The Effect of Alloying Elements on the Structural Stability, Mechanical Properties, and Debye Temperature of Al3Li: A First-Principles Study

Abstract
:
1. Introduction
2. Computational Studies
3. Results and Discussion
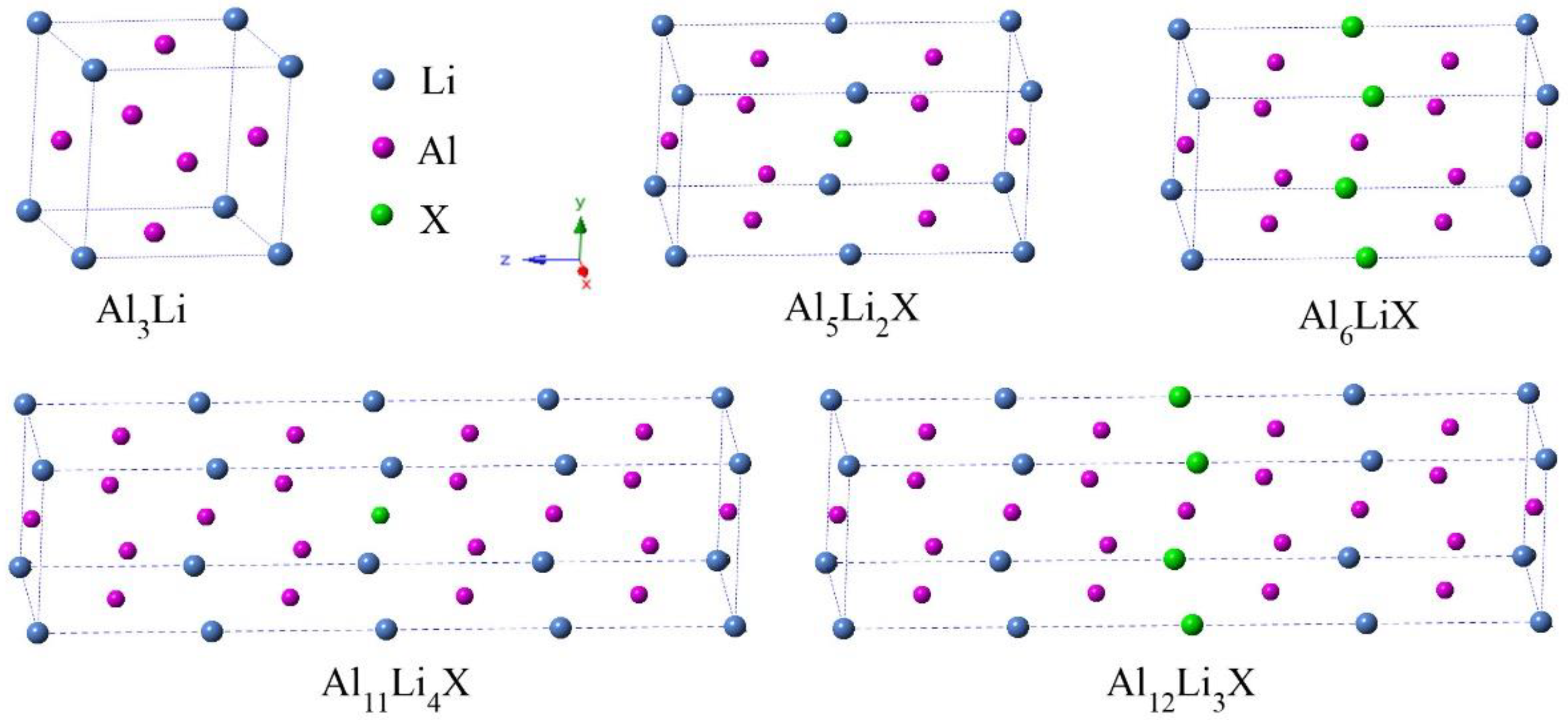
3.1. Site Preference and Phase Stability
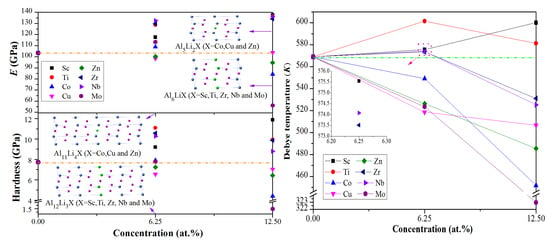
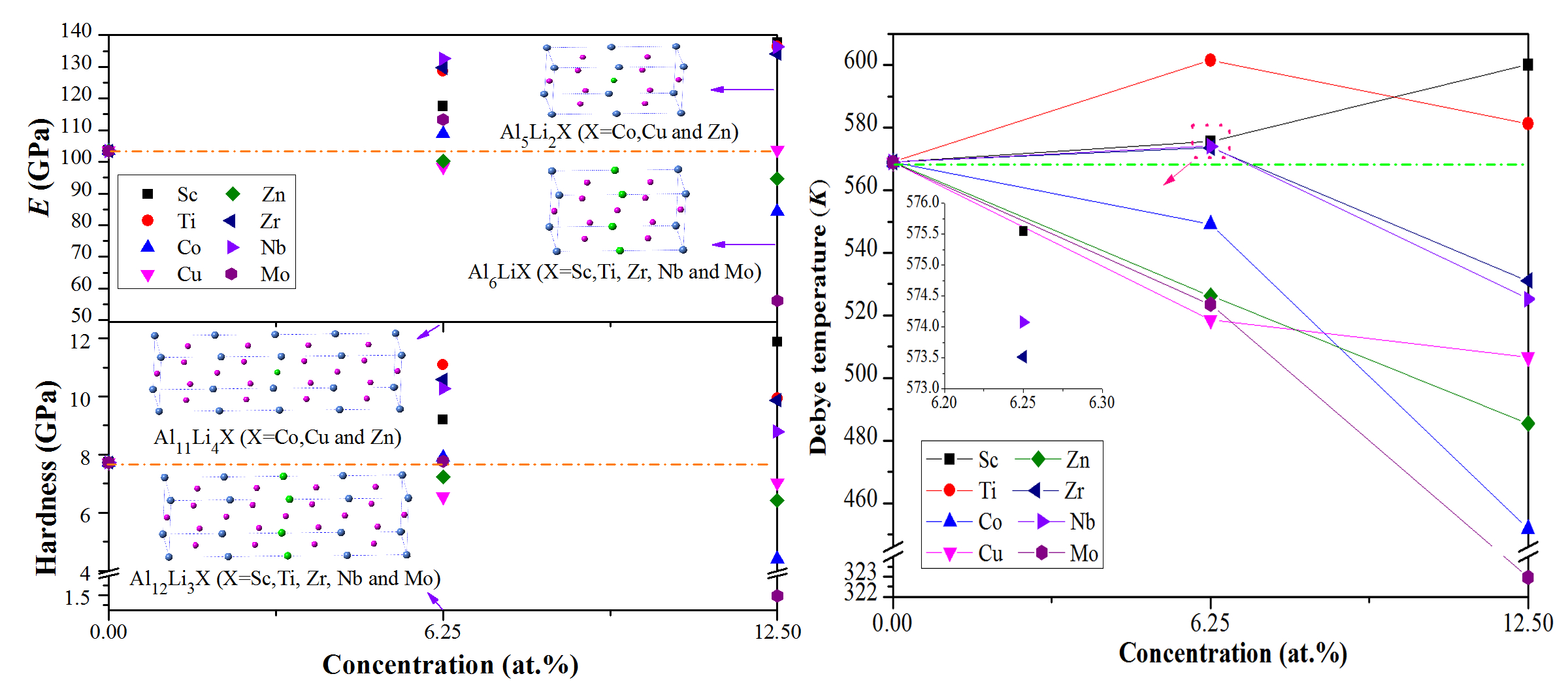
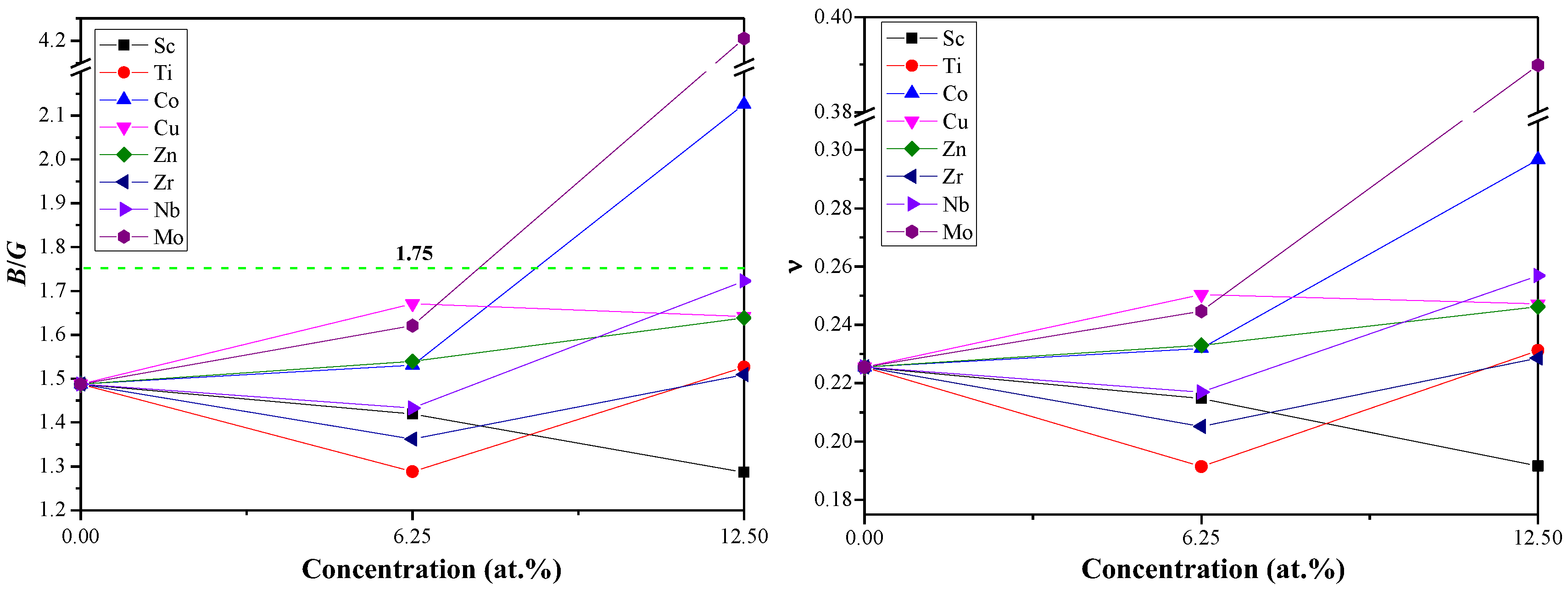
3.2. Mechanical Properties
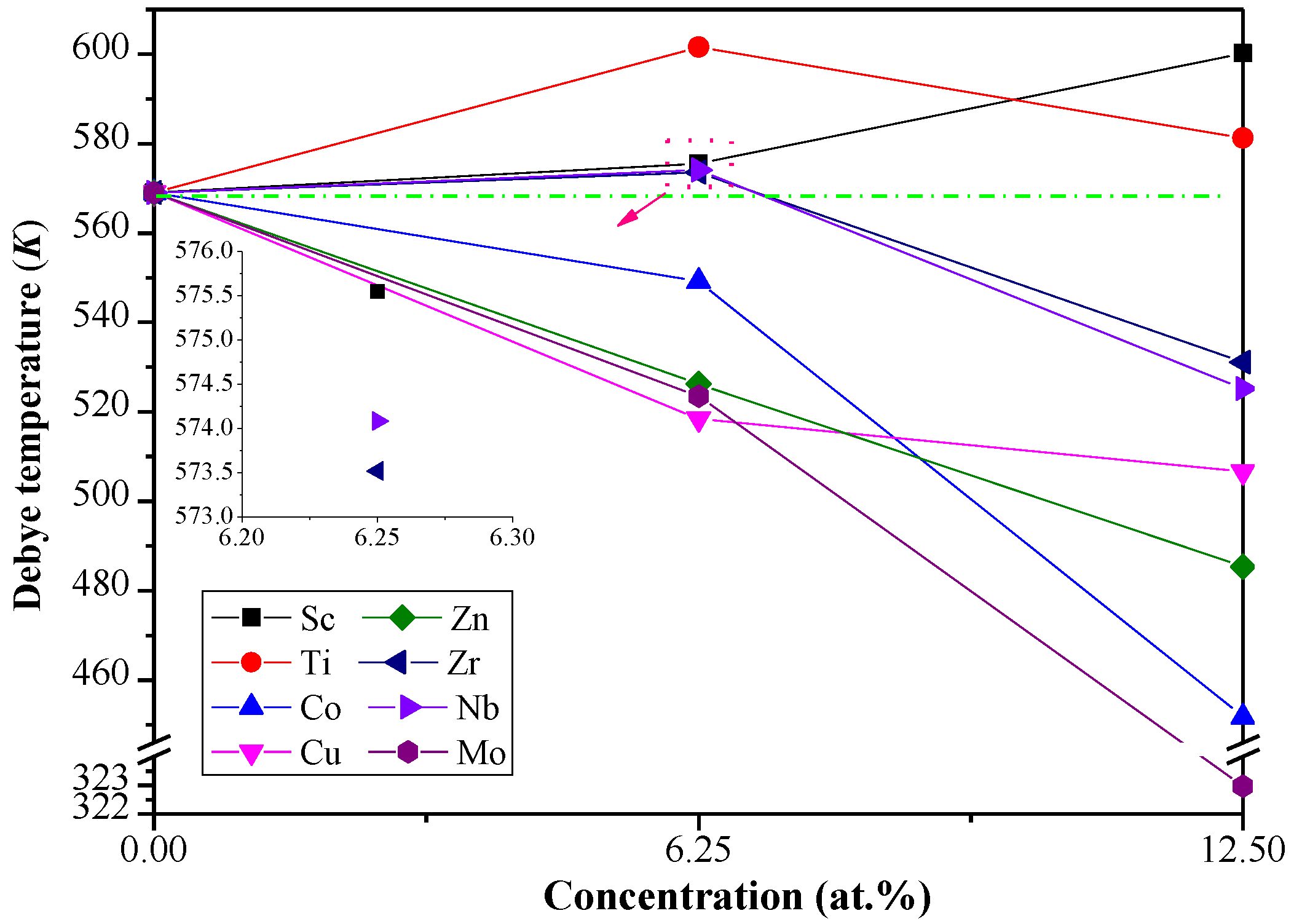
3.3. Debye Temperature
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ning, J.; Zhang, L.J.; Bai, Q.L.; Yin, X.Q.; Niu, J.; Zhang, J.X. Comparison of the microstructure and mechanical performance of 2A97 Al-Li alloy joints between autogenous and non-autogenous laser welding. Mater. Des. 2017, 120, 144–156. [Google Scholar] [CrossRef]
- Bairwa, M.L.; Date, P.P. Effect of heat treatment on the tensile properties of Al-Li alloys. J. Mater. Process. Technol. 2004, 153–154, 603–607. [Google Scholar] [CrossRef]
- Ovri, H.; Lilleodden, E.T. New insights into plastic instability in precipitation strengthened Al-Li alloys. Acta Mater. 2015, 89, 88–97. [Google Scholar] [CrossRef]
- Rösner, H.; Kalogeridis, A.; Liu, W.; Pesicka, J.; Nembach, E. Dislocation mechanisms in Al-rich Al-Li alloys. Mater. Sci. Eng. A 1997, 234–236, 298–301. [Google Scholar] [CrossRef]
- Flower, H.; Gregson, P. Solid state phase transformations in aluminium alloys containing lithium. Mater. Sci. Technol. 1987, 3, 81–90. [Google Scholar] [CrossRef]
- Williams, D.; Edington, J. The precipitation of δ′(Al3Li) in dilute aluminium–lithium alloys. Met. Sci. 1975, 9, 529–532. [Google Scholar] [CrossRef]
- Laverock, J.; Dugdale, S.B.; Alam, M.A.; Roussenova, M.V.; Wensley, J.R.; Kwiatkowska, J.; Shiotani, N. Fermi surface of an important nanosized metastable phase: Al3Li. Phys. Rev. Lett. 2010, 105, 236401. [Google Scholar] [CrossRef] [PubMed]
- Pletcher, B.A.; Wang, K.G.; Glicksman, M.E. Experimental, computational and theoretical studies of δ′ phase coarsening in Al-Li alloys. Acta Mater. 2012, 60, 5803–5817. [Google Scholar] [CrossRef]
- Mogucheva, A.; Kaibyshev, R. Microstructure and Mechanical Properties of an Al-Li-Mg-Sc-Zr Alloy Subjected to ECAP. Metals 2016, 6, 254. [Google Scholar] [CrossRef]
- Chabala, J.M.; Levi-Setti, R.; Soni, K.K.; Williams, D.B.; Newbury, D.E. Secondary ion imaging of the distribution of δ′ (Al3Li) in Al-Li alloys. Appl. Surf. Sci. 1991, 51, 185–192. [Google Scholar] [CrossRef]
- Gu, B.P.; Liedl, G.L.; Kulwicki, J.H.; Sanders, T.H., Jr. Coarsening of δ′ (Al3Li) precipitates in an Al-2.8Li0.3Mn alloy. Mater. Sci. Eng. 1985, 70, 217–228. [Google Scholar] [CrossRef]
- Hoyt, J.J.; Spooner, S. The surface energy of metastable Al3Li precipitates from coarsening kinetics. Acta Metall. Et Mater. 1991, 39, 689–693. [Google Scholar] [CrossRef]
- Lee, B.C.; Park, J.K. Effect of the addition of Ag on the strengthening of Al3Li phase in Al-Li single crystals. Acta Mater. 1998, 46, 4181–4187. [Google Scholar] [CrossRef]
- Mao, Z.; Seidman, D.N.; Wolverton, C. The effect of vibrational entropy on the solubility and stability of ordered Al3Li phases in Al-Li alloys. APL Mater. 2016, 4, 144202. [Google Scholar] [CrossRef]
- Poduri, R.; Chen, L.Q. Computer simulation of morphological evolution and coarsening kinetics of δ′ (Al3Li) precipitates in Al-Li alloys. Acta Mater. 1998, 46, 3915–3928. [Google Scholar] [CrossRef]
- Li, Z.; Tse, J.S. Ab initio studies on the vibrational and thermal properties of Al3Li. Phys. Rev. B 2000, 61, 14531–14536. [Google Scholar] [CrossRef]
- Yu, H.; Duan, X.; Ma, Y.; Zeng, M. First Principles Study of Al-Li Intermetallic Compounds. Chin. J. Chem. Phys. 2012, 25, 659–665. [Google Scholar] [CrossRef]
- Sluiter, M.; De, F.D.; Guo, X.Q.; Podloucky, R.; Freeman, A.J. First-principles calculation of phase equilibria in the aluminum lithium system. Phys. Rev. B 1990, 42, 10460–10476. [Google Scholar] [CrossRef]
- Hu, W.C.; Liu, Y.; Li, D.J.; Zeng, X.Q.; Xu, C.S. Mechanical and thermodynamic properties of Al3Sc and Al3Li precipitates in Al-Li-Sc alloys from first-principles calculations. Phys. B Condens. Matter 2013, 427, 85–90. [Google Scholar] [CrossRef]
- Wolverton, C.; Ozoliņš, V. First-principles aluminum database: Energetics of binary Al alloys and compounds. Phys. Rev. B 2006, 73, 144104. [Google Scholar] [CrossRef]
- Guo, X.; Podloucky, R.; Xu, J.; Freeman, A.J. Cohesive, electronic, and structural properties of Al3Li: An important metastable phase. Phys. Rev. B 1990, 41, 12432. [Google Scholar] [CrossRef]
- Yao, J.; Zhang, C.; Jiang, Y.; Tao, H.; Yin, D. Prediction on elastic properties of off-stoichiometric L12-Al3Li intermetallic due to point defects. Comput. Mater. Sci. 2015, 107, 184–189. [Google Scholar] [CrossRef]
- Makineni, S.K.; Sugathan, S.; Meher, S.; Banerjee, R.; Bhattacharya, S.; Kumar, S.; Chattopadhyay, K. Enhancing elevated temperature strength of copper containing aluminium alloys by forming L12 Al3Zr precipitates and nucleating θ″ precipitates on them. Sci. Rep. 2017, 7, 11154. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Wu, X.; Wang, R.; Jia, Z.; Li, W.; Liu, Q. Structural stability, mechanical properties and stacking fault energies of TiAl3 alloyed with Zn, Cu, Ag: First-principles study. J. Alloys Compd. 2016, 666, 185–196. [Google Scholar] [CrossRef]
- Gu, J.; Bai, J.; Zhu, Y.; Qin, Y.; Gu, H.; Zhai, Y.; Ma, P. First-principles study of the influence of doping elements on phase stability, crystal and electronic structure of Al2Cu (θ) phase. Comput. Mater. Sci. 2016, 111, 328–333. [Google Scholar] [CrossRef]
- Kubouchi, M.; Hayashi, K.; Miyazaki, Y. Electronic structure and thermoelectric properties of boron doped Mg2Si. Scr. Mater. 2016, 123, 59–63. [Google Scholar] [CrossRef]
- Segall, M.; Lindan, P.J.; Probert, M.A.; Pickard, C.; Hasnip, P.; Clark, S.; Payne, M. First-principles simulation: Ideas, illustrations and the CASTEP code. J. Phys. Condens. Matter 2002, 14, 2717–2744. [Google Scholar] [CrossRef]
- Perdew, J.P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B 1986, 33, 8822. [Google Scholar] [CrossRef]
- Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 1990, 41, 7892–7895. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Cohen, A.J.; Mori-Sánchez, P.; Yang, W. Insights into current limitations of density functional theory. Science 2008, 321, 792–794. [Google Scholar] [CrossRef] [PubMed]
- Yoshiyama, T.; Hasebe, K.; Mannami, M. Al3Li Superlattice in Al-4.5 wt % Li Alloy. J. Phys. Soc. Jpn. 2007, 25, 908. [Google Scholar] [CrossRef]
- Sahu, B.R. Electronic structure and bonding of ultralight LiMg. Mater. Sci. Eng. B 1995, 49, 74–78. [Google Scholar] [CrossRef]
- Wang, J.-H.; Lu, Y.; Zhang, X.-L.; Shao, X.-H. The elastic behaviors and theoretical tensile strength of γ-TiAl alloy from the first principles calculations. Intermetallics 2018, 101, 1–7. [Google Scholar] [CrossRef]
- Mouhat, F.; Coudert, F.-X. Necessary and sufficient elastic stability conditions in various crystal systems. Phys. Rev. B 2014, 90, 224104. [Google Scholar] [CrossRef]
- Tian, J.; Zhao, Y.; Wang, B.; Hou, H.; Zhang, Y. The structural, mechanical and thermodynamic properties of Ti-B compounds under the influence of temperature and pressure: First-principles study. Mater. Chem. Phys. 2018, 209, 200–207. [Google Scholar] [CrossRef]
- Xiao, B.; Feng, J.; Zhou, C.T.; Jiang, Y.H.; Zhou, R. Mechanical properties and chemical bonding characteristics of Cr7C3 type multicomponent carbides. J. Appl. Phys. 2011, 109, 083521. [Google Scholar] [CrossRef]
- Qi, Y.Y.; Mu, Y.; Cheng, Y.; Ji, G.F. Pressure effect on electronic, elastic and optical properties of Eu:CaF2 crystal: A first-principles study. Philos. Mag. 2015, 95, 2974–2989. [Google Scholar] [CrossRef]
- Mao, Z.; Chen, W.; Seidman, D.N.; Wolverton, C. First-principles study of the nucleation and stability of ordered precipitates in ternary Al-Sc-Li alloys. Acta Mater. 2011, 59, 3012–3023. [Google Scholar] [CrossRef]
- Hill, R. The elastic behaviour of a crystalline aggregate. Proc. Phys. Soc. 1952, 65, 349–354. [Google Scholar] [CrossRef]
- Watt, J.P.; Peselnick, L. Clarification of the Hashin-Shtrikman bounds on the effective elastic moduli of polycrystals with hexagonal, trigonal, and tetragonal symmetries. J. Appl. Phys. 1980, 51, 1525–1531. [Google Scholar] [CrossRef]
- Gao, F.M.; Gao, L.H. Microscopic models of hardness. J. Superhard Mater. 2010, 32, 148–166. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.Q.; Niu, H.; Li, D.; Li, Y. Modeling hardness of polycrystalline materials and bulk metallic glasses. Intermetallics 2011, 19, 1275–1281. [Google Scholar] [CrossRef] [Green Version]
- Yousef, E.S.; El-Adawy, A.; El-Kheshkhany, N. Effect of rare earth (Pr2O3, Nd2O3, Sm2O3, Eu2O3, Gd2O3 and Er2O3) on the acoustic properties of glass belonging to bismuth–borate system. Solid State Commun. 2006, 139, 108–113. [Google Scholar] [CrossRef]
- Zhang, W.; Chai, C.; Song, Y.; Fan, Q.; Yang, Y. Structural, Mechanical, Anisotropic, and Thermal Properties of AlAs in oC12 and hP6 Phases under Pressure. Materials 2018, 11, 740. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Shang, S.; Wang, Y.; Saengdeejing, A.; Chen, L.; Liu, Z. First-principles calculations of the elastic, phonon and thermodynamic properties of Al12Mg17. Acta Mater. 2010, 58, 4012–4018. [Google Scholar] [CrossRef]
- Nong, Z.-S.; Zhu, J.-C.; Yang, X.-W.; Cao, Y.; Lai, Z.-H.; Liu, Y.; Sun, W. First-principles calculations of the stability and hydrogen storage behavior of C14 Laves phase compound TiCrMn. Solid State Sci. 2014, 32, 1–7. [Google Scholar] [CrossRef]
- Haines, J.; Leger, J.; Bocquillon, G. Synthesis and design of superhard materials. Ann. Rev. Mater. Res. 2001, 31, 1–23. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, J.; Huang, J.; Wang, W.; Ye, Z.; Chen, S.; Zhao, Y. First-principles calculations on physical properties of Ni3Snx binary system intermetallic compounds and Ni/Ni3Sn interfaces in Nickel-Tin TLPS bonding layer. Intermetallics 2018, 101, 27–38. [Google Scholar] [CrossRef]
- Ledbetter, H.; Migliori, A. A general elastic-anisotropy measure. J. Appl. Phys. 2006, 100, 063516. [Google Scholar] [CrossRef]
- Ranganathan, S.I.; Ostoja-Starzewski, M. Universal elastic anisotropy index. Phys. Rev. Lett. 2008, 101, 055504. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Wu, X.; Wang, R.; Li, W.; Liu, Q. Phase stability, mechanical properties and electronic structure of TiAl alloying with W, Mo, Sc and Yb: First-principles study. J. Alloys Compd. 2016, 658, 689–696. [Google Scholar] [CrossRef]
- Ravindran, P.; Fast, L.; Korzhavyi, P.A.; Johansson, B.; Wills, J.; Eriksson, O. Density functional theory for calculation of elastic properties of orthorhombic crystals: Application to TiSi2. J. Appl. Phys. 1998, 84, 4891–4904. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | a | V | Mass Density | Hform |
|---|---|---|---|---|
| Present | 4.034 | 65.65 | 2.223 | −0.097 |
| Cal. [17] | 4.030 | 65.45 | 2.221 | - |
| Cal. [14] | 4.029 | 65.40 | - | −0.100 |
| Exp. [32] | 4.01 | 64.48 | 2.260 | - |
| Element X | ΔHf | Site Preference | Element X | ΔHf | Site Preference | ||
|---|---|---|---|---|---|---|---|
| Al6LiX | Al5Li2X | Al12Li3X | Al11Li4X | ||||
| Sc | −0.267 | −0.154 | Li | Sc | −0.180 | −0.127 | Li |
| Ti | −0.232 | −0.127 | Li | Ti | −0.169 | −0.108 | Li |
| Co | −0.185 | −0.202 | Al | Co | −0.140 | −0.152 | Al |
| Cu | −0.057 | −0.128 | Al | Cu | −0.075 | −0.111 | Al |
| Zn | −0.017 | −0.099 | Al | Zn | −0.057 | −0.098 | Al |
| Zr | −0.276 | −0.148 | Li | Zr | −0.191 | −0.118 | Li |
| Nb | −0.188 | −0.098 | Li | Nb | −0.150 | −0.094 | Li |
| Mo | −0.100 | −0.059 | Li | Mo | −0.107 | −0.075 | Li |
| Phase | Species | C11 | C33 | C44 | C66 | C12 | C13 | B | G | E | H | AU |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Al3Li | Present | 129.7 | - | 37.7 | - | 29.4 | - | 62.8 | 42.2 | 103.5 | 7.73 | 0.099 |
| Cal. [17] | 128 | - | 39 | - | 30 | - | 63.3 | 42.8 | 116.8 | - | - | |
| Exp. [39] | 123.6 | - | 42.8 | - | 37.2 | - | 66 | 43 | 105.9 | - | - | |
| Al12Li3Sc | Present | 136.2 | 130.4 | 46.1 | 51.7 | 31.0 | 38.3 | 68.7 | 48.4 | 117.6 | 9.20 | 0.019 |
| Al12Li3Ti | Present | 141.3 | 147.7 | 52.4 | 54.4 | 35.2 | 31.2 | 69.5 | 54.0 | 128.6 | 11.10 | 0.007 |
| Al11Li4Co | Present | 138.0 | 118.9 | 39.4 | 48.1 | 29.0 | 39.2 | 67.7 | 44.2 | 108.9 | 7.90 | 0.085 |
| Al11Li4Cu | Present | 126.7 | 116.2 | 37.0 | 37.5 | 37.7 | 37.0 | 65.8 | 39.4 | 98.5 | 6.55 | 0.032 |
| Al11Li4Zn | Present | 124.3 | 121.8 | 36.9 | 38.9 | 33.8 | 31.2 | 62.5 | 40.6 | 100.1 | 7.23 | 0.049 |
| Al12Li3Zr | Present | 143.4 | 147.5 | 53.1 | 55.9 | 42.2 | 35.2 | 73.3 | 53.8 | 129.7 | 10.58 | 0.008 |
| Al12Li3Nb | Present | 147.8 | 163.5 | 53.1 | 54.9 | 50.8 | 35.5 | 78.1 | 54.5 | 132.6 | 10.28 | 0.041 |
| Al12Li3Mo | Present | 118.7 | 147.0 | 52.0 | 51.5 | 66.9 | 36.6 | 73.8 | 45.4 | 113.3 | 7.77 | 0.426 |
| Al6LiSc | Present | 146.6 | 136.8 | 61.3 | 62.0 | 35.2 | 41.3 | 74.3 | 57.7 | 137.6 | 11.88 | 0.040 |
| Al6LiTi | Present | 153.7 | 157.5 | 55.0 | 62.0 | 51.8 | 48.2 | 84.6 | 55.4 | 136.4 | 9.93 | 0.020 |
| Al5Li2Co | Present | 144.9 | 76.5 | 35.9 | 34.0 | 15.6 | 58.2 | 69.1 | 32.5 | 84.3 | 4.41 | 1.534 |
| Al5Li2Cu | Present | 134.6 | 122.0 | 37.3 | 41.8 | 39.3 | 36.6 | 68.4 | 41.6 | 103.8 | 7.03 | 0.055 |
| Al5Li2Zn | Present | 123.2 | 116.4 | 33.5 | 36.4 | 37.0 | 30.9 | 62.2 | 38.0 | 94.7 | 6.42 | 0.082 |
| Al6LiZr | Present | 153.9 | 143.0 | 55.7 | 60.5 | 51.6 | 47.2 | 82.4 | 54.6 | 134.2 | 9.88 | 0.026 |
| Al6LiNb | Present | 162.0 | 156.2 | 54.2 | 64.0 | 56.4 | 61.9 | 93.4 | 54.2 | 136.3 | 8.88 | 0.047 |
| Al6LiMo | Present | 90.3 | 146.3 | 39.6 | 50.9 | 86.8 | 67.9 | 85.0 | 20.2 | 56.1 | 1.48 | 17.33 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tian, J.; Zhao, Y.; Hou, H.; Wang, B. The Effect of Alloying Elements on the Structural Stability, Mechanical Properties, and Debye Temperature of Al3Li: A First-Principles Study. Materials 2018, 11, 1471. https://doi.org/10.3390/ma11081471
Tian J, Zhao Y, Hou H, Wang B. The Effect of Alloying Elements on the Structural Stability, Mechanical Properties, and Debye Temperature of Al3Li: A First-Principles Study. Materials. 2018; 11(8):1471. https://doi.org/10.3390/ma11081471
Chicago/Turabian StyleTian, Jinzhong, Yuhong Zhao, Hua Hou, and Bing Wang. 2018. "The Effect of Alloying Elements on the Structural Stability, Mechanical Properties, and Debye Temperature of Al3Li: A First-Principles Study" Materials 11, no. 8: 1471. https://doi.org/10.3390/ma11081471
APA StyleTian, J., Zhao, Y., Hou, H., & Wang, B. (2018). The Effect of Alloying Elements on the Structural Stability, Mechanical Properties, and Debye Temperature of Al3Li: A First-Principles Study. Materials, 11(8), 1471. https://doi.org/10.3390/ma11081471