2.1. Oxidation
Niobium is a group 5 transition element in the periodic table. It has high solubility for oxygen, about 9 at.% at 1950 °C [
19]. Alloying aims to reduce this and to slow down the diffusion of oxygen. At 800 °C and 1200 °C the diffusion distances of oxygen in Nb after 100 h are about 2.75 and 15.8 mm, respectively [
49]. Titanium reduces the diffusivity of oxygen in Nb, for example the diffusivity of oxygen in Nb-25Ti is 1/20 that in pure Nb [
50]. The effect of alloying on oxygen transport kinetics in pure Nb and Nb-34Hf-21Al (at.%) at 1300 °C was demonstrated in [
51].
The other two transition metals in group 5, namely V and Ta, also have high solubilities for oxygen, which respectively are about 15 at.% and 6 at.% but the elements in group 6 (Cr, Mo and W) have significantly lower oxygen solubilities [
19]. The aforementioned elements can be in solution in bcc Nb together with simple metals and metalloids. Also, they substitute Nb in Nb
5Si
3. An example of Nb
ss with no Si is the solid solution 63.4Nb-11.7Ti-2.3Hf-11.1Mo-5.7W-6.1Al (see introduction and [
6] for more compositions of Nb
ss in Nb-silicide based alloys). For an example of an actual chemical composition of a Nb
5Si
3 silicide see introduction.
Atomic size is important in diffusion in alloys. The diffusion of solutes to the substrate/oxide interface is one of the factors that control oxidation. Wagner showed that the oxidation rate of Ni-Pt alloys at 850 °C and 1100 °C was essentially determined by the diffusion of Ni to the alloy-NiO interface [
52]. The low oxidation rate of Ni alloys with Be or Si additions compared with the high oxidation rates of Ni alloys with Mo or Cu additions was attributed to differences in the atomic size of solute and solvent [
52]. Silicon and Be, and Cu and Mo respectively have smaller and larger atomic size than Ni. The different oxidation behavior was attributed to enhanced diffusion of Ni due to lattice distortion that increased oxidation rate [
52]. The relationship between solute atomic size and diffusivity in Nb was discussed in [
6].
Research has linked oxygen solubility in binary Nb alloys with the electron per atom ratio (
e/α) [
53]. The latter is often used to discuss phase stability. Minimum oxygen solubility in Nb-X (X = Mo, Re) alloys was reported for (
e/α) ratios of 5.7 and 5.75 for Mo and Re respectively, and was suggested that minimum oxygen solubility occurs when (
e/α) = 5.7 [
54]. The latter was disputed in [
55]. The author’s research group has shown that in Nb-silicide based alloys the concentration of Mo in the alloy is very important for the oxidation behavior [
15].
Oxygen dissolves interstitially in octahedral holes in bcc Nb. The different solubilities of oxygen in metals arise from differences in the binding energy of an oxygen atom to an interstitial site. An oxygen atom in the lattice of an element will cause electron redistribution. As an oxygen atom creates a charge density hole, electrons are excluded from the latter and then some of the excluded electrons go back to the hole. The electrons that were displaced from the excluded region will be accommodated in unoccupied metal orbitals and thus the Fermi level will change.
Phase stability can be considered in terms of (
e/α) (averaged valence of alloying elements in an alloy) and VEC (number of valence electrons per atom filled into the valence band). The (
e/α) ratio is the parameter in the Hume–Rothery rules [
53] and the VEC is essential to determining the Fermi level in the valence band [
56]. The (
e/α) ratio is difficult to use as a universal parameter in alloy design because its value cannot be uniquely assigned to a transition metal as it depends on the surrounding environment. Instead, VEC is a more important parameter in transition metal alloys [
56].
Electronegativity (χ) describes the tendency of an atom to attract electrons. The activity of a metal is correlated with its electronegativity, and metals are categorized as very electropositive (χ < 1.4), electropositive (1.4 < χ < 1.9) and electronegative (1.9 < χ < 2.54). Very electropositive metals oxidize easily. Electropositive metals can form protective metal oxide and their surfaces tarnish in oxygen. Chromium, Ge, Sn, Ti, V belong in this category. Electronegative metals do not form very stable oxides and the latter can decompose on heating. Niobium, Mo, Ta, W belong in this category. Electropositive and electronegative elements are present in the three types of bcc Nb
ss [
6], in the Nb
5Si
3 silicide, where they substitute Nb or Si [
47], in the C14-NbCr
2 Laves phase where they substitute Nb or Si and in A15-Nb
3X compounds [
48], see examples of compositions given in the introduction.
The oxidation of Nb-silicide based alloys starts with the oxidation of Nb
ss grains in the surface, this is followed by the oxidation of Nb
5Si
3 grains (and other intermetallics) in the surface. The oxidation promotes the surface segregation of Nb that subsequently oxidizes [
15,
57]. This is followed by oxidation of Nb
ss and Nb
5Si
3 below the surface and even in the bulk [
15] as well as of other intermetallic phases such as the C14-NbCr
2 Laves and A15-Nb
3X phases (this is often referred to as (phase) contamination by oxygen). The oxidation of the solid solution is more sever compared with the silicide(s) and other intermetallics [
14,
15,
57]. The oxidation of all phases depends on their chemical composition and on the alloy microstructure (volume fractions of phases, shape, size and spatial distributions of phases) that results from processing. The oxidation includes the dissolution of oxygen and the formation of sub-oxides and of several crystalline oxides in various oxidation states and leads to a mixture of oxides or oxygen-saturated metal + oxides [
14,
15].
It is important to consider the types and structure(s) of the oxides forming the scales on Nb-silicide based alloys. Oxides in which Al, Cr, Hf, Mo, Nb, Si, Ta, Ti, V or W can participate must be considered because these elements are key for achieving a balance of properties in Nb-silicide based alloys. Not all these elements are simultaneously or necessarily present in every alloy. However, Si and Ti are always present in the alloys, both tend to segregate to Nb
ss grains in the surface and the segregation of Ti is more severe [
58,
59]. The latter was suggested to promote the selective oxidation of Ti and the early formation of TiO
2 in the scale [
58].
The oxide scales formed on Nb-silicide based alloys contain TiO
2 (rutile), SiO
2, Nb
2O
5 and niobates [
14,
15]. In the latter, TM, RM and simple metals can be present [
14,
15]. The early formation of rutile and Nb oxides in the scale of Nb-silicide based alloys is critical because many of the mixed oxides that form have structures that show analogy with the rutile structure. This is briefly discussed below.
Nb can form the oxides NbO, NbO
2 and Nb
2O
5 [
19], where the valence of Nb respectively is Nb
2+, Nb
3+ and Nb
5+. The latter oxide is the most stable. The most commonly encountered polymorph of the Nb
2O
5 is the monoclinic H-Nb
2O
5, which is the stable form in air above 900 °C. The stoichiometry of all polymorphs is maintained by NbO
6 octahedra. In stoichiometric and slightly reduced Nb
2O
5, the diffusion coefficients for oxygen are up to 200 times greater parallel to the b axis than perpendicular to this direction [
60].
Rutile forms a series of “shear-type” Ti
nO
2n−1 structures (Magneli phases) with 4 ≤ n ≤ 10 [
61]. The structures are made up of layers of rutile structure with a width of n TiO
6 octahedra. The TiO
6 octahedra share faces across the discontinuity or crystallographic shear plane.
Binary (mixed) oxides of Nb and solute elements like Al, or TM or RM can form. Examples include the Al, Cr and V niobates AlNbO
4, CrNbO
4 and VNbO
4 and the binary (mixed) oxides of Nb
2O
5 with TiO
2, Ta
2O
5, V
2O
5, MoO
3 and WO
3. Rutile can also form binary oxides with HfO
2 [
62] and Ta
2O
5 [
63]. There are similarities in the structures of such binary oxides that are important for the diffusion of oxygen and solute elements. Up to about 5 mol % hafnia can be in solution in rutile [
62].
The AlNbO
4 niobate crystallizes in a monoclinic system and in its crystal structure blocks of ReO
3 type can be distinguished, built from distorted MO
6 octahedra. The CrNbO
4 niobate has tetragonal structure (tP6) with prototype TiO
2 (rutile) and its structure consists of (Cr,Nb)O
6 octahedra. The VNbO
4 also has tetragonal structure (tP6) with prototype TiO
2 (rutile). Adherence of CrNbO
4 scale (with small amount of Cr
2O
3) on NbCr
2 Laves phase has been reported after cyclic oxidation in air at 1200 °C that gave a rate of metal loss of 4.9 µm/h [
64].
A very wide range of Ti
(1−x)Nb
xO
2 compositions have the ideal rutile structure [
65]. Binary oxides between Nb
2O
5 and TiO
2 include TiO
2-Nb
2O
5 (TiNb
2O
7) [
66], TiO
2-3Nb
2O
5 (Ti
2Nb
10O
29) [
66,
67,
68], and TiNb
24O
62. Disorder is created by Nb
5+ as a donor dopant in TiO
2. The crystal structure of TiNb
2O
7 contains ReO
3 blocks of corner-sharing MO
6 octahedra. The different stoichiometries and structures of the above oxides are a consequence of variations in the size of these ReO
3 blocks and the way they are joined together.
The metal positions in the structures are occupied by one or other of these elements in a random manner. Each metal atom (Ti and Nb) is coordinated to six oxygen atoms [
69,
70] forming an octahedral grouping (TiO
6 and NbO
6). In TiNb
2O
7 these blocks contain MO
6 octahedra and form a linear column along the b-axis of the unit cell. Perpendicular to the b-axis the columns are bound by crystallographic shear planes. Across the shear planes the MO
6 octahedra share edges [
70]. In all cases, the b axis (the short axis of the structures) is around 3.8 Å, the length of the octahedral body diagonal of the structures. The smaller and lower-charged Ti
4+ ion has a preference for the octahedra at the corners and edges of the blocks. TiNb
2O
7 is very friable, whereas the TiNb
2O
6.42 is very resistant to fracture. These properties are reversible as the binary oxide composition can change in the oxidizing atmosphere.
The structures of binary oxides in the Nb
2O
5-WO
3 system [
70] also derive from blocks of the ReO
3 type, with networks of octahedral MO
6 groups linked by sharing the oxygen atoms at their vertices. The growth axes of the Nb-W oxides are parallel to the infinite extension of the blocks, with a periodicity of 3.8 Å. The structure of Nb
14Mo
3O
44 and Nb
12MoO
33 (isostructural oxides respectively with Nb
14W
3O
44 and Nb
12WO
33) also consists of blocks of ReO
3 type, built up from deformed NbO
6 octahedra sharing only corners. Oxides in the V
2O
5–Nb
2O
5 system consist of corner sharing metal-oxygen polyhedra. The structure of VNb
9O
25 consists of block structures of NbO
6 octahedra, which are shared with VO
4 tetrahedra at each corner of those blocks [
71].
The columbite-tantalite mineral group is the most common group of Nb and Ta bearing minerals. Tantalum is found together with Nb and Ti in at least 15 minerals, and with Nb in 12 minerals. Tantalum is the main impurity in commercial purity Nb. Its oxide, Ta
2O
5, has two polymorphs, a low temperature one (known as L-Ta
2O
5) and the high temperature H-Ta
2O
5. Their structure involves highly distorted TaO
7 and TaO
6 polyhedra [
65] and the periodicity of the structure is sensitive to small concentrations of dopants [
72]. Nb
2O
5 can form a solid solution with Ta
2O
5 and (Ta
1−xNb
x)
2O
5 solid solutions crystallize at a lower temperature than Ta
2O
5.
To summarize, the atomic sizes of the elements that participate in the mixed oxides formed on Nb-silicide based alloys are important because the oxide structures consist of blocks of the ReO3 type with networks of octahedral MO6 groups.
Ta
2O
5 contains a large number of oxygen vacancies [
73]. The loss of oxygen can be suppressed by TiO
2 substitution [
74]. The TiO
2 addition significantly slows down the H- to L- phase reversion [
72]. Ti substitution in the octahedra is more energy favorable [
72]. By doping of TiO
2 in Ta
2O
5, oxygen vacancies are compensated by Ti
4+ ions that are substitutionally incorporated at Ta
5+ sites [
72]. The Ti
4+ has radius similar to Ta
5+, which allows for dense packing in the Ta and O matrix.
The TiTa
2O
7 is isostructural with TiNb
2O
7 and Nb and Ta are compatible in rutile. The latter is often considered as stoichiometric TiO
2 and Nb
5+ and Ta
5+ replace Ti
4+ in the octahedral Ti sites. The charge balance in these sites and interstitial sites is provided by oxygen vacancies. The diffusivities of Ti and oxygen in rutile are significantly higher than those of Nb and Ta that are also higher than the diffusivity of Hf in rutile. Furthermore, the diffusivity of Nb in rutile is four times that of Ta [
75]. Marschall et al. [
75] attributed the high D
NbTiO2/D
TaTiO2 ratio to differences in electronegativity rather than to mass difference after considering the factors that influence the substitution of elements in minerals, namely ionic charge (the same for Nb and Ta), ionic radii (very similar for Nb and Ta) and electronegativity (1.6 for Nb vs. 1.5 for Ta). The latter influences bond strength and thus the potential for substitution in rutile [
75].
The above discussion shows that in the oxidation of Nb-silicide based alloys there is inter-dependence of solubility and diffusivity of oxygen, oxidation and diffusion of Nb and solute elements and chemistry and structure of the oxides on atomic size, electronegativity and VEC.
2.2. Creep
In polycrystalline alloys, creep is controlled by diffusion-controlled dislocation creep, grain boundary sliding and diffusional creep. The creep rate έ is related to the stress σ with the power law relation έ ∝ σ
n. Harper-Dorn creep occurs when n = 1. The exponent n can be approximately equal to 3 (viscous glide of dislocations, activation energy Q
creep approximately equal to the activation energy for interdiffusion of solute atoms Q
interdiffusion), or 5 (climb of dislocations) or 8 (dislocation climb creep under constant microstructure). For the latter two cases Q
creep is approximately equal to the activation energy for self-diffusion Q
self-diffusion. Creep data for dispersion hardened alloys provides support for n ≈ 8. However, the latter alloys can have Q
creep ≠ Q
self-diffusion. Metal matrix composites can have high and variable stress exponent and activation energy, but when a threshold stress (stress below which creep cannot occur) is considered the values of the above parameters are similar to those of n and Q
creep of pure metals and solid solution alloys [
76].
For Nb and the group 5 and 6 transition metals in the periodic table that can be in solution in Nb
ss, often Q
creep is noticeably less than Q
self-diffusion for homologous temperatures less than 0.6 and often much higher at higher homologous temperatures, and the values of n can be in the range 4 to 7 [
77]. For intermetallics that can be present in the microstructures of Nb-silicide based alloys the following is known regarding creep. The compressive creep of NbCr
2 at 1200 °C and σ = 69 MPa gave n = 2 and Q
creep ≈ 130 kJ/mol [
64]. The compressive creep of Nb
3Al at 1000 °C was comparable with that of Ni based superalloys at the same temperature and gave n = 2.86 and Q = 350 kJ/mol [
78]. The latter is close to the activation energy for chemical inter-diffusion in Nb
3Al (Q ≈ 366 kJ/mol [
79]) and the activation energy for the diffusion of Al in Nb (335 kJ/mol [
6]). For the creep of Nb
3Al at 1200 °C the stress exponent n was 2.19 [
78]. For Nb
3Al, nano-indentation creep gave creep exponent 4.2 and Q = 531 kJ/mol, or about 1.5Q
self-diffusion [
79]. The compressive creep of Nb
3Sn (for 25 at.% Sn) at 1400 °C gave n = 4.5 and Q ≈ 450 kJ/mol [
78] (compared with 320 kJ/mol for the activation energy for the diffusion of Sn in Nb [
6]). The creep at 850 and 1000 °C of dual phase alloys based on (Ti,Nb)
5(Si,Al)
3 and (Ti,Nb)
3(Si,Al) could be described in terms of power law creep [
80].
The Nb-silicide based alloys are also known as in situ composites where the two most important phases are considered to be the bcc Nb
ss and tetragonal Nb
5Si
3 (see introduction). Experimental data has given n ≈ 1 for the Nb
5Si
3 silicide and n ≈ 3 for the Nb
ss [
16,
17] (see previous paragraph for n values of other intermetallics that can be present in the microstructures of Nb-silicide based alloys). The volume fractions of these phases can differ between composites. When the matrix is the strong phase (silicide) the composite is an (ductile phase toughened) intermetallic matrix composite (IMC). When the matrix is the weak phase (Nb
ss) the composite is a metal matrix composite (MMC). Either composite type can have more than one type of bcc Nb
ss (see [
6]) and Nb
5Si
3 silicide (meaning βNb
5Si
3 and/or αNb
5Si
3 and/or γNb
5Si
3, see introduction), the silicide can have complex composition with Nb and Si substituted by other solute elements (see example given in the introduction and [
47]) and there is partitioning of Ti and Hf at the interface between Nb
ss and silicide [
9,
47]. The latter interface is rich in Ti and Hf, during exposure to high temperature only the solid solution is homogenized but the interface between solid solution and Nb
5Si
3silicide can continue to be rich in Ti but only on the silicide side [
9]. Furthermore, there can be precipitation of second phase(s) in the silicide [
5,
7,
9,
81,
82]. Alloying affects the creep of all phases (see below), can stabilize the hexagonal γNb
5Si
3 silicide (which is considered undesirable for creep [
1]), can result to significant changes of the properties of tetragonal Nb
5Si
3 [
47] and can promote or slow down the transformation of βNb
5Si
3 to αNb
5Si
3 (for example, see [
5,
7,
9,
81,
82]). A consensus has not been reached about the type(s) of other intermetallic phases that are desirable for the creep of Nb-silicide based alloys. Research on how different microstructures affect the creep of Nb-silicide based alloys is also extremely limited, owing (i) to the limited resources worldwide for the processing of these materials and (ii) the limited availability of material for experimental work (see introduction).
The parameters δ, Δχ and VEC, which are based respectively on atomic size, electronegativity and valence electron concentration and describe the alloying behavior of Nb-silicide based alloys and the phases that are present in their microstructures [
6,
41,
46,
47,
48], depend on electronic structure. Could these parameters be related to creep rate?
Creep refers to the plastic deformation of a material with time at constant stress. Creep theories consider the motion of dislocations in the microstructure. The creep rate έ is related to the dislocation velocity υ (έ = bρυ, υ = Bτ
effmexp(−U/
kT), where ρ is the density of mobile dislocations, b is Burger’s vector, τ
eff is an effective shear stress acting on the dislocation, B is a constant and m and U describe the stress and temperature dependence of the dislocation velocity [
83]). What is the role played by electronic structure in the motion of dislocations in plastic flow? Below, deformation is briefly considered in the context of dislocations and electronic structure.
The deformation of materials can be dislocation mobility controlled (intrinsic resistance to dislocation motion) or dislocation obstacle controlled (extrinsic resistance). Materials in which flow is limited by dislocation mobility are strain rate sensitive and are characterized by a low stress exponent n and a large strain rate exponent. In such materials dislocations propagate by the motion of kinks, in other words the latter limits dislocation mobility. In materials where flow is dislocation obstacle controlled, deformation occurs by the rapid motion of highly mobile dislocations that are free to glide until they become obstructed by extrinsic resistances such as the dislocation substructure, precipitates and phase interfaces and other obstacles (see below), at which point the generation of new dislocations is required to continue deformation. Extrinsic resistances are those in which dislocation lines as a whole interact with some structural feature of a material and include the cutting or bypassing of precipitates (Orowan bowing) by dislocations.
The deformation of bcc metals and Class I solid solutions is controlled by dislocation mobility, i.e., by the motion of kinks. In contrast, in pure fcc metals and Class II solid solution alloys dislocation mobility is controlled by obstacles such as dislocation substructure. The activation volume (V
activation) and the stress exponent n can be used to identify the microstructure mechanisms that control deformation. For the bcc metals Nb and W the stress exponent n respectively is in the range 5 to 7 and 5 to 9 and the activation volume V
activation respectively is 50
b3 and 5
b3 (b is Burger’s vector) [
84] compared with the significantly higher n and V
activation values for metals whose flow is dislocation obstacle controlled. For example, for Cu, n = 100 and V
activation = 2000
b3 [
85]. For Si and Ge (covalent bonding) the V
activation values at 900 °C were 23.4
b3 and 18
b3 respectively and 9
b3 for Ge at 600 °C [
86].
The effect of symmetry on bonding is very important. The band gap between the conduction and valence energy bands forms a basis for properties such as elastic stiffness, plastic flow resistance (dislocation mobility), and crystal structure stability. The larger the band gap the more stable the crystal structure. Dislocation mobilities determine rates of plastic shape change. The arrays of atoms at the cores of dislocations are important for mechanical behavior. The symmetry changes as material passes into and then out of the core of a moving dislocation. Dislocation lines move through kinks that lie along their lengths. The atomic configuration at a kink is very different than it is in the normal crystal structure. Kink mobility is directly related to electronic structure. The activation energy for dislocation mobility in Si, Ge and Sn is proportional to the band gap. In Si (covalently bound element) kinks are very localized [
87]. The mobility of dislocations is very small in covalently bonded crystals like Si.
The range of dislocation mobilities is very large when measured by the stress needed to move a dislocation. This range is from very small in a perfect metal to about G/4π for a covalently bonded crystal (G is the appropriate shear modulus). Extrinsic resistance to dislocation mobility arises from dislocations, stacking faults, vacancies, interstitials, grain boundaries, anti-phase boundaries, twins, precipitates, free surfaces and others. Theories of creep in metals and alloys consider extrinsic resistances and grain size. Dislocations move by becoming intermittently trapped by various obstacles followed by “free run”. The former is caused by extrinsic factors (see above) that create obstacles to dislocation movement. The latter is limited by intrinsic factors, i.e., by the interaction of dislocations with conduction electrons and phonons.
Dislocation mobility depends strongly on chemical bonding. In simple metals the theoretical width of a dislocation is a few atomic spaces but the bonding is very delocalized, thus the energy of a dislocation is nearly independent of its position. In covalently bonded crystals the bonding is highly localized to the regions between pairs of atoms (less than one atomic distance) and this depends strongly on the position of the center of a dislocation. In the transition metals the electrons that contribute most to the cohesion are localized in spd-hybrid bonds. Thus, the cohesive energy is not nearly independent of the atomic configuration.
In bcc metals slip occurs in the <111> direction and the Burger’s vector is α/2<111>. The bonding in bcc metals gives the screw dislocations a non-planar core structure. Dislocations move through the nucleation and propagation of double kinks. There is disagreement about the fundamental slip planes in bcc metals, i.e., the planes where kinks form. According to slip trace analyses at low temperatures slip always occurs on {110} planes and as the temperature increases slip is observed on {110}, {112} and {123} planes in order of increasing rarity. The continuum theory predicts slip on {110} planes at low temperatures and on {112} for temperatures above about 100 K but atomistic simulations show {110} slip at 0 K and at finite temperatures [
88].
For edge and screw dislocations the bonding disregistry respectively is perpendicular to and along the dislocation line direction. The elastic energy of a dislocation is independent of the sense of its Burger’s vector but its mobility need not be because its core may not have mirror symmetry about its mid-point. In bcc metals the mechanical properties are governed by ½<111> screw dislocations, and asymmetry appears when the glide direction is <111>. The nature of the dislocation core dictates the types of kinks that can form on a dislocation line. Dislocation kink mechanisms are linked with symmetry breaking that can be caused by mixed character of dislocation, the symmetry of the crystal lattice and atomic core reconstruction. The latter is dependent on the details of interatomic interaction [
89]. Atomistic simulations of screw dislocation cores in Mo and Ta have shown them to be non-degenerate cores. There is no data for the screw dislocation cores in Cr, Nb and V but it is expected that they are also non-degenerate [
88]. In other words, dislocation cores in bcc metals are non-degenerate and spatially spread. The spreading into {110} planes varies locally depending on local atomic composition [
90]. An atomistic modelling study of kinks on screw dislocation in Si concluded that “the structure of a single kink is characterized by a narrow core and highly stretched bonds between some of the atoms”. Ge has similarities with Si regarding dislocation properties [
91].
Correlations exist between the cohesive properties and electronic structure band. Covalently bonded solids possess intrinsic plastic resistance. The motion of dislocations is limited by the motion of their cores and the core motion is limited by the motion of kinks along the cores. In materials with localized bonding, dislocations are expected to move bond by bond. At a kink the chemical structure is severely disrupted (a chemical bond is broken). This disruption is very localized.
In the microstructures of Nb-silicide based alloys the bcc Nb solid solution(s) co-exist with covalently bound compounds (silicides and other intermetallic compounds, see introduction). The importance of electronic structure for the properties of intermetallic compounds in Nb-silicide based alloys was demonstrated in [
47,
48] using the parameters VEC and Δχ. The latter is related to electronegativity and the former, which gives the number of valence electrons per atom filled into the valence band, is key to determining the Fermi level in the valence band [
56]. Changes in the properties of the intermetallic phases were related to the parameters VEC and Δχ [
47,
48]. For example, the worsening of the creep of alloyed Nb
5Si
3 compared with the unalloyed Nb
5Si
3 was accompanied by decrease of VEC and increase or decrease of Δχ depending on alloying addition(s) [
47], and the better creep of Nb(Cr,Si)
2 Laves phase compared with the unalloyed NbCr
2 Laves phase was related to the decrease of the VEC and Δχ parameters [
48]. In [
41] it was shown that the hardness of microstructures containing both Nb
ss and Nb
5Si
3 or Nb
ss, Nb
5Si
3 and A15-Nb
3X increased as the VEC parameter of such microstructures increased. The same trend between hardness and VEC was observed for the hardness of the A15-Nb
3X phases in the Nb-silicide based alloys [
48], and for β(Nb,Ti)
5Si
3 and alloyed tetragonal Nb
5Si
3 [
41]. The strong relationship between the hardness and VEC of eutectics with Nb
ss and Nb
5Si
3 was attributed to the covalent bonded intermetallic phase(s) in the eutectics, with the latter being the key phases that determined the hardness of the eutectics [
41]. Most importantly, the trends between the C
44 and VEC and hardness and VEC of α(Nb,Ti)
5Si
3 were the same as those reported for transition metal covalently bonded carbonitrides [
41]. Also the trend between the C
44 and VEC of β(Nb,Ti)
5Si
3 was the same as that reported for covalently bonded M
2AlC compounds [
41].
Dislocation mobility is fundamental to mechanical behavior and plastic deformation of materials including creep. How atomic size, electronegativity and valence electron concentration can elucidate the creep of Nb-silicide based alloys? What role can the electronic structure play in the creep of Nb-silicide based alloys? Are mechanisms related to electronic structure (i.e., intrinsic mechanisms) important in the creep of Nb-silicide based alloys? The following discussion will make a case that the link between the creep of Nb-silicide based alloys and their parameters δ (related to atomic size), Δχ and VEC, which relate to the electronic structure of alloys, is attributed primarily to the covalently bound intermetallics in their microstructures and to the increase of the covalency of the Nb
ss with alloying. In
Section 2.3 it will be shown that there exist relationships between creep rate and each of the above parameters.
The electronic structure that underlies the structural geometry of elements plays a key role in determining the mechanical behavior of metals and alloys. Chemical bonding, which is provided by electronic structure, is crucial to mechanical behavior. The sizes of atoms are determined by energies of electrons in occupied quantum states. Interactions between atoms provide cohesion via the redistribution of the bonding electrons between the atoms. The spatial distribution of bonding electrons is key to mechanical behavior.
The elastic constants depend on structural geometry and the corresponding electronic structure. The shear coefficients are the most important sub-sets of the elastic constants. The Young’s modulus is a function of the bulk modulus B and the shear modulus G. The latter rather than the former appears in the equations of dislocation theory. Both B and G are fundamental coefficients for describing mechanical behavior.
The primary factor that determines elastic stiffness is chemical constitution because the latter determines bonding. The covalent bonds are the stiffest. A key parameter in bonding is the size of atoms (bond length). A key parameter for elastic stiffness is the valence electron concentration. In covalent bonds the charge associated with electron pairs is localized. In metallic bonds the bonding electrons are delocalized.
The shear moduli depend on both the shear plane and the shear direction and the structures of both of these depend on crystal symmetries and local atomic structure. For simple metals (those bonded primarily by s- and p-level electrons) as the number of valence electrons increases the bulk stiffness increases, and decreases as the atomic size increases. Only the s and p quantum states contribute to the cohesion of these elements. For the transition metals the occupied d and f states play an important role in the cohesion. The valence electron concentration is important for the bulk modulus of the covalently bonded Ge, Si and Sn. In covalently bonded materials the shear moduli can be significantly larger compared with the bulk moduli.
The most characteristic shear moduli for cubic symmetry are the C
44 (shear on (100) planes), (C
11-C
12)/2 (shear on (110) planes in the face diagonal directions) and 3C
44(C
11-C
12)/[4C
44 + (C
11-C
12)] (shear on (111) planes).
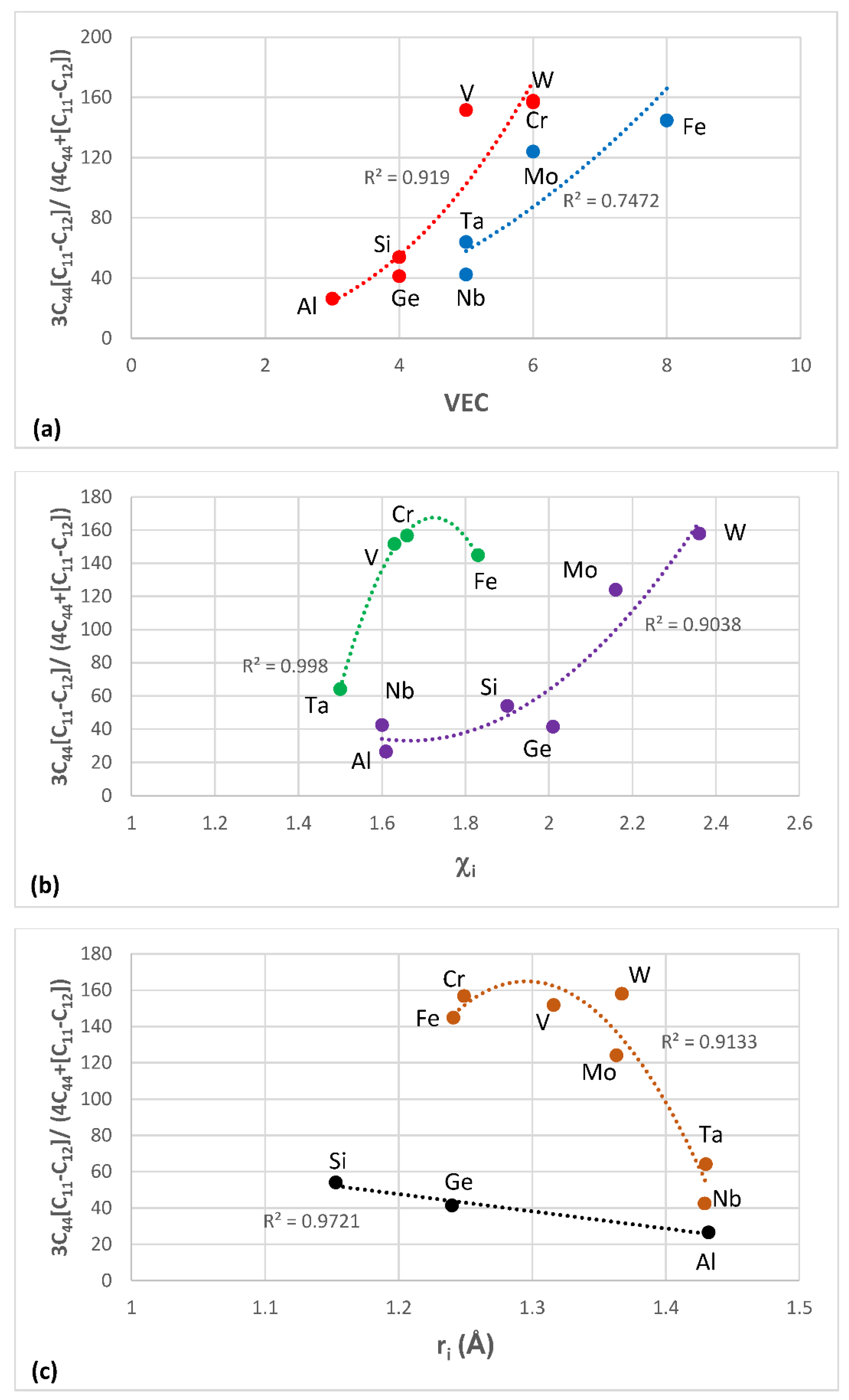
Figure 1,
Figure 2,
Figure 3 and
Figure 4 show data for shear moduli, Zener anisotropy factor (see below) and G/B ratios of Nb and other cubic and hexagonal metals in Nb-silicide based alloys, 5-3 silicides and A15 compounds. The lines in these figures are provided to highlight trends for different groups of metals and compounds.
The shear moduli of metals of cubic symmetry that are alloying additions in Nb-silicide based alloys belong in different groups when the aforementioned characteristic shear moduli are plotted versus VEC, Pauling electronegativity χ
i and atomic size r
i.
Figure 1 shows plots of the latter of the above moduli parameters versus VEC, χ
i and r
i. The same groups of elements as in
Figure 1a–c respectively were in plots of C
44 versus VEC and (C
11-C
12)/2 versus VEC (not shown), in plots of C
44 versus χ
i and (C
11-C
12)/2 versus χ
i (not shown) and in plots of C
44 versus r
i and (C
11-C
12)/2 versus r
i (not shown).
The G/B ratio is positive, is small for ductile materials and increases as the materials becomes increasingly rigid and more brittle. For covalently bonded solids the ratios C
44/B, [C
11-C
12]/[2B] and 3C
44(C
11-C
12)/[B(4C
44 + (C
11-C
12))] are high and are greater than one for diamond (about 1.3, 1.08 and 1.14 respectively [
92]). In such solids the dislocation mobility is significantly reduced compared with elements with metallic bonding. Increased covalency due to alloying would result to decreased dislocation mobility.
Figure 2 shows plots of the second of the above ratios versus VEC, χ
i and r
i. The same groups of elements as in
Figure 2a–c respectively were in plots of C
44/B versus VEC and 3C
44(C
11-C
12)/[B(4C
44 + (C
11-C
12))] versus VEC (not shown), in plots of C
44/B versus χ
i and 3C
44(C
11-C
12)/[B(4C
44 + (C
11-C
12))] versus χ
i (not shown) and in plots of C
44/B versus r
i and 3C
44(C
11-C
12)/[B(4C
44 + (C
11-C
12))] versus r
i (not shown). Note that the colours indicate the same groups of elements in
Figure 1a and
Figure 2a,
Figure 1b and
Figure 2b and
Figure 1c and
Figure 2c.
The Zener anisotropy constant A = 2C
44/[C
11-C
12] gives the deviation from isotropy for cubic, tetragonal and hexagonal structures [
93].
Figure 3a is a plot of this parameter versus atomic radius r
i of cubic and hexagonal symmetry elements in Nb-silicide based alloys. Note that the hexagonal metals Hf, Ti, Y and Zr are in the same group with Al, Ge, Si and Ta. Also note that the grouping of elements in
Figure 1,
Figure 2 and
Figure 3a is in accordance with the separation of Nb-silicide based alloys in different groups that was discussed in [
46]. The grouping of elements would also indicate that if there were to be relationships between mechanical properties and alloy parameters VEC, δ and Δχ, they would apply for specific groups of elements in Nb-silicide based alloys. The data for C
11, C
12 and C
44 for the elements in
Figure 1,
Figure 2 and
Figure 3a is from [
94] (Al), [
95] (Cr), [
96] (Fe, V), [
97] (Ge), [
98] (Mo), [
99] (Nb, Si), [
100] (Ta), [
101] (W) and [
102] (Hf, Ti, Y, Zr).
Data for the Zener parameter A for 5-3 silicides with prototypes W
5Si
3 or Cr
5B
3 is plotted versus atomic radius respectively in
Figure 3b,c and for A15-Nb
3X compounds in
Figure 3d. The R
2 values in
Figure 3b,c correspond to all the data. Note that the plots based on atomic radius show that the effect of alloying on the Zener parameter of tetragonal Nb
5Si
3 is weak. The metalloid elements form covalent bonds with metals in intermetallic compounds, which tend to reduce the dislocation mobility in these compounds. The latter have negligible ductilities because of the low mobilities of dislocations.
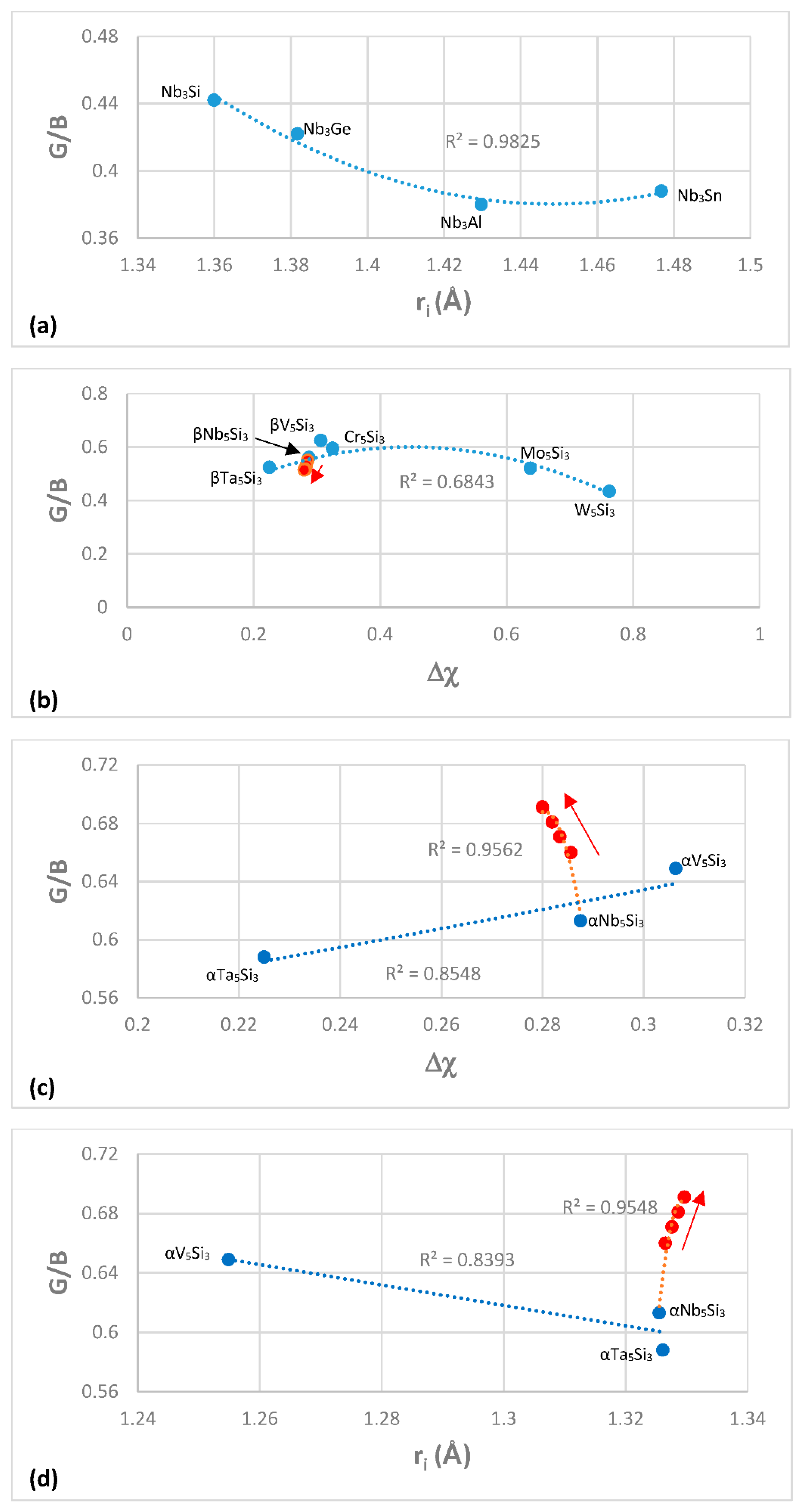
Figure 4 shows plots of G/B for A15-Nb
3X compounds and 5-3 silicides with prototypes W
5Si
3 or Cr
5B
3. The G/B data is plotted versus atomic radius in
Figure 4a,d and versus Δχ in
Figure 4b,c. Note similar trends in
Figure 3d and
Figure 4a for the A15 compounds. Also note that the plots of G/B versus r
i or Δχ show clearly the strong effect of alloying with Ti on increasing the G/B ratio and thus the covalency of α(Nb,Ti)
5Si
3.
Figure 4c,d also show that alloying with Ti has a stronger effect on the covalency of αNb
5Si
3 compared with βNb
5Si
3. The data for G/B and C
11, C
12 and C
44 for the 5-3 silicides and A15-Nb
3X compounds is from [
103] (Cr
5Si
3), [
99] (Nb
5Si
3), [
104] ((Nb,Ti)
5Si
3), [
100] (Mo
5Si
3, Ta
5Si
3), [
105] (W
5Si
3, V
5Si
3), [
106] (Nb
3Sn) and [
107] (Nb
3Si).
Elastic properties like Young’s modulus and shear modulus depend on actual composition. Chan reported that the Young’s modulus of tetragonal unalloyed Nb
5Si
3 is reduced when Nb is substituted by Ti and as the concentration of Ti in (Nb,Ti)
5Si
3 increases (the structure changes to hexagonal (Ti,Nb)
5Si
3) the modulus decreases further towards that of Ti
5Si
3 [
108]. Papadimitriou et al. [
104] showed that this is the case only of βNb
5Si
3 and that the substitution of Nb by Ti in αNb
5Si
3 increases the Young’s modulus.
The shear moduli increase relatively more than the bulk moduli; thus, the G/B ratio is greater for intermetallic compounds compared with metals. For example, for Nb the G/B ratio is 0.228, for βNb
5Si
3 this ratio is 0.54 and for αNb
5Si
3 is 0.613. In other words, the tetragonal Nb
5Si
3 is more than twice as rigid as Nb. For the Nb solid solutions with no Sn or B additions that were given in the Table 1 in reference [
6] the G/B ratio increases with alloying and is 54% higher than that of pure Nb for the 40.4Nb-31.5Ti-1.6Si-2.7Hf-1Ge-15Cr-7.8Al solid solution, which is about 90% of the G/B ratio of γNb5Si3 [
104]. Alloying can increase further the G/B ratio of the Nb
ss, for example in one alloy recently studied in our group [
109] the G/B ratio of the Nb
ss with no Si was 0.403, i.e., higher than that of γNb
5Si
3 (the G/B ratios of the aforementioned Nb
ss were calculated using the rule of mixtures). These high G/B ratios would also suggest that the Nb
ss becomes increasingly rigid and more brittle as the ratio is increased. The larger G/B ratios of the tetragonal Nb
5Si
3 and alloyed solid solution indicate that the dislocation core energies depend strongly on their positions.
The location of the Fermi level is indicative of phase stability. For intermetallics, a pseudo-gap in the density of states is observed close to the Fermi level owing to the combined effects of charge transfer and hybridization. For example, when there is large electronegativity difference between elements, the redistribution of electrons changes the shape of the band, the screening electrons are assigned to low states in the band and this gives the minimum in the density of states curve. The intermetallic is stable when the Fermi level is exactly at the pseudo-gap and unstable in the antibonding region (Fermi level to the right of the pseudo-gap). Alloying may stabilize a metastable intermetallic phase. When the Fermi level is to the left of the pseudo-gap (bonding states) not all bonding states are completely filled and additional electrons are needed to increase stability. If the Fermi level falls on a peak in the density of state curve of the intermetallic the D
self-diffusion of the latter is increased. Considering the Ti and Hf rich Nb
ss/Nb
5Si
3 interfaces in Nb silicide-based alloys and their contamination by oxygen and formation of hafnia near Hf rich areas of the Nb
5Si
3 [
15,
57,
81], the changes of the Fermi level resulting from alloying will affect the stability of phases and their properties, both of which are important in creep.
In reference [
47] it was shown that the alloying of Nb
5Si
3 changed the position of the Nb
5Si
3 silicide in Δχ versus VEC maps and that the changes (meaning increase or decrease) of these parameters depended on specific alloying addition(s). The effect of the substitution of Nb by Ti was clearly demonstrated, as was the substitution of Si by Ge or Sn and the alloying of the silicide with B in the Figures 5 and 6 in reference [
47]. The specific case of the substitution of Nb only with Ti in Nb
5Si
3 silicides was studied using first-principles calculations in [
104] to find out how the stability of different Nb
5Si
3 silicides and their elastic properties are affected with increasing Ti concentration. It was shown that for all 5-3 silicide structures (meaning tetragonal αNb
5Si
3 and βNb
5Si
3 and hexagonal γNb
5Si
3) the main contribution to the total electronic density of states (TDOS) was the partial electronic density of states (PDOS) of d electron states, followed by the p electron states. The s electron states contributed the least to the TDOS of all structures. For the unalloyed Nb
5Si
3 silicide the gradual decrease of phase stability from tetragonal to hexagonal Nb
5Si
3 silicide was explained by the location of the Fermi level of each silicide. The hexagonal γNb
5Si
3 silicide became stable compared with the tetragonal αNb
5Si
3 and βNb
5Si
3 when the Ti concentration reached 50 at.%. For the αNb
5Si
3 and γNb
5Si
3 silicides the shear and Young’s moduli increased with increasing Ti addition and decreased in the case of βNb
5Si
3. The substitution of Nb by Ti strengthened atomic bonding in αNb
5Si
3 and γNb
5Si
3, and decreased bond strength in βNb
5Si
3. The above discussion shows that the Nb
ss/Nb
5Si
3 interface in Nb-silicide based alloys is dynamic, meaning as the local chemistry changes during exposure to high temperature so do the mechanical properties of the interface.
The creep properties of Nb-silicide based alloys are key for their application at high homologous temperatures where diffusion is important. Diffusivities in the solid solution and silicide will depend on composition and in the case of the silicide will also depend on crystal structure. The latter can change as solutes partition to the silicide. The case for Ti was demonstrated in [
104]. These changes also will be important in oxidation. Activation energies for creep may be strongly dependent on concentration(s) of impurities, for example contamination of Nb
ss (mainly) and silicide(s) by oxygen near the surface and below it and even in the bulk alloy microstructure is possible in Nb-silicide based alloys, depending on alloy composition [
14,
15,
57]. Relationships between atomic radius and electronegativity of solute elements in Nb and their activation energies for diffusion and diffusivity at 1200 °C were discussed in [
6].
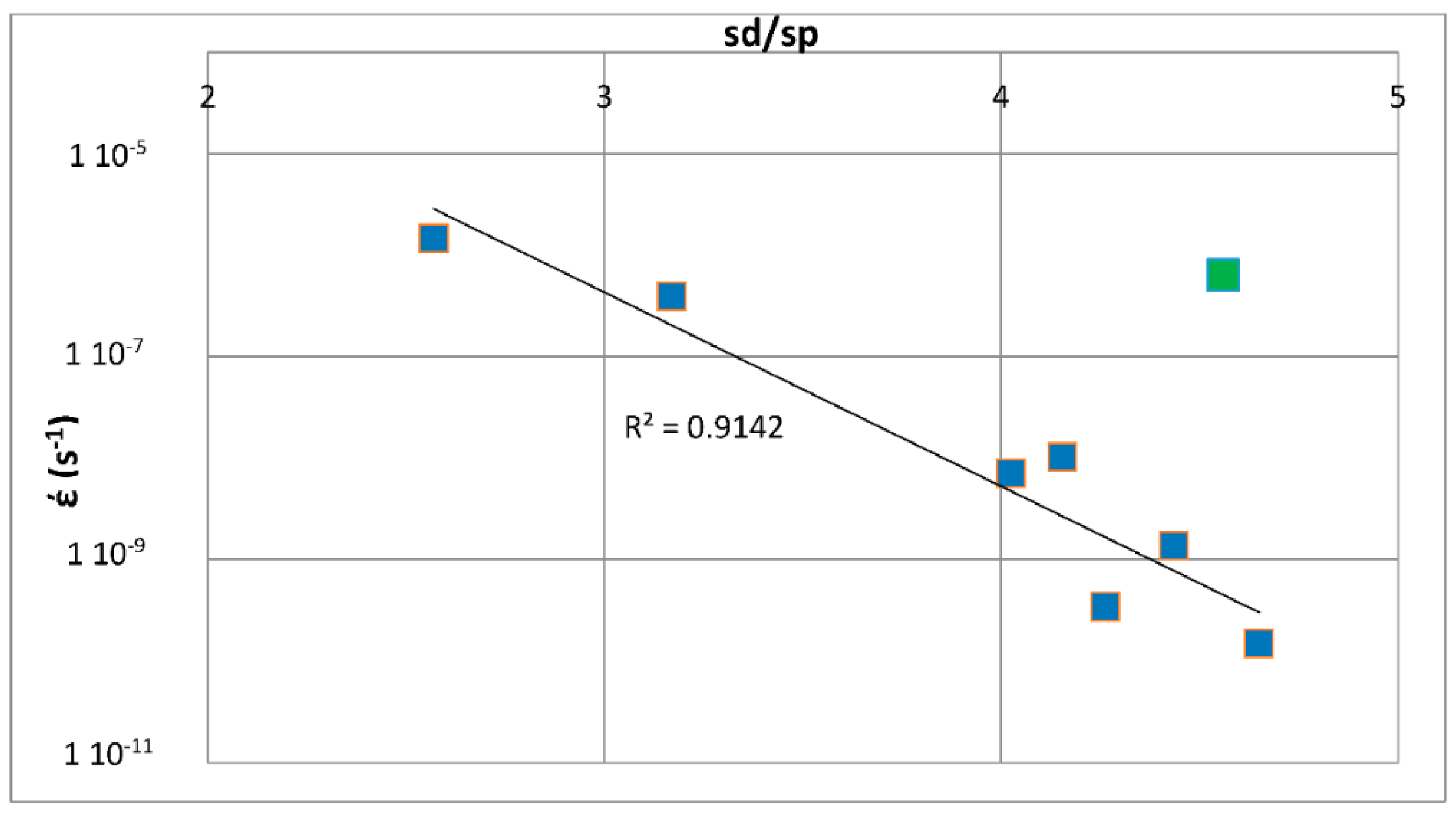
The above discussion has indicated that if there were to be relationships between the creep of Nb-silicide based alloys and their parameters δ, Δχ and VEC, such relationships (i) would be about the contributions to creep rates made by intrinsic resistances not extrinsic ones and (ii) would not give the contributions made separately by intrinsic resistances to each parameter. In other words, any relationships between creep rate and parameters would be averaging the contributions of intrinsic resistances to creep that are expressed by a relationship between creep rate and a parameter. In the following parts of this paper it will be shown that as a matter of fact the available experimental data does give relationships between creep rates and each of the alloy parameters δ, Δχ and VEC.
Phases in Nb-Silicide Based Alloys: Δχ versus VEC Maps and Creep
The alloying behavior of bcc Nb solid solutions, tetragonal Nb
5Si
3, eutectics with Nb
ss and Nb
5Si
3, hexagonal C14-NbCr
2 Laves phases and cubic A15-Nb
3X compounds that are formed in Nb-silicide based alloys was studied, respectively in [
6,
41,
47,
48] and the data is summarized in the Δχ versus VEC maps in
Figure 5.
Figure 5a shows the phases without the eutectic. The data for the eutectic is included in
Figure 5b, where the data for Nb
ss and Nb
5Si
3 alloyed with B has been excluded (eutectics with solid solution and 5-3 silicide are formed in B containing Nb-silicide based alloys, but currently there is no data about the actual chemical composition of these eutectics). Note that some data in
Figure 5b for the eutectic is in the areas that were occupied by the data for B containing Nb
ss and Nb
5Si
3 in
Figure 5a. Details of the solid solution area in the map are shown in the Δχ versus VEC map in the
Figure 6.
Figure 6A has the data for all solid solutions in cast and heat-treated alloys [
6].
Figure 6B shows the data for the Ti rich Nb
ss and the Nb
ss with no Si [
6]. The former is not stable after heat treatment.
There is a gap in Δχ values of the Nb
ss in
Figure 6, which cannot be easily recognized in
Figure 5, and no solid solutions fall in the range 0.13 < Δχ < 0.18. This gap in the Δχ values of the Nb
ss was discussed in [
6]. The solid solutions with no B, Ta and V have Δχ > 0.18 and the solid solutions with no W have Δχ < 0.13. In
Figure 6B in the bottom ellipse are the Ti rich Nb
ss with no refractory metals (RMs) (series 2). These solid solutions have 30 < Ti < 47 at.%, 9 < Cr < 16 at.% and 2 < Ti/Cr < 4. The Ti rich Nb
ss with RMs are in the bottom of the top ellipse. These solid solutions are lean in Mo and W and have 5 < (Mo + W) < 10 at.%, 3 < (Mo/W) < 5 and 2 < {Ti/(Mo + W)} < 6. In
Figure 6B the Nb
ss with no Si (series 3) has 0.23 < Δχ < 0.33. This type of solid solution has 1 < (Mo/W) < 3, 0 < {Ti/(Mo+W)} < 1 and (Mo + W) > 14 at.%.
Data for the creep of bcc Nb solid solutions, tetragonal Nb
5Si
3, hexagonal C14-NbCr
2 Laves phases and cubic A15-Nb
3Al are shown in
Figure 7A and details of the Nb
5Si
3 silicide data and the Nb
ss data are shown respectively in
Figure 7B,C. Note that the phases in
Figure 5a are represented in
Figure 7A. The creep data for the intermetallics was discussed in [
47,
48].
Figure 7A shows the gradual decrease of the value of the exponent n and the shift towards lower creep rates and higher stresses from the left-hand side (occupied by the solid solution) to the right hand side of the figure (occupied by the Nb
5Si
3 silicide). The values of the creep exponent n of the silicide and solid solution are given in
Figure 7B,C, respectively. The n values for Nb
3Al, NbCr
2 and Nb-55Cr-15Si, respectively are 3.39, 1.8 and 0.99.
The Nb solid solutions for which there is creep data for 1200 °C (
Figure 7C) are shown in
Figure 6B. In
Figure 7A,C the creep data for the Nb-1Si and Nb-46Ti-1Si solid solutions shows the adverse effect that Ti has on the creep of the solid solution. This is further supported by the data for the leaner in Ti solid solution of composition Nb-27Ti-5Hf-2Al-2Cr-0.9Si. The positions of the Ti rich solid solutions Nb-46Ti-1Si and Nb-27Ti-5Hf-2Al-2Cr-0.9Si with regard to the Ti rich Nb
ss are shown in
Figure 6B. Notice that the solid solutions Nb-1Si and Nb-5.4Hf-2Ti are outside the area for the Ti rich Nb
ss.
In
Figure 7C there are two sets of data for the solid solution alloy Nb-5.4Hf-2Ti [
113], which, as indicated in the caption, correspond to two different activation energies in έ = A(σ/E)
nexp(−Q/RT), where E is the Young’s modulus [
114]. The higher activation energy is closer to the activation energy for the diffusion of Hf in Nb [
6]. The solid solution alloy Nb-5.4Hf-2Ti (known commercially as alloy C103) is considered as a Class I solid solution regarding its creep (n = 3). In this alloy, the Hf atoms rather than the Ti atoms were considered to be responsible for the solute effects on dislocation motion [
113]. It has been suggested [
113] that there is a critical concentration of Hf beyond which this element does not contribute significantly to strengthening at high temperatures. If the data for (d) and (e) in
Figure 7C is correct, it would indicate a positive effect of Hf for the creep of Nb at low concentrations.
Under creep conditions the constituent phases in Nb-silicide based alloys, namely the Nb
ss and intermetallics (silicides and others) can be rigid and/or creeping. Models [
16] of the creep behavior of these alloys and comparison of the results of modelling with experimental data for alloys with about 37% volume fraction silicide, (i.e., with composites with weak matrix, see above) have shown that rigid Nb silicides have high creep exponent and poor creep. Chan [
16] concluded (a) that creeping silicides with low creep exponent (n ≈ 1) are desirable, (b) that creeping or rigid phases with n ≥ 3 are undesirable, (c) that desirable phases for optimum creep should be rigid at low stresses and creep with low n at higher stresses and (d) that diffusional creep and Harper-Dorn creep in intermetallics are preferred over power law creep with n > 1. The creep of composites with strong (Nb
5Si
3) matrix has been simulated by Henshall et al. [
17] using data for bulk unalloyed Nb
5Si
3 and Nb-1.25Si solid solution. This work under-predicted steady state creep rates and over-predicted primary creep strains compared with experimental data for Nb-10Si.
Elastic constants depend on interatomic bonding and are important to understanding how the material will deform (see previous section). In reference [
6] it was shown that the solutes in Nb belong in different groups when activation energy for diffusion (Q) and diffusivity (D) were plotted against atomic size or electronegativity.
Figure 8 shows the Young’s (E) moduli of solute elements in Nb-silicide based alloys against atomic radius (
Figure 8a) and electronegativity (
Figure 8b). The solute elements fall in three groups with Boron in one of these groups (series c in both parts of
Figure 8), consistent with the data for Nb-silicide based alloys in [
46]. The transition metal (TM) and refractory metal (RM) elements in the series a, b and c in
Figure 8 are in agreement with the ranking of substitutional solutes in binary Nb-X alloys in terms of their effect on creep strength [
115].
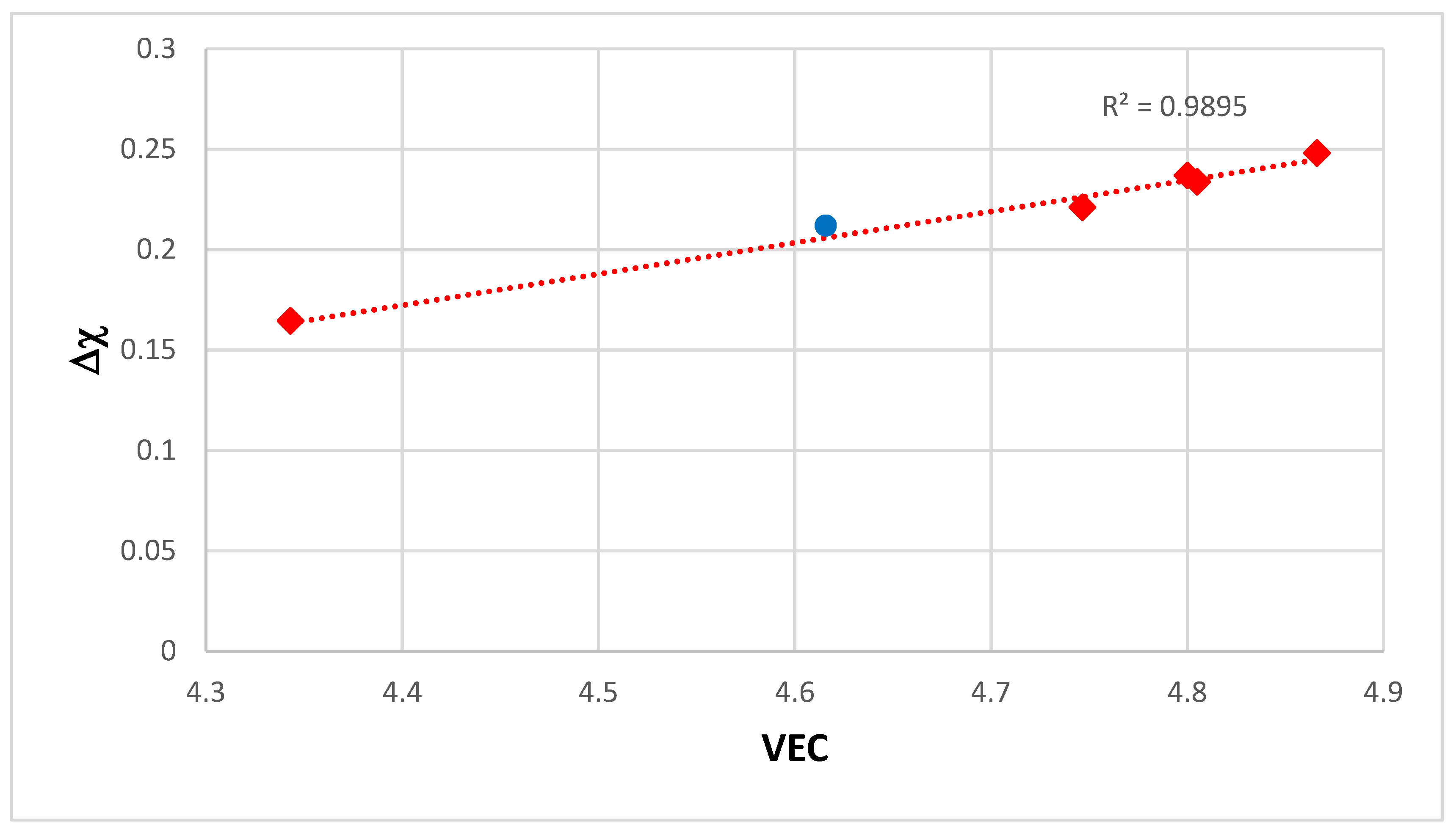
2.3. Relationships between Alloy Parameters, Properties and Solute Concentrations
Correlations were found between weight gain per unit area (ΔW/A) after 100 h isothermal oxidation at 800 °C or 1200 °C and the alloy parameters Δχ, δ or VEC. Different functions (ΔW/A) = f
1(Δχ), (ΔW/A) = f
2(δ) or (ΔW/A) = f
3(VEC) and plots like the one shown in
Figure 9 were established for each temperature. In
Figure 9 the R
2 value for the linear fit of the data is 0.9359. In similar plots (not shown) of (ΔW/A) versus δ and (ΔW/A) versus Δχ the R
2 values respectively were 0.932 and 0.949 for oxidation at 800 °C. For the oxidation at 1200 °C the R
2 values for the (ΔW/A) versus Δχ, (ΔW/A) versus δ and (ΔW/A) versus VEC plots (not shown) were 0.935, 0.95 and 0.951, respectively. The oxidation data (isothermal weight gain) was from [
14,
15,
59,
109,
116,
117,
118,
119,
120,
121].
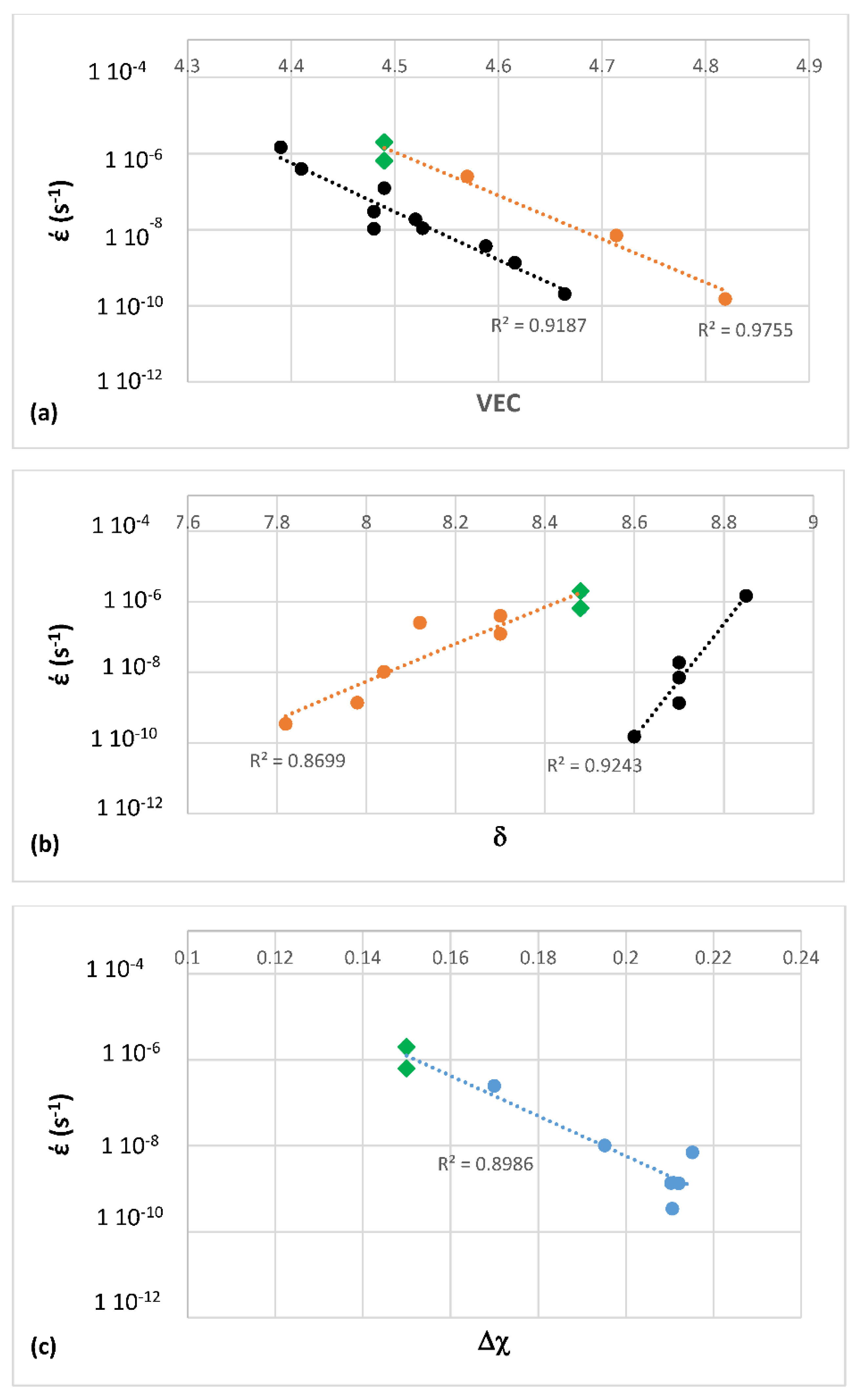
Figure 10 shows compressive creep data at 1050 °C and 100 MPa of the MASC alloy and other Nb-silicide based alloys [
122]. The creep rate is plotted versus the parameters VEC, δ or Δχ. The creep rate decreases with increasing VEC or Δχ and increases with increasing δ. Creep rate of Nb-silicide based alloys at 1050 °C and 1200 °C for stresses higher than 100 and up to 300 MPa [
122] also was found to be related to the alloy parameters δ, VEC and Δχ. The R
2 value for the fit of data for creep rate at 1200 °C and 170 MPa was 0.9299 in έ = g
1(δ) (figure not shown). The R
2 value for the fit of data for creep rate at 1200 °C and 170 MPa in έ = g
2(Δχ) and έ = g
3(VEC) was 0.9586 and 0.9149, respectively (figures not shown). The size of the phases in the studied alloys was similar [
7,
9].
The macrosegregation of Si (MACSi) in cast Nb-silicide based alloys was discussed in [
13].
Figure 11 shows such a relationship between MACSi = [C
maxSi − C
minSi] and ΔH
msd with R
2 = 0.9033. Similar relationships were found for the macrosegregation of Si versus ΔH
msd/ΔH
msp or T
msd/T
msp with R
2 = 0.946 and R
2 = 0.996, respectively (figures not shown). For the definition of the parameters ΔH
msd, ΔH
msp, T
msd and T
msp see [
13].
Having established relationships between properties and parameters of Nb-silicide based alloys, the next step was to find out whether the property goals and said relationships could be used to assist the design (selection) of Nb-silicide based alloys. Relationships between the alloy parameters and solute additions were sought. It was discovered that the concentrations of all solute elements in Nb-silicide based alloys can be expressed as functions of the alloy parameter Δχ. An example is shown in
Figure 12a for the linear relationship Hf = ψ
1(Δχ) for the concentration of Hf in Nb-silicide based alloys. The R
2 value for the fit of the data in
Figure 12a was 0.9521. Similar relationships were discovered for the solutes Al, Ge, Cr, Mo, Si, Sn, Ti, and W with R
2 values 0.957, 0.926, 0.9513, 0.930, 0.916, 0.930, 0.915 and 0.973, respectively (figures not shown). The concentrations of solutes also could be expressed as functions of other alloy parameters; an example is shown in
Figure 12b for W versus the alloy parameter VEC, where R
2 = 0.9831.
The relationships between alloy weight gain in isothermal oxidation at 800 °C and 1200 °C and the alloy parameters VEC, δ and Δχ (for example, see
Figure 9), and between the concentrations of elements in Nb-silicide based alloys and the alloy parameters were used to find out the contribution each element makes in weight gain in Nb-silicide based alloys at each temperature by expressing weight gain as a function of solute element concentration. The relevant equations were of the type [ΔW/A]
i (g/cm
2) = a
i + b
iC
i where i = Al, B, Cr, Ge, Hf, Mo, Nb, Si, Sn, Ti, W, and C is the concentration (at.%) of element i and a and b are constants. For example, for oxidation at 1200 °C the above constants for Al were a
Al = 6.22·10
−4 g/cm
2 and b
Al = 0.0105 g/cm
2 at.% for Nb-silicide based alloys without Boron. The grouping of elements was based on the results in [
46] and the available experimental data for isothermal oxidation.
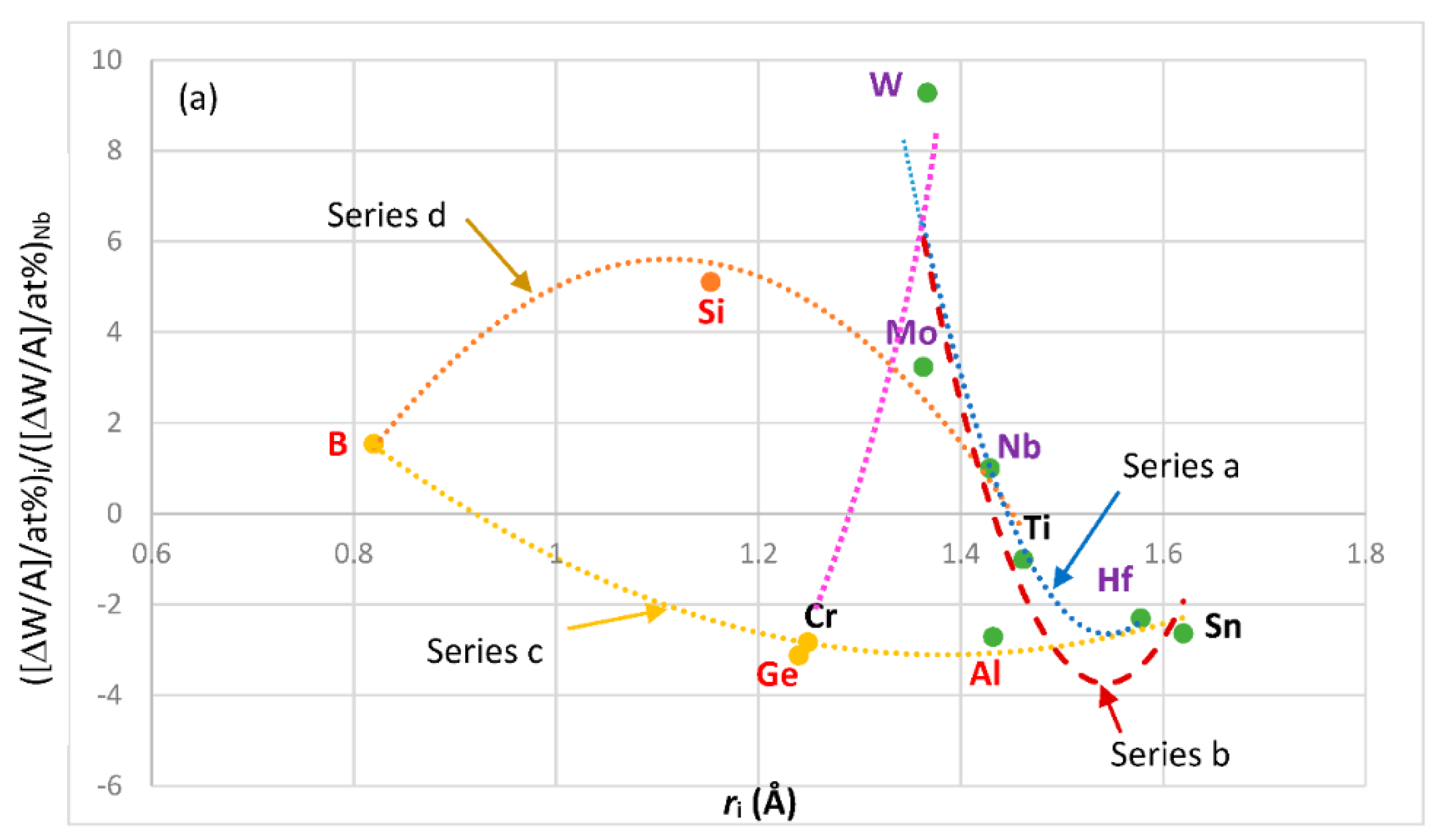
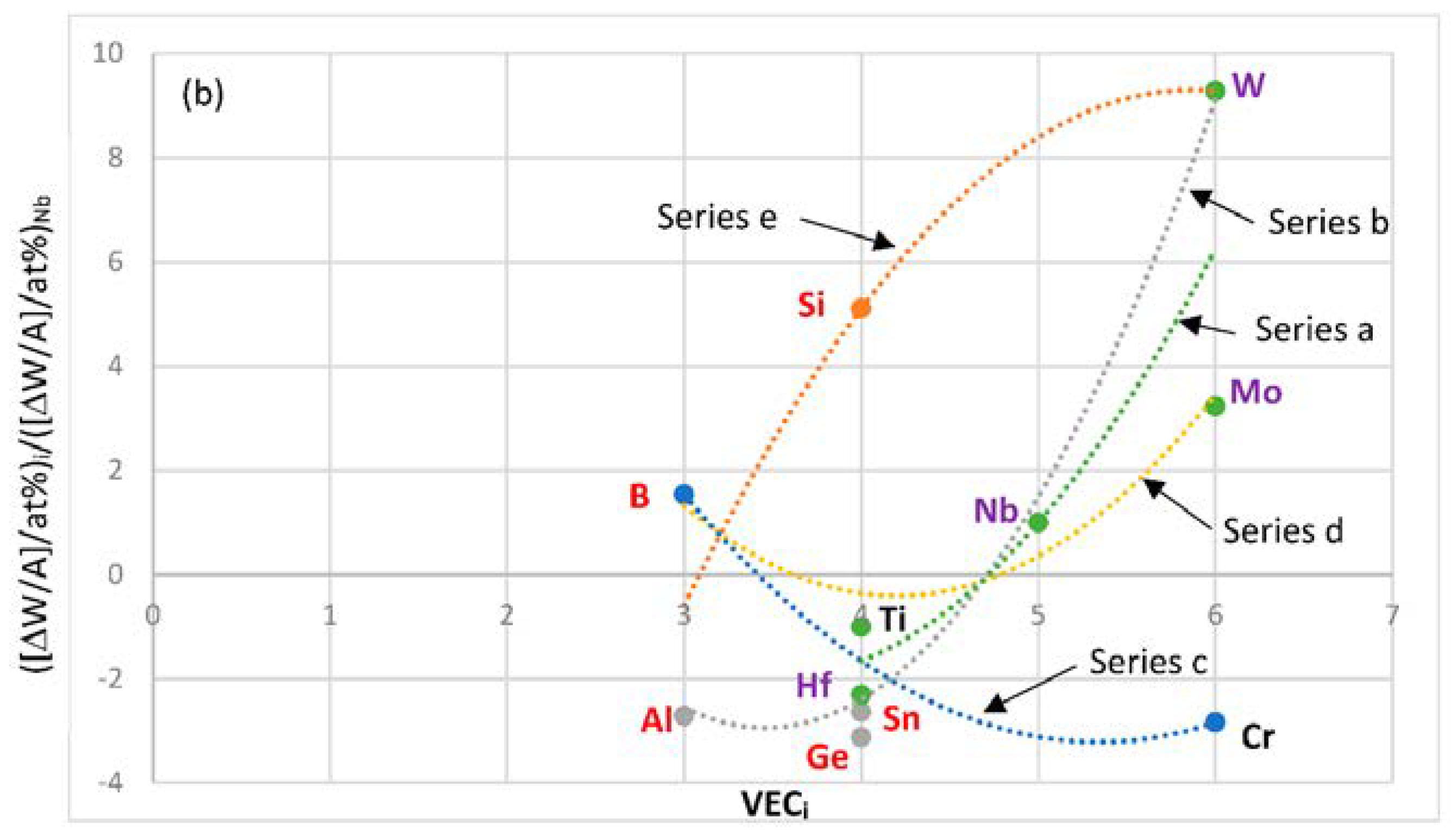
Figure 13 shows the contribution to weight gain of element i at 1200 °C normalized against the weight gain of Nb, versus atomic radius and VEC of element i. Positive values of ([ΔW/A]/at.%)
i/([ΔW/A]/at.% )
Nb mean reduction in weight gain. The solutes with atomic size
ri <
rNb, namely B, Mo, Si and W, reduce weight gain, the solutes with atomic size
ri >
rNb, namely Al, Hf, Sn and Ti, increase weight gain. The solutes Cr and Ge, which have
ri <
rNb, fall in the same group as Mo and W but increase weight gain. It should be noted that the data in
Figure 13 is only relevant to the concentrations of solute additions used in Nb-silicide based alloys. The solutes Al, B, Cr, Ge and Sn that are used to control the oxidation of Nb-silicide based alloys fall in the same group (series c in
Figure 13a). Ti and Hf, which improve oxidation, belong in a separate group. The majority of the elements in series b, c and d in
Figure 13b are the same as those in the same series in
Figure 13a. Also, it should be noted that the same elements belong in the series a in the
Figure 8a and
Figure 13a,b and that there are similarities in the elements in series a, b and c in
Figure 8b and
Figure 13b.
The creep goal was given in the introduction. Experimental work has shown that the density of Nb-silicide based alloys can be around 7 g/cm
3 [
123] and that these alloys should have 16 < Si < 22 at.% [
124] to meet this goal.
The relationships between creep rate at 1200 °C and 170 MPa and the alloy parameters VEC, δ and Δχ and between the concentrations of elements in Nb-silicide based alloys and the alloy parameters were used to find out the contribution each element makes in creep rate for the creep goal conditions by expressing creep rate as a function of solute element concentration. The relevant equations were of the type lnέ = c
i + d
iC
i where i = Al, B, Cr, Ge, Hf, Mo, Nb, Si, Sn, Ti, W, and C is the concentration (at.%) of element i and c and d are constants. For example, for Cr, c
Cr = −23.62 s
−1 and d
Cr = 2.63 s
−1 per at.%.
Figure 14 shows the contribution to creep rate at 1200 °C and 170 MPa of element i normalized against the contribution to creep rate of Nb, versus Pauling electronegativity χ
i of element i. Positive values of [lnέ/at.%]
i/[lnέ/at.%]
Nb mean decrease in creep rate. The elements Mo, Si and W contribute to decrease creep rate while Al, B, Cr, Ge, Hf, Sn and Ti increase creep rate. The ranking of RM alloying additions is in agreement with [
115]. There are similarities in the elements in series c in the
Figure 8b,
Figure 13 and
Figure 14, and in series c in
Figure 8a and series a in
Figure 14.
2.4. Approaches to Alloy Design and Selection
The final objective of the research was to use the above data to develop a design methodology. The first approach to design a Nb-silicide based alloy was based on selecting a Si concentration, for example this could be from the range of the concentration of Si for “best” creep (16 < Si < 22 at.% [
124]), say 18 at.%. Next, this Si concentration was used to calculate the value of the parameter Δχ
alloy from the Si = ψ
1(Δχ
alloy) equation. The calculated value of Δχ
alloy was then used to calculate the concentrations of all other solute elements, for example Hf = ψ
1(Δχ
alloy) for Hf, see
Figure 12a. The calculated concentrations of the solutes gave an alloy composition for which the values of the alloy parameters ΔH
calmix,alloy, ΔS
calmix,alloy, δ
calalloy, Δχ
calalloy, VEC
calalloy, Ω
calalloy (sd/sp)
cal and {Nb/(Ti + Hf)}
cal were calculated as discussed in [
46]. To proceed with the calculation (prediction) of properties (creep, macrosegregation and oxidation (weight gain after isothermal oxidation for 100 h at 800 °C and 1200 °C)) all the alloy parameters of the calculated alloy composition were checked to find out if they were in the ranges given in [
46]. If the latter was the case, the creep rate at 1200 °C and 170 MPa was calculated using the function έ = g
2(δ
calalloy) or έ = g
1(Δχ
calalloy) or έ = g
3(VEC
calalloy). The weight gain at 800 °C was calculated using the function ΔW/A = f
3(VEC
calalloy), for example see
Figure 9, or ΔW/A = f
2(δ
calalloy) or ΔW/A = f
1(Δχ
calalloy), and the weight gain at 1200 °C was calculated using similar functions for this temperature. The macrosegregation of Si in the cast alloy was predicted using relationships like that shown in
Figure 11. The outlined procedure is shown in
Figure 15.
An example of a Nb-silicide based alloy designed using the approach described above is the alloy Nb-21.93Ti-18Si-4.3Cr-4.06Al-4.9Hf-3.43Mo-1.03W-4.5Ge (at.%). We shall call this alloy A. This alloy has VEC
calalloy = 4.513, δ
calalloy = 8.79 and Δχ
calalloy = 0.2. The parameters of this alloy are in the ranges for Nb-silicide based alloys [
46]. The calculated (predicted) weight gains at 800 °C and 1200 °C, respectively are 8.9 mg/cm
2 and 64.5 mg/cm
2 and the macrosegregation of Si is 5.5 at.%. The predicted creep rate at 1200 °C and 170 MPa is lower than the creep goal (see below). The weight gain after 100 h at 1200 °C is significantly higher than that of the single crystal Ni based superalloy CMSX-4 (1 to 4 mg/cm
2).
Another approach was to use έ = g
1(Δχ
alloy) and a selected value of the creep rate, for example use the creep goal (see
Section 2.3) to set έ, and then to solve the equation for Δχ
alloy and then use the latter to calculate the concentrations of the solutes and thus the alloy composition as described above, see
Figure 15.
An alternative approach to alloy design (selection) could be to use the dependence of weight gain at 800 °C and 1200 °C on the alloy parameters VEC and Δχ (i.e., use the functions [ΔW/A]
3 = f
3(VEC) and [ΔW/A]
1 = f
1(Δχ)) and the fact that the concentration of Si in the alloy is related to each of these two parameters with functions Si = ψ
1(Δχ) and Si = ψ
3(VEC). The weight gain functions can thus be re-written as functions of Si concentration in the alloy, and from [ΔW/A]
1 = [ΔW/A]
3 the Si concentration can be calculated at each temperature. The Si concentration in the alloy is then taken as the average of the two concentrations calculated for 800 °C and 1200 °C (the difference between the two calculated Si concentrations is ≤1 at.%). The Si concentration is accepted if it is in the range required for “best” creep (16 < Si < 22 at.% [
124]). The accepted Si concentration is then used to get Δχ
alloy and the latter to calculate the concentrations of the other solute elements, following the same procedure as described previously (see
Figure 15). An example of an Nb-silicide based alloy designed using this approach is the alloy Nb-19.3Ti-18.5Si-4.3Mo-3.8Hf-3.8Sn-3.7Ge-3.3Cr-3.1Al-1.3W (at.%). We shall call this alloy B. This alloy has VEC
calalloy = 4.536, δ
calalloy = 9.4 and Δχ
calalloy = 0.2086. The parameters of this alloy are in the ranges for Nb-silicide based alloys [
46]. The calculated (predicted) weight gains for isothermal oxidation for 100 h at 800 °C and 1200 °C, respectively are 5 mg/cm
2 and 39 mg/cm
2 and the macrosegregation of Si is 5.1 at.%. The calculated creep rate at 1200 °C and 170 MPa does not meet the creep goal (see below).
Another approach to alloy design was to use the relationship of creep rate with the alloy parameter Δχ and write έ = g
1(Δχ) as a function of weight gains at 800 °C and 1200 °C given that [ΔW/A]
1,i = f
1,i(Δχ) (i = 800, 1200) and then calculate the weight gains at 800 °C and 1200 °C for the creep goal. Next, from the weight gain equation, the value of Δχ
alloy is calculated, and the latter is subsequently used to calculate the concentrations of all solute elements, as described previously (see
Figure 11).
Given that the weight gain for oxidation at a particular temperature is related to more than one of the studied parameters, as a first approximation, the calculated alloy weight gain at 800 °C or 1200 °C was taken to be the average of the values calculated using each of the three alloy parameters δ, Δχ and VEC. The weight gains calculated using each parameter at a given temperature were not significantly different from each other. The same approach was used for macrosegregation, meaning the macrosegregation of Si was the average of the values calculated using each of the parameters discussed above and in [
13].
In the case of creep there can be at least an order of magnitude difference between the values calculated from the relationships with atomic size, electronegativity and valence electron concentration (VEC). For alloys that do not oxidize catastrophically in pest oxidation and at high temperatures the creep rate calculated from the relationship with VEC is higher than 10
−7 s
−1 and the relationships between the atomic size or electronegativity with alloy creep rate give the latter respectively lower than 10
−6 and 10
−7 s
−1. This is attributed (a) to different contributions to creep from each parameter given that electronegativity links with all the solutes in Nb-silicide based alloys, the atomic size describes the alloying behavior of the Nb
ss, C14-NbCr
2 Laves and A15-Nb
3X phases, and electronegativity and VEC describe the alloying behavior of all phases in Nb-silicide based alloys [
6,
41,
46,
47,
48], (b) to the different contributions of elements to creep (
Figure 7B,
Figure 8b and
Figure 14), (c) to the anticipated changes in the properties (i) of Nb
5Si
3 silicides because of the shift of Nb
5Si
3 silicides in the Δχ versus VEC maps depending on solute elements substituting Nb and Si in the silicide (see [
47] and
Figure 7B), and (ii) of the Nb
ss because of changes in the composition of the solid solution (
Figure 6 and
Figure 7C) and (d) to changes of the elastic properties of the Nb
5Si
3 silicides with alloying [
104].
Not the same values of the parameters δ, Δχ and VEC are calculated if one were to use the creep goal and some isothermal oxidation (weight gain) targets, say 1 mg/cm
2 and 20 mg/cm
2, respectively for 800 °C and 1200 °C. (Note that these targets are not the same as the oxidation goal. The latter was given as “a recession rate of less than 0.25 μm/hour at 1315 °C” [
1]. This goal was derived from the requirement of achieving the oxidation life at 1315 °C that 2nd generation single crystal Ni-based superalloys presently exhibit at 1150 °C). The former oxidation target is achieved in Nb-silicide based alloys that do not pest and the latter is considered realistic and has been achieved. The alloy VEC values decrease from the creep goal to weight gain goal for oxidation at 800 and at 1200 °C, and the opposite is the case for the values of the alloy parameter δ. The alloy parameter Δχ has the highest value for the oxidation weight gain target at 800 °C and the lowest for the creep goal. This means that for the current property goals it is unlikely that an alloy could be designed where both creep and oxidation property goals can be met simultaneously. However, it is possible (i) to design (select) alloys where one of the property goals is surpassed and the other is met very closely or (ii) to design alloys where both goals are met closely. Both (i) and (ii) have been achieved in the author’s research group. It is the opinion of the author that attaining a weight gain of 1 to 4 mg/cm
2, which is the weight gained by the single crystal Ni based superalloy CMSX-4 alloy after 100 h isothermal oxidation in air at 1200 °C, in polycrystalline Nb-silicide based alloys at 1200 °C is unrealistic unless the vol % of Nb
ss is extremely low in the latter.
In the alloy design (selection) methodology outlined in this paper, the criterion used to select an alloy for further study considering the predicted creep rate at the creep goal conditions is the following. “If the calculated creep rates from the relationships based on atomic size, electronegativity and VEC are higher than 10
−7 s
−1 the alloy is considered highly unlikely to meet the creep goal”, which is “the creep strength should be greater than 170 MPa at a creep rate of 2 10
−8 s
−1 at 1200 °C” [
1]. Using this criterion, the alloys A and B (see above) were predicted not to meet the creep goal.
Each approach to alloy design (selection) that has been discussed above would give an alloy composition for which solidification path, volume fractions of phases and phase equilibria at different temperatures could be calculated, provided thermodynamic data was available. This task is not possible currently, for the reasons discussed in the introduction. Using knowledge gained from on-going research it is possible to make some informed predictions for the selected alloys. The latter and the calculated (predicted) properties can be tested experimentally. For example, the alloy A should have βNb5Si3 as its primary phase, in its microstructure tetragonal Nb5Si3 and Nbss should be stable at 1500 °C but not the C14 NbCr2 Laves and A15 intermetallic phases and the alloy should not pest. The alloy B should have βNb5Si3 as its primary phase, eutectic with Nbss and Nb5Si3 should form (see below), in its microstructure tetragonal Nb5Si3, Nbss (most likely solid solution with no Si) and A15 intermetallic should be stable at 1500 °C but not C14 NbCr2 Laves phase and the alloy should not pest.
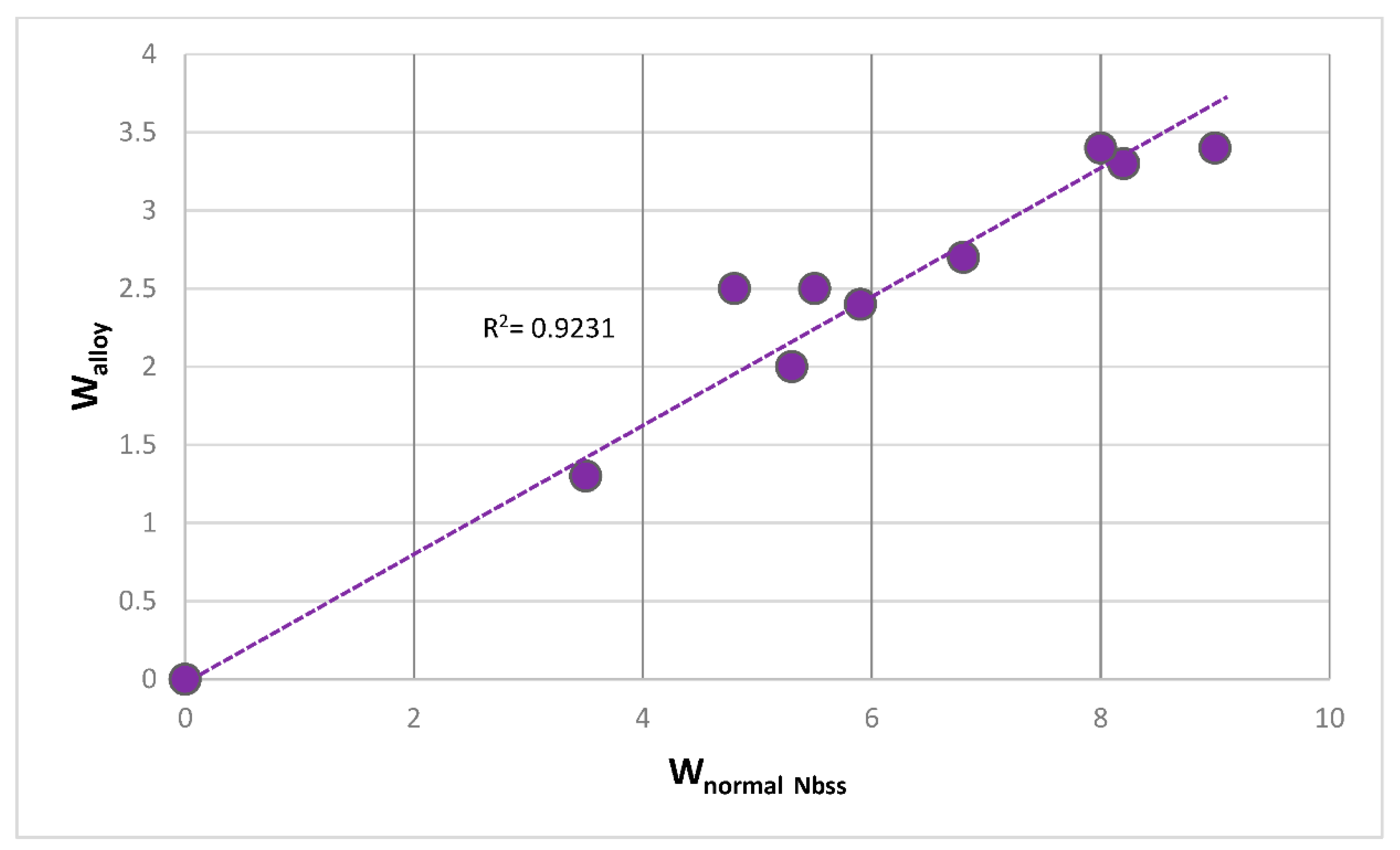
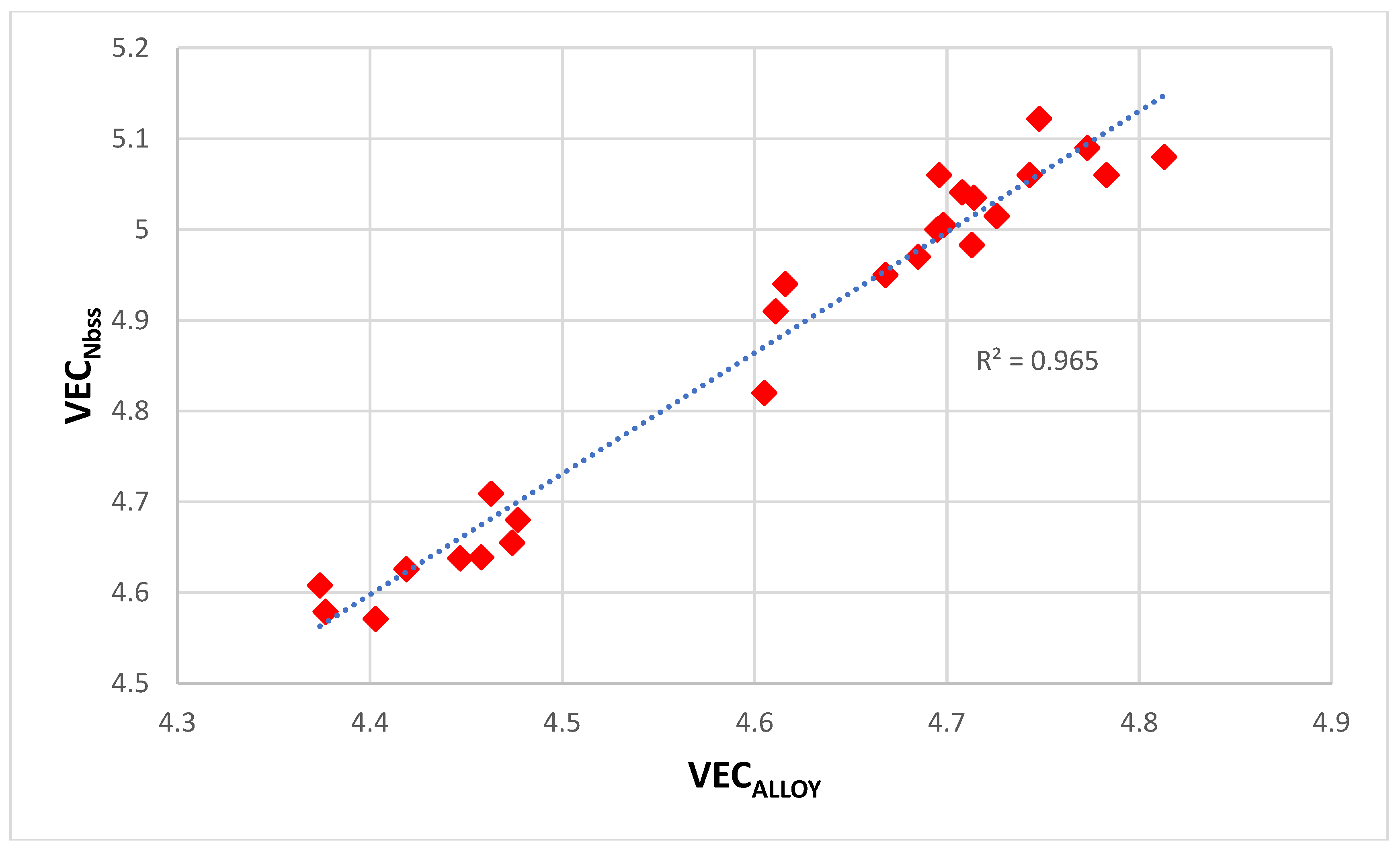
The parameters VEC, Δχ and δ also are important for the types of bcc Nb
ss that form in Nb-silicide based alloys, see [
6]. It is known that the solubility of Cr in bcc Nb
ss depends on that of Ti in the solid solution [
9,
40]. Relationships between solute elements in the alloy and solid solution exist, for example see
Figure 16 for the relationship (R
2 = 0.9231) between W in the alloy and W in the normal Nb
ss. There are also relationships between Δχ
alloy and Δχ
Nbss, δ
alloy and δ
Nbss and VEC
alloy and VEC
Nbss, for example see
Figure 17 for the relationship between VEC
alloy and VEC
Nbss (R
2 = 0.965) Such relationships make it possible to calculate (predict) the composition of the solid solution in a designed (selected) alloy, see
Figure 15. There are also relationships between the parameters of the alloy and Nb
5Si
3, for example Δχ
alloy = h(Δχ
Nb5Si3) with R
2 = 0.9081 for alloying elements Al, B, Cr, Ge, Hf, Si, Sn, Ta, Ti, from which the Δχ
Nb5Si3 is calculated and the latter is then used to get concentrations of solute additions in the silicide from, say C
iNb5Si3= ψ
i(Δχ
Nb5Si3), see
Figure 15.
It is possible to predict the presence or not of a eutectic with Nb
ss and Nb
5Si
3 and whether it will be a rich or poor in Ti eutectic in a Nb-silicide based alloy designed using the alloy design methodology described in this paper. The parameters Δχ and VEC of the eutectics with Nb
ss and Nb
5Si
3 [
41] can account for their alloying behavior and are related to the parameters Δχ and VEC of the Nb-silicide based alloys with linear relationships with R
2 = 0.9325 and R
2 = 0.9517, respectively (figures not shown). For an alloy selected using the methodology described in this paper, the Δχ
eutectic and VEC
eutectic will be calculated from the aforementioned relationships and Figure 5 in reference [
41] could be used to find out whether a eutectic with Nb
ss and Nb
5Si
3 would form. Then Figure 4 in reference [
41] would be used to predict whether the eutectic will be rich or poor in Ti. The relevant part of Figure 5 in reference [
41] is reproduced in
Figure 18 in the present paper. The alloy B will be used to demonstrate the above procedure. The composition of the alloy B is Nb-19.3Ti-18.5Si-4.3Mo-3.8Hf-3.8Sn-3.7Ge-3.3Cr-3.1Al-1.3W (at.%), see above. The calculated parameters for a eutectic with Nb
ss and Nb
5Si
3 in alloy B are VEC
eutectic = 4.602 and Δχ
eutectic = 0.2156.
Figure 18 shows that a eutectic with Nb
ss and Nb
5Si
3 can form in this alloy (the data point is very close to the line for eutectics with the same elements as those in the alloy B (series e in Figure 5 in reference [
41]). Figure 4 in reference [
41] shows that in the alloy B a eutectic with Nb
ss and Nb
5Si
3 and the above Δχ
eutectic and VEC
eutectic values would be poor in Ti. Thus, the alloy B is predicted to have a poor in Ti eutectic with Nb
ss and Nb
5Si
3.
























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}