwardane, R. Mechanism of Methane Chemical Looping Combustion with Hematite Promoted with CeO2. Energy Fuels 2013, 27, 4087–4096. [Google Scholar] [CrossRef]
- Luo, M.; Yi, Y.; Wang, S.; Wang, Z.; Du, M.; Pan, J.; Wang, Q. Review of hydrogen production using chemical-looping technology. Renew. Sustain. Energy Rev. 2018, 81, 3186–3214. [Google Scholar] [CrossRef]
- Protasova, L.; Snijkers, F. Recent developments in oxygen carrier materials for hydrogen production via chemical looping processes. Fuel 2016, 181, 75–93. [Google Scholar] [CrossRef]
- Urasaki, K.; Tanimoto, N.; Hayashi, T.; Sekine, Y.; Kikuchi, E.; Matsukata, M. Hydrogen production via steam−iron reaction using iron oxide modified with very small amounts of palladium and zirconia. Appl. Catal. A Gen. 2005, 288, 143–148. [Google Scholar] [CrossRef]
- Otsuka, K.; Kaburagi, T.; Yamada, C.; Takenaka, S. Chemical storage of hydrogen by modified iron oxides. J. Power Sources 2003, 122, 111–121. [Google Scholar] [CrossRef]
- Corbella, B.M.; Palacios, J.M. Titania-supported iron oxide as oxygen carrier for chemical-looping combustion of methane. Fuel 2007, 86, 113–122. [Google Scholar] [CrossRef]
- Rihko-Struckmann, L.K.; Datta, P.; Wenzel, M.; Sundmacher, K.; Dharanipragada, N.V.R.A.; Poelman, H.; Galvita, V.V.; Marin, G.B. Hydrogen and Carbon Monoxide Production by Chemical Looping over Iron-Aluminium Oxides. Energy Technol. 2016, 4, 304–313. [Google Scholar] [CrossRef]
- Galinsky, N.L.; Shafiefarhood, A.; Chen, Y.; Neal, L.; Li, F. Effect of support on redox stability of iron oxide for chemical looping conversion of methane. Appl. Catal. B Environ. 2015, 164, 371–379. [Google Scholar] [CrossRef]
- Hu, J.; Galvita, V.V.; Poelman, H.; Detavernier, C.; Marin, G.B. A core-shell structured Fe2O3/ZrO2@ZrO2 nanomaterial with enhanced redox activity and stability for CO2 conversion. J. CO2 Util. 2017, 17, 20–31. [Google Scholar] [CrossRef]
- Dharanipragada, N.V.R.A.; Galvita, V.V.; Poelman, H.; Buelens, L.C.; Detavernier, C.; Marin, G.B. Bifunctional Co- and Ni- ferrites for catalyst-assisted chemical looping with alcohols. Appl. Catal. B Environ. 2018, 222, 59–72. [Google Scholar] [CrossRef]
- Galvita, V.; Sundmacher, K. Redox behavior and reduction mechanism of Fe2O3–CeZrO2 as oxygen storage material. J. Mater. Sci. 2007, 42, 9300–9307. [Google Scholar] [CrossRef]
- Zafar, Q.; Mattisson, T.; Gevert, B. Integrated Hydrogen and Power Production with CO2 Capture Using Chemical-Looping ReformingRedox Reactivity of Particles of CuO, Mn2O3, NiO, and Fe2O3 Using SiO2 as a Support. Ind. Eng. Chem. Res. 2005, 44, 3485–3496. [Google Scholar] [CrossRef]
- Leion, H.; Mattisson, T.; Lyngfelt, A. The use of petroleum coke as fuel in chemical-looping combustion. Fuel 2007, 86, 1947–1958. [Google Scholar] [CrossRef]
- Shulman, A.; Linderholm, C.; Mattisson, T.; Lyngfelt, A. High Reactivity and Mechanical Durability of NiO/NiAl2O4 and NiO/NiAl2O4/MgAl2O4 Oxygen Carrier Particles Used for more than 1000 h in a 10 kW CLC Reactor. Ind. Eng. Chem. Res. 2009, 48, 7400–7405. [Google Scholar] [CrossRef]
- Johansson, M.; Mattisson, T.; Lyngfelt, A. Investigation of Fe2O3 with MgAl2O4 for Chemical-Looping Combustion. Ind. Eng. Chem. Res. 2004, 43, 6978–6987. [Google Scholar] [CrossRef]
- Galvita, V.V.; Poelman, H.; Bliznuk, V.; Detavernier, C.; Marin, G.B. CeO2-Modified Fe2O3 for CO2 Utilization via Chemical Looping. Ind. Eng. Chem. Res. 2013, 52, 8416–8426. [Google Scholar] [CrossRef]
- Tang, M.; Xu, L.; Fan, M. Progress in oxygen carrier development of methane-based chemical-looping reforming: A review. Appl. Energy 2015, 151, 143–156. [Google Scholar] [CrossRef]
- Zhu, X.; Li, K.; Wei, Y.; Wang, H.; Sun, L. Chemical-Looping Steam Methane Reforming over a CeO2–Fe2O3 Oxygen Carrier: Evolution of Its Structure and Reducibility. Energy Fuels 2014, 28, 754–760. [Google Scholar] [CrossRef]
- Zeng, T.; Chen, W.-W.; Cirtiu, C.M.; Moores, A.; Song, G.; Li, C.-J. Fe3O4 nanoparticles: A robust and magnetically recoverable catalyst for three-component coupling of aldehyde, alkyne and amine. Green Chem. 2010, 12, 570–573. [Google Scholar] [CrossRef]
- Hudson, R.; Riviere, A.; Cirtiu, C.M.; Luska, K.L.; Moores, A. Iron-iron oxide core-shell nanoparticles are active and magnetically recyclable olefin and alkyne hydrogenation catalysts in protic and aqueous media. Chem. Commun. 2012, 48, 3360–3362. [Google Scholar] [CrossRef] [PubMed]
- Yfanti, V.-L.; Vasiliadou, E.S.; Sklari, S.; Lemonidou, A.A. Hydrodeoxygenation of glycerol with in situ H2 formation over Pt catalysts supported on Fe modified Al2O3: Effect of Fe loading. J. Chem. Technol. Biotechnol. 2017, 92, 2236–2245. [Google Scholar] [CrossRef]
- De Vos, Y.; Jacobs, M.; Van Driessche, I.; Van Der Voort, P.; Snijkers, F.; Verberckmoes, A. Processing and characterization of Fe-based oxygen carriers for chemical looping for hydrogen production. Int. J. Greenhouse Gas Control 2018, 70, 12–21. [Google Scholar] [CrossRef]
- Tu, Y.; Tian, S.; Kong, L.; Xiong, Y. Co-catalytic effect of sewage sludge-derived char as the support of Fenton-like catalyst. Chem. Eng. J. 2012, 185–186, 44–51. [Google Scholar] [CrossRef]
- Luo, M.; Yuan, S.; Tong, M.; Liao, P.; Xie, W.; Xu, X. An integrated catalyst of Pd supported on magnetic Fe3O4 nanoparticles: Simultaneous production of H2O2 and Fe2+ for efficient electro-Fenton degradation of organic contaminants. Water Res. 2014, 48, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Amanatidou, E.; Samiotis, G.; Trikoilidou, E.; Tsikritzis, L. Particulate organics degradation and sludge minimization in aerobic, complete SRT bioreactors. Water Res. 2016, 94, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Bolt, P.H.; Habraken, F.H.P.M.; Geus, J.W. Formation of Nickel, Cobalt, Copper, and Iron Aluminates from α- and γ-Alumina-Supported Oxides: A Comparative Study. J. Solid State Chem. 1998, 135, 59–69. [Google Scholar] [CrossRef]
- Reshetenko, T.V.; Avdeeva, L.B.; Khassin, A.A.; Kustova, G.N.; Ushakov, V.A.; Moroz, E.M.; Shmakov, A.N.; Kriventsov, V.V.; Kochubey, D.I.; Pavlyukhin, Y.T.; et al. Coprecipitated iron-containing catalysts (Fe-Al2O3, Fe-Co-Al2O3, Fe-Ni-Al2O3) for methane decomposition at moderate temperatures: I. Genesis of calcined and reduced catalysts. Appl. Catal. A 2004, 268, 127–138. [Google Scholar] [CrossRef]
- Martins, N.; Martins, L.; Amorim, C.; Amaral, V.; Pombeiro, A. Solvent-Free Microwave-Induced Oxidation of Alcohols Catalyzed by Ferrite Magnetic Nanoparticles. Catalysts 2017, 7, 222. [Google Scholar] [CrossRef]
- Martins, N.; Martins, L.; Amorim, C.; Amaral, V.; Pombeiro, A. First-Row-Transition Ion Metals(II)-EDTA Functionalized Magnetic Nanoparticles as Catalysts for Solvent-Free Microwave-Induced Oxidation of Alcohols. Catalysts 2017, 7, 335. [Google Scholar] [CrossRef]
- Baldychev, I.; Vohs, J.M.; Gorte, R.J. The effect of thermodynamic properties of zirconia-supported Fe3O4 on water-gas shift activity. Appl. Catal. A 2009, 356, 225–230. [Google Scholar] [CrossRef]
- Cabello, A.; Dueso, C.; García-Labiano, F.; Gayán, P.; Abad, A.; de Diego, L.F.; Adánez, J. Performance of a highly reactive impregnated Fe2O3/Al2O3 oxygen carrier with CH4 and H2S in a 500Wth CLC unit. Fuel 2014, 121, 117–125. [Google Scholar] [CrossRef] [Green Version]
- Dharanipragada, N.V.R.A.; Buelens, L.C.; Poelman, H.; De Grave, E.; Galvita, V.V.; Marin, G.B. Mg-Fe-Al-O for advanced CO2 to CO conversion: Carbon monoxide yield vs. oxygen storage capacity. J. Mater. Chem. A 2015, 3, 16251–16262. [Google Scholar] [CrossRef]
- Fan, L.-S.; Zeng, L.; Luo, S. Chemical-looping technology platform. AICHE J. 2015, 61, 2–22. [Google Scholar] [CrossRef]
- Galvita, V.V.; Poelman, H.; Marin, G.B. Combined chemical looping for energy storage and conversion. J. Power Sources 2015, 286, 362–370. [Google Scholar] [CrossRef]
- Iliuta, I.; Tahoces, R.; Patience, G.S.; Rifflart, S.; Luck, F. Chemical-looping combustion process: Kinetics and mathematical modeling. AICHE J. 2010, 56, 1063–1079. [Google Scholar] [CrossRef]
- Dharanipragada, N.V.R.A.; Galvita, V.V.; Poelman, H.; Buelens, L.C.; Marin, G.B.; Longo, A. Insight in kinetics from pre-edge features using time resolved in situ XAS. AICHE J. 2017. [Google Scholar] [CrossRef]
- Wang, J.; Li, C. A study of surface and inner layer compositions of Mg-Fe-Al-O mixed spinel sulfur-transfer catalyst using Auger electron spectroscopy. Mater. Lett. 1997, 32, 223–227. [Google Scholar] [CrossRef]
- Saravanan, G.; Jayasree, K.P.; Divya, Y.; Pallavi, M.; Nitin, L. Ordered intermetallic Pt-Fe nano-catalysts for carbon monoxide and benzene oxidation. Intermetallics 2018, 94, 179–185. [Google Scholar] [CrossRef]
- Jiang, G.; Li, X.; Lv, X.; Chen, L. Core/shell FePd/Pd catalyst with a superior activity to Pt in oxygen reduction reaction. Sci. Bull. 2016, 61, 1248–1254. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, Q.; Wang, Y. Rh-catalyzed syngas conversion to ethanol: Studies on the promoting effect of FeOx. Catal. Today 2011, 171, 257–265. [Google Scholar] [CrossRef]
- Dimitrakopoulou, M.; Huang, X.; Krohnert, J.; Teschner, D.; Praetz, S.; Schlesiger, C.; Malzer, W.; Janke, C.; Schwab, E.; Rosowski, F.; et al. Insights into structure and dynamics of (Mn,Fe)Ox-promoted Rh nanoparticles. Faraday Discuss. 2017. [Google Scholar] [CrossRef]
- Palomino, R.M.; Magee, J.W.; Llorca, J.; Senanayake, S.D.; White, M.G. The effect of Fe–Rh alloying on CO hydrogenation to C2+ oxygenates. J. Catal. 2015, 329, 87–94. [Google Scholar] [CrossRef] [Green Version]
- Yang, N.; Medford, A.J.; Liu, X.; Studt, F.; Bligaard, T.; Bent, S.F.; Nørskov, J.K. Intrinsic Selectivity and Structure Sensitivity of Rhodium Catalysts for C2+ Oxygenate Production. J. Am. Chem. Soc. 2016, 138, 3705–3714. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Göeltl, F.; Ro, I.; Ball, M.R.; Sener, C.; Aragão, I.B.; Zanchet, D.; Huber, G.W.; Mavrikakis, M.; Dumesic, J.A. Synthesis Gas Conversion over Rh-Based Catalysts Promoted by Fe and Mn. ACS Catal. 2017, 7, 4550–4563. [Google Scholar] [CrossRef]
- Phan, T.N.; Ko, C.H. Synergistic effects of Ru and Fe on titania-supported catalyst for enhanced anisole hydrodeoxygenation selectivity. Catal. Today 2017. [Google Scholar] [CrossRef]
- Tharamani, C.N.; Beera, P.; Jayaram, V.; Begum, N.S.; Mayanna, S.M. Studies on electrodeposition of Fe–W alloys for fuel cell applications. Appl. Surf. Sci. 2006, 253, 2031–2037. [Google Scholar] [CrossRef]
- Shi, X.; Yu, H.; Gao, S.; Li, X.; Fang, H.; Li, R.; Li, Y.; Zhang, L.; Liang, X.; Yuan, Y. Synergistic effect of nitrogen-doped carbon-nanotube-supported Cu–Fe catalyst for the synthesis of higher alcohols from syngas. Fuel 2017, 210, 241–248. [Google Scholar] [CrossRef]
- Koike, M.; Hisada, Y.; Wang, L.; Li, D.; Watanabe, H.; Nakagawa, Y.; Tomishige, K. High catalytic activity of Co-Fe/α-Al2O3 in the steam reforming of toluene in the presence of hydrogen. Appl. Catal. B 2013, 140–141, 652–662. [Google Scholar] [CrossRef]
- Unmuth, E.E.; Schwartz, L.H.; Butt, J.B. Iron alloy Fischer-Tropsch catalysts: I. Oxidation-reduction studies of the Fe-Ni system. J. Catal. 1980, 61, 242–255. [Google Scholar] [CrossRef]
- Okamoto, H. Handbook of Binary Alloy Phase Diagrams; ASM International®: Materials Park, OH, USA, 1990. [Google Scholar]
- Kustov, A.L.; Frey, A.M.; Larsen, K.E.; Johannessen, T.; Nørskov, J.K.; Christensen, C.H. CO methanation over supported bimetallic Ni–Fe catalysts: From computational studies towards catalyst optimization. Appl. Catal. A 2007, 320, 98–104. [Google Scholar] [CrossRef]
- Kim, S.M.; Abdala, P.M.; Margossian, T.; Hosseini, D.; Foppa, L.; Armutlulu, A.; van Beek, W.; Comas-Vives, A.; Copéret, C.; Müller, C. Cooperativity and Dynamics Increase the Performance of NiFe Dry Reforming Catalysts. J. Am. Chem. Soc. 2017, 139, 1937–1949. [Google Scholar] [CrossRef] [PubMed]
- Galvita, V.V.; Poelman, H.; Detavernier, C.; Marin, G.B. Catalyst-assisted chemical looping for CO2 conversion to CO. Appl. Catal. B 2015, 164, 184–191. [Google Scholar] [CrossRef]
- Zhou, L.; Enakonda, L.R.; Saih, Y.; Loptain, S.; Gary, D.; Del-Gallo, P.; Basset, J.M. Catalytic Methane Decomposition over Fe-Al2O3. ChemSusChem 2016, 9, 1243–1248. [Google Scholar] [CrossRef] [PubMed]
- Reshetenko, T.V.; Avdeeva, L.B.; Ushakov, V.A.; Moroz, E.M.; Shmakov, A.N.; Kriventsov, V.V.; Kochubey, D.I.; Pavlyukhin, Y.T.; Chuvilin, A.L.; Ismagilov, Z.R. Coprecipitated iron-containing catalysts (Fe-Al2O3, Fe-Co-Al2O3, Fe-Ni-Al2O3) for methane decomposition at moderate temperatures: Part II. Evolution of the catalysts in reaction. Appl. Catal. A 2004, 270, 87–99. [Google Scholar] [CrossRef]
- Kong, J.; Soh, H.T.; Cassell, A.M.; Quate, C.F.; Dai, H. Synthesis of individual single-walled carbon nanotubes on patterned silicon wafers. Nature 1998, 395, 878–881. [Google Scholar] [CrossRef]
- Boskovic, B.O.; Stolojan, V.; Khan, R.U.A.; Haq, S.; Silva, S.R.P. Large-area synthesis of carbon nanofibres at room temperature. Nat. Mater. 2002, 1, 165–168. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Yang, G.; Ma, Q.; Wu, M.; Tan, Y.; Yoneyama, Y.; Tsubaki, N. Confinement Effect of Carbon Nanotubes: Copper Nanoparticles Filled Carbon Nanotubes for Hydrogenation of Methyl Acetate. ACS Catal. 2012, 2, 1958–1966. [Google Scholar] [CrossRef]
- Iijima, S. Helical microtubules of graphitic carbon. Nature 1991, 354, 56–58. [Google Scholar] [CrossRef]
- Pan, X.; Bao, X. Reactions over catalysts confined in carbon nanotubes. Chem. Commun. 2008. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Jin, Y.; Liu, G.; Li, Y. Production of Hydrogen and Nanocarbon from Catalytic Decomposition of Methane over a Ni–Fe/Al2O3 Catalyst. Energy Fuels 2013, 27, 4448–4456. [Google Scholar] [CrossRef]
- Lobo, L.S.; Figueiredo, J.L.; Bernardo, C.A. Carbon formation and gasification on metals. Bulk diffusion mechanism: A reassessment. Catal. Today 2011, 178, 110–116. [Google Scholar] [CrossRef]
- Lobo, L.S. Carbon Formation from Hydrocarbons on Metals. Ph.D. Thesis, Imperial College of Science and Technology, London, UK, 1971. [Google Scholar]
- Rostrup-Nielsen, J.; Trimm, D.L. Mechanisms of carbon formation on nickel-containing catalysts. J. Catal. 1977, 48, 155–165. [Google Scholar] [CrossRef]
- Chesnokov, V.V.; Buyanov, R.A. The formation of carbon filaments upon decomposition of hydrocarbons catalysed by iron subgroup metals and their alloys. Russ. Chem. Rev. 2000, 69, 623–638. [Google Scholar] [CrossRef]
- Shah, N.; Panjala, D.; Huffman, G.P. Hydrogen Production by Catalytic Decomposition of Methane. Energy Fuels 2001, 15, 1528–1534. [Google Scholar] [CrossRef]
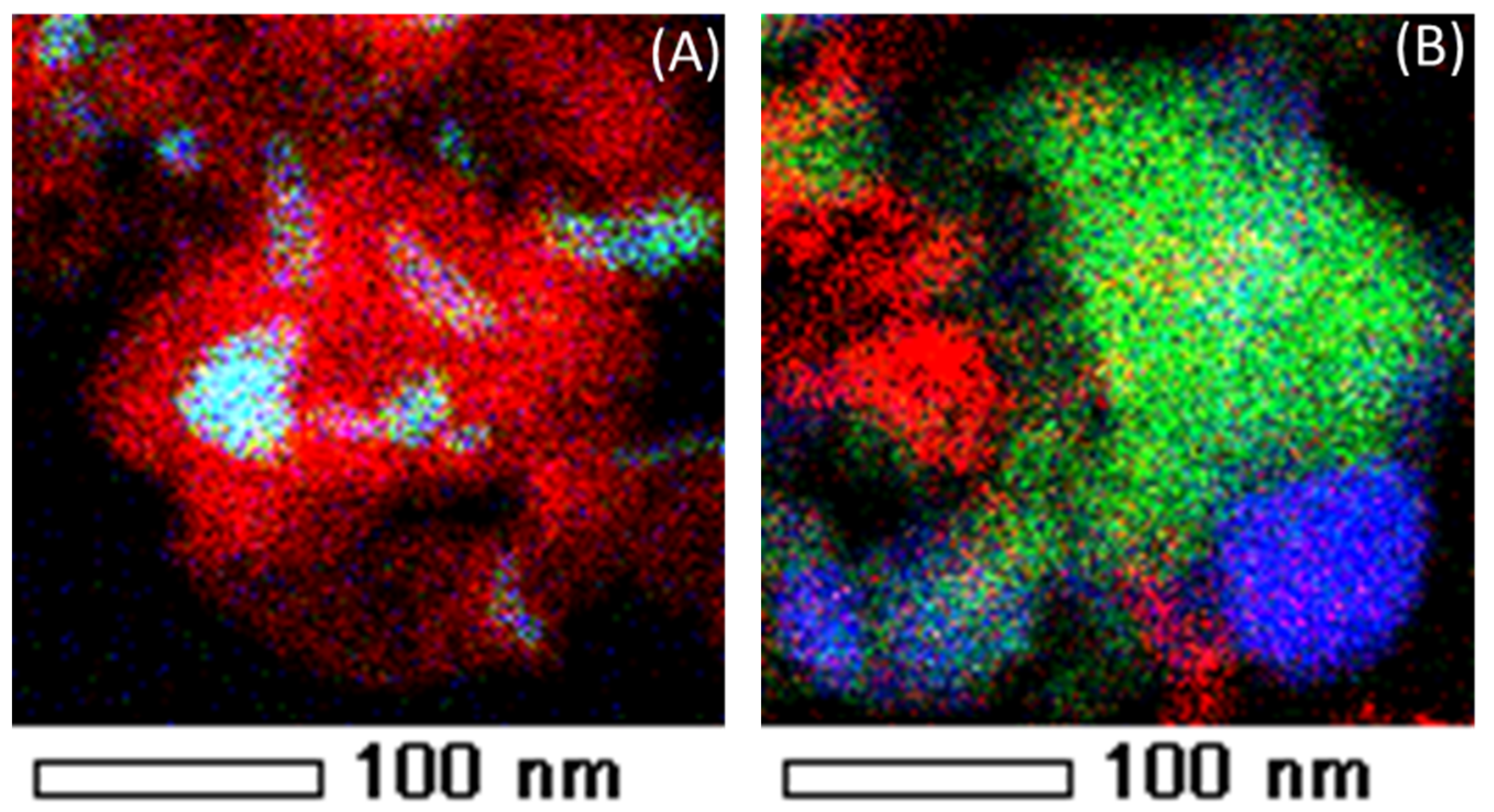
- Theofanidis, S.A.; Galvita, V.V.; Sabbe, M.; Poelman, H.; Detavernier, C.; Marin, G.B. Controlling the stability of a Fe–Ni reforming catalyst: Structural organization of the active components. Appl. Catal. B 2017, 209, 405–416. [Google Scholar] [CrossRef]
- Heidebrecht, P.; Sundmacher, K. Thermodynamic analysis of a cyclic water gas-shift reactor (CWGSR) for hydrogen production. Chem. Eng. Sci. 2009, 64, 5057–5065. [Google Scholar] [CrossRef]
- Theofanidis, S.A.; Batchu, R.; Galvita, V.V.; Poelman, H.; Marin, G.B. Carbon gasification from Fe–Ni catalysts after methane dry reforming. Appl. Catal. B 2016, 185, 42–55. [Google Scholar] [CrossRef]
- Lobo, L.S. Intrinsic kinetics in carbon gasification: Understanding linearity, “nanoworms” and alloy catalysts. Appl. Catal. B Environ. 2014, 148–149, 136–143. [Google Scholar] [CrossRef]
- Lobo, L.S. Catalytic Carbon Gasification: Review of Observed Kinetics and Proposed Mechanisms or Models—Highlighting Carbon Bulk Diffusion. Catal. Rev. 2013, 55, 210–254. [Google Scholar] [CrossRef]
- Zong, N.; Liu, Y. Learning about the mechanism of carbon gasification by CO2 from DSC and TG data. Thermochim. Acta 2012, 527, 22–26. [Google Scholar] [CrossRef]
- Pakhare, D.; Spivey, J. A review of dry (CO2) reforming of methane over noble metal catalysts. Chem. Soc. Rev. 2014, 43, 7813–7837. [Google Scholar] [CrossRef] [PubMed]
- Gardner, T.H.; Spivey, J.J.; Kugler, E.L.; Pakhare, D. CH4–CO2 reforming over Ni-substituted barium hexaaluminate catalysts. Appl. Catal. A 2013, 455, 129–136. [Google Scholar] [CrossRef]
- Gac, W.; Denis, A.; Borowiecki, T.; Kępiński, L. Methane decomposition over Ni–MgO–Al2O3 catalysts. Appl. Catal. A 2009, 357, 236–243. [Google Scholar] [CrossRef]
- Al–Fatish, A.S.A.; Ibrahim, A.A.; Fakeeha, A.H.; Soliman, M.A.; Siddiqui, M.R.H.; Abasaeed, A.E. Coke formation during CO2 reforming of CH4 over alumina-supported nickel catalysts. Appl. Catal. A 2009, 364, 150–155. [Google Scholar] [CrossRef]
- Guo, J.; Lou, H.; Zheng, X. The deposition of coke from methane on a Ni/MgAl2O4 catalyst. Carbon 2007, 45, 1314–1321. [Google Scholar] [CrossRef]
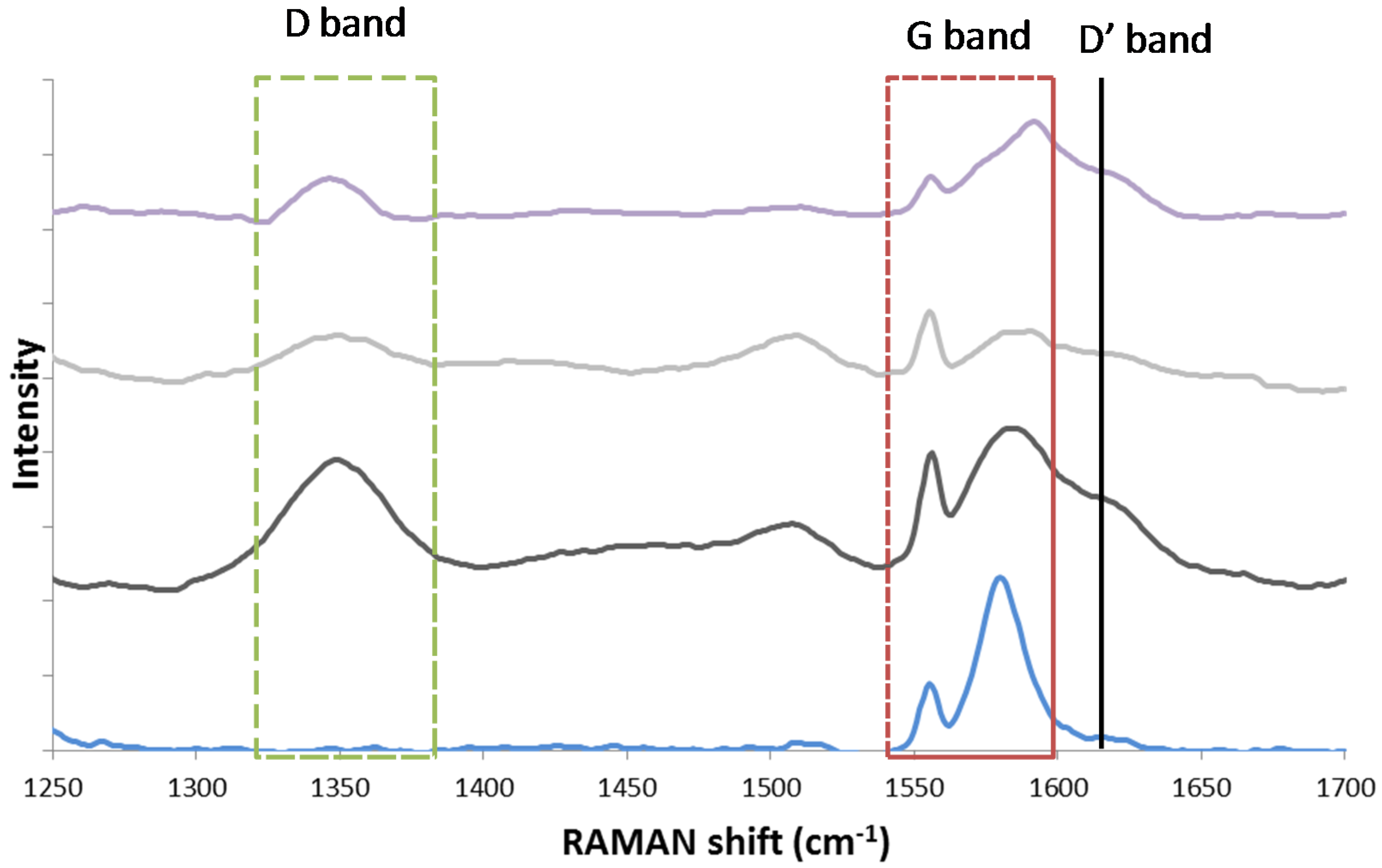
- Espinat, D.; Dexpert, H.; Freund, E.; Martino, G.; Couzi, M.; Lespade, P.; Cruege, F. Characterization of the coke formed on reforming catalysts by laser raman spectroscopy. Appl. Catal. 1985, 16, 343–354. [Google Scholar] [CrossRef]
- Darmstadt, H.; Sümmchen, L.; Ting, J.M.; Roland, U.; Kaliaguine, S.; Roy, C. Effects of surface treatment on the bulk chemistry and structure of vapor grown carbon fibers. Carbon 1997, 35, 1581–1585. [Google Scholar] [CrossRef]
- Jawhari, T.; Roid, A.; Casado, J. Raman spectroscopic characterization of some commercially available carbon black materials. Carbon 1995, 33, 1561–1565. [Google Scholar] [CrossRef]
- Ferrari, A.C. Raman spectroscopy of graphene and graphite: Disorder, electron–phonon coupling, doping and nonadiabatic effects. Solid State Commun. 2007, 143, 47–57. [Google Scholar] [CrossRef]
- De Miguel, S.R.; Jablonski, E.L.; Castro, A.A.; Scelza, O.A. Highly selective and stable multimetallic catalysts for propane dehydrogenation. J. Chem. Technol. Biotechnol. 2000, 75, 596–600. [Google Scholar] [CrossRef]
- Siddiqi, G.; Sun, P.; Galvita, V.; Bell, A.T. Catalyst performance of novel Pt/Mg(Ga)(Al)O catalysts for alkane dehydrogenation. J. Catal. 2010, 274, 200–206. [Google Scholar] [CrossRef]
- Jablonski, E.L.; Castro, A.A.; Scelza, O.A.; de Miguel, S.R. Effect of Ga addition to Pt/Al2O3 on the activity, selectivity and deactivation in the propane dehydrogenation. Appl. Catal. A 1999, 183, 189–198. [Google Scholar] [CrossRef]
- Nawaz, Z.; Wei, F. Hydrothermal study of Pt–Sn-based SAPO-34 supported novel catalyst used for selective propane dehydrogenation to propylene. J. Ind. Eng. Chem. 2010, 16, 774–784. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhou, Y.; Huang, L.; Zhou, S.; Sheng, X.; Wang, Q.; Zhang, C. Structure and catalytic properties of the Zn-modified ZSM-5 supported platinum catalyst for propane dehydrogenation. Chem. Eng. J. 2015, 270, 352–361. [Google Scholar] [CrossRef]
- Coq, B.; Tijani, A.; Figuéras, F. Influence of alloying platinum for the hydrogenation of p-chloronitrobenzene over PtM/Al2O3 catalysts with M: Sn, Pb, Ge, Al, Zn. J. Mol. Catal. 1992, 71, 317–333. [Google Scholar] [CrossRef]


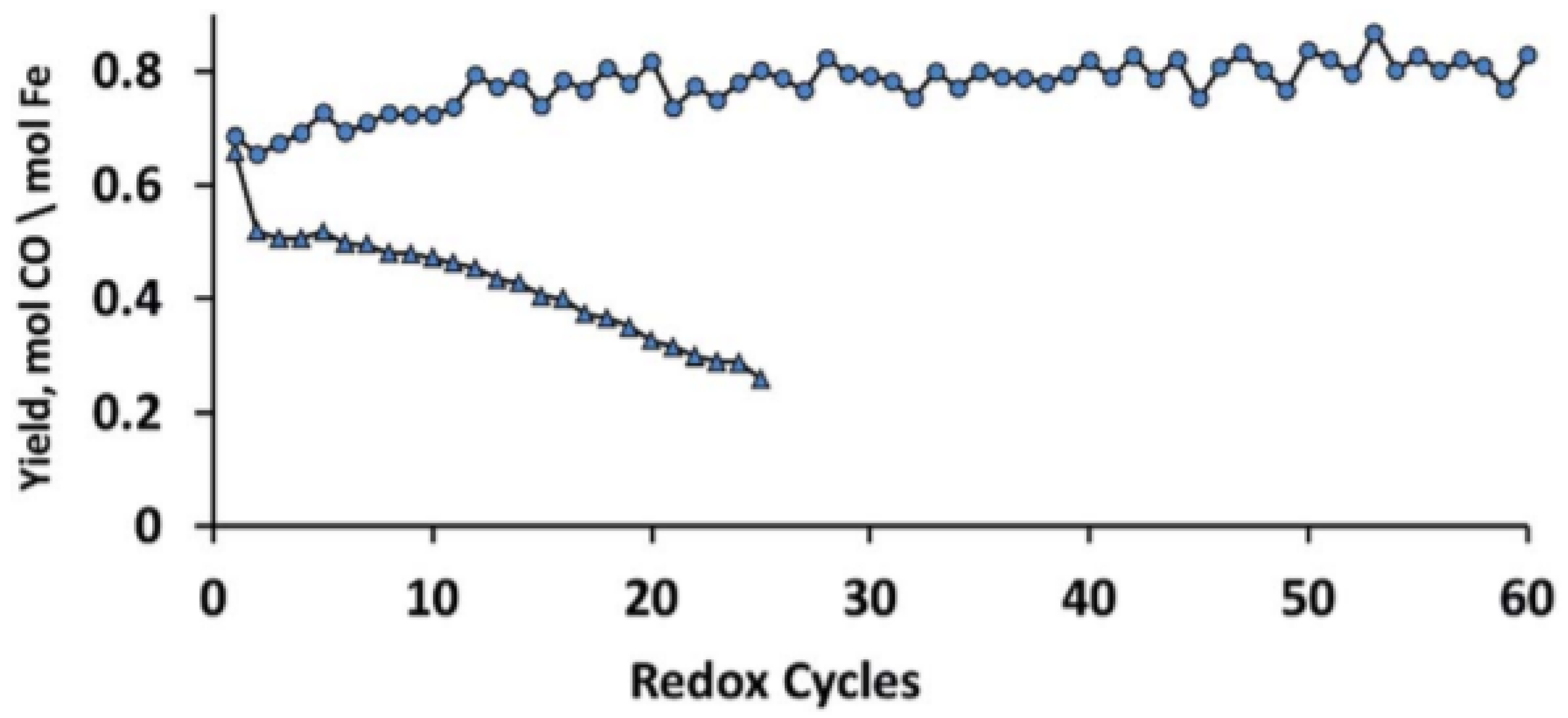
 : iron incorporated in spinel structure;
: iron incorporated in spinel structure;  : separate Fe2O3 phase. Obtained from [81].
: iron incorporated in spinel structure; : separate Fe2O3 phase. Obtained from [81].
: separate Fe2O3 phase. Obtained from [81].
: iron incorporated in spinel structure; : separate Fe2O3 phase. Obtained from [81].













 H2/H2O;
H2/H2O;  H2/CO/H2O/CO2 (equimolar amount of C and H2 corresponding with a feed of CH4 + CO2);
H2/CO/H2O/CO2 (equimolar amount of C and H2 corresponding with a feed of CH4 + CO2);  CO/CO2. Obtained from [49].
H2/H2O; H2/CO/H2O/CO2 (equimolar amount of C and H2 corresponding with a feed of CH4 + CO2); CO/CO2. Obtained from [49].
CO/CO2. Obtained from [49].
H2/H2O; H2/CO/H2O/CO2 (equimolar amount of C and H2 corresponding with a feed of CH4 + CO2); CO/CO2. Obtained from [49].








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ΔEseg (kJ/mol) | Ni3Fe | Ni2PdFe | |
|---|---|---|---|
| Adsorbate Overlayer | Fe ↔ Ni | Fe ↔ Ni | Fe ↔ Pd |
| 0% (vacuum) | +55 | +53 | +104 |
| 100% H | +49 | +52 | +43 |
| 100% CO | +29 | +42 | +3 |
| 100% O | −94 | −76 | −218 |
| 50% CO, 50% O | −25 | −8 | −92 |
| 50% CO, 25% O, 25% H | +1 | +11 | +38 |
| 25% of CH, CO, O, H | +2 | +9 | +143 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Theofanidis, S.A.; Galvita, V.V.; Konstantopoulos, C.; Poelman, H.; Marin, G.B. Fe-Based Nano-Materials in Catalysis. Materials 2018, 11, 831. https://doi.org/10.3390/ma11050831
Theofanidis SA, Galvita VV, Konstantopoulos C, Poelman H, Marin GB. Fe-Based Nano-Materials in Catalysis. Materials. 2018; 11(5):831. https://doi.org/10.3390/ma11050831
Chicago/Turabian StyleTheofanidis, Stavros Alexandros, Vladimir V. Galvita, Christos Konstantopoulos, Hilde Poelman, and Guy B. Marin. 2018. "Fe-Based Nano-Materials in Catalysis" Materials 11, no. 5: 831. https://doi.org/10.3390/ma11050831
APA StyleTheofanidis, S. A., Galvita, V. V., Konstantopoulos, C., Poelman, H., & Marin, G. B. (2018). Fe-Based Nano-Materials in Catalysis. Materials, 11(5), 831. https://doi.org/10.3390/ma11050831