Synthesis, Nanomechanical Characterization and Biocompatibility of a Chitosan-Graft-Poly(ε-caprolactone) Copolymer for Soft Tissue Regeneration

 ,
,  ,
, 

Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials and Synthesis
2.2. Disc Fabrication
2.3. Characterization Techniques
2.4. Discoid Sample Degradation
2.5. Scanning Electron Microscope (SEM)
2.6. Nanoindentation Testing
2.7. Cell Culture
2.8. Cell Viability and Proliferation
2.9. Cell Morphology on CS-g-PCL
2.10. Extracellular Collagen Production
3. Results and Discussion
3.1. Synthesis and Physicochemical Characteristics of CS-g-PCL
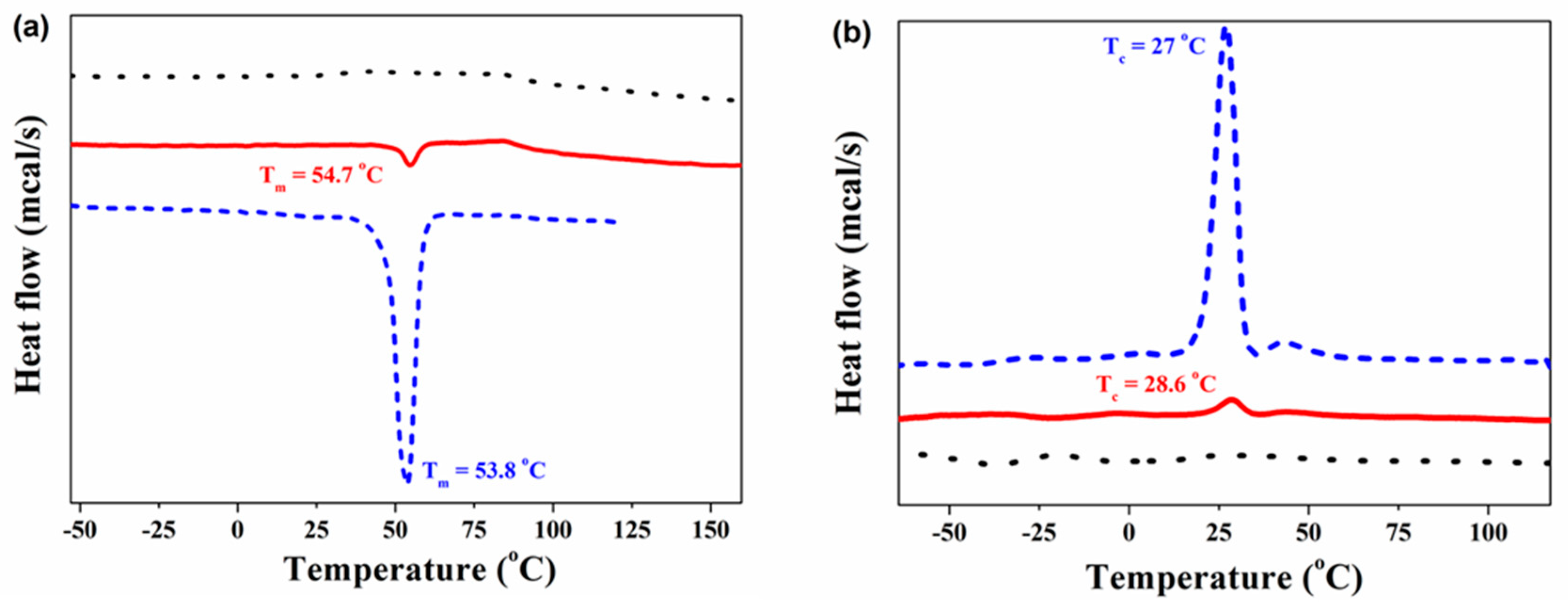
3.2. Thermal Properties
3.3. Degradation Experiments
3.4. Nanoindentation Data of as-Prepared Samples
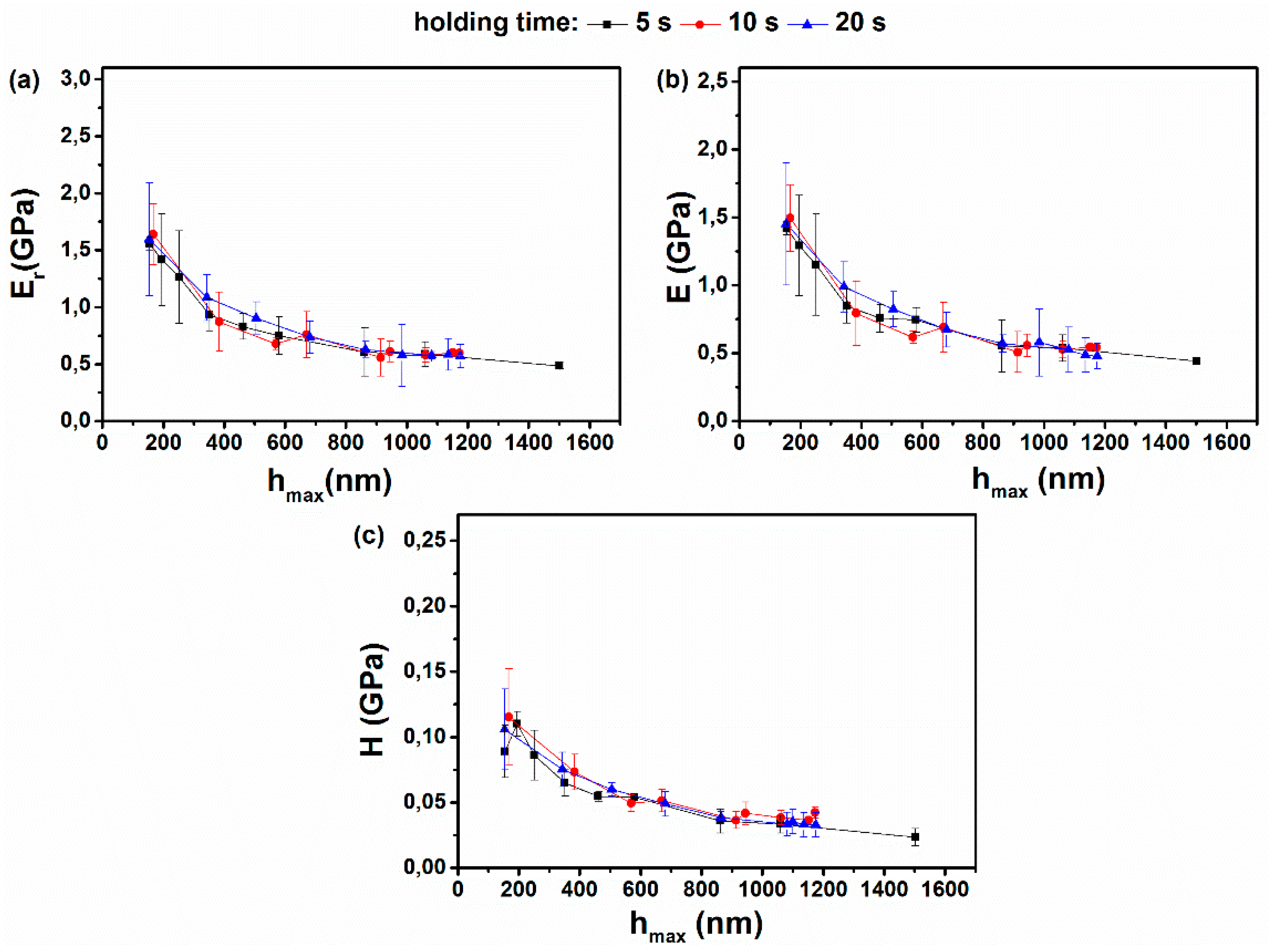
3.5. Influence of the Creep Time on the Elastic Modulus and Hardness Values
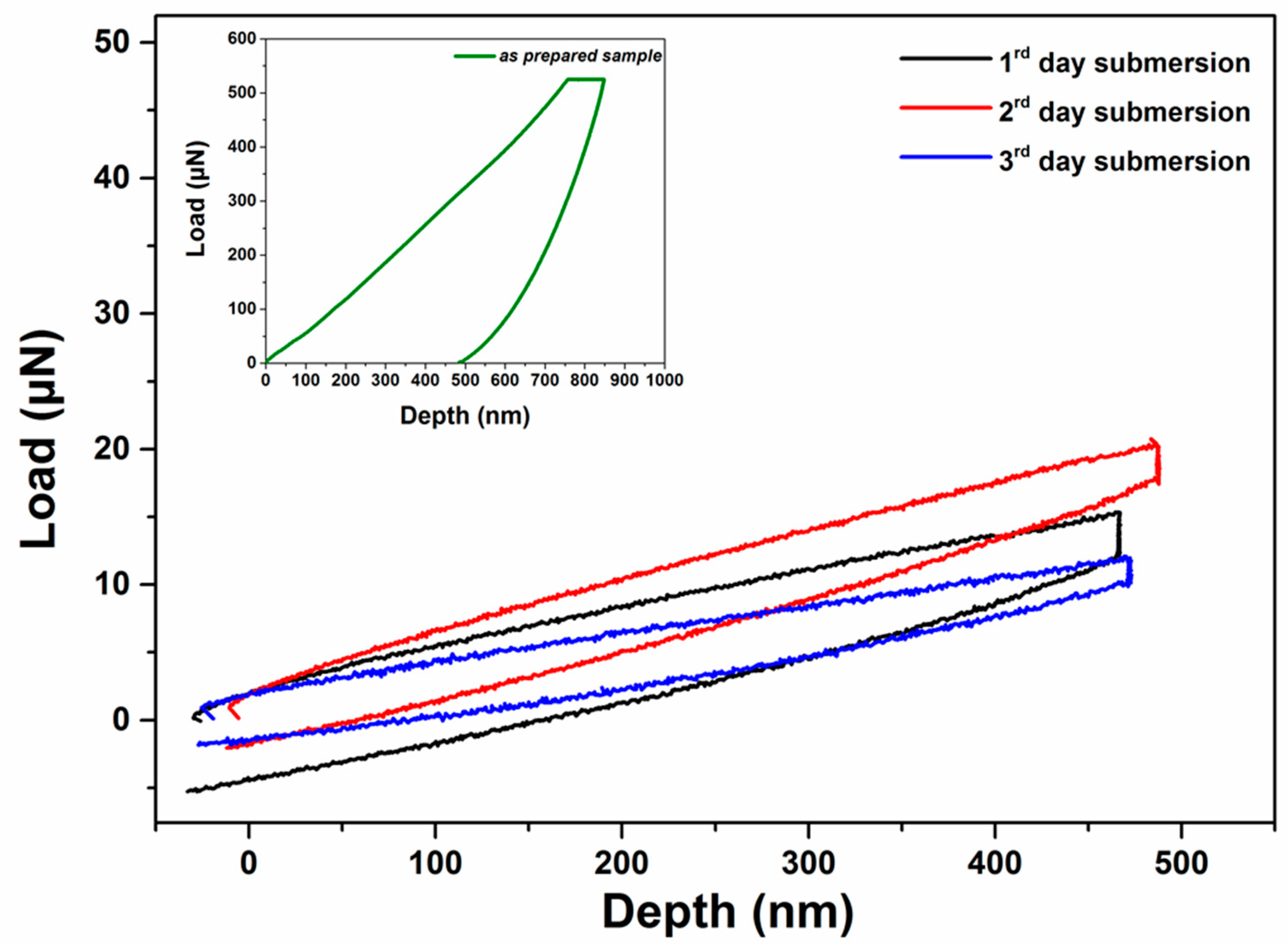
3.6. Nanoindentation Analysis of the Samples Following Immersion in a-MEM
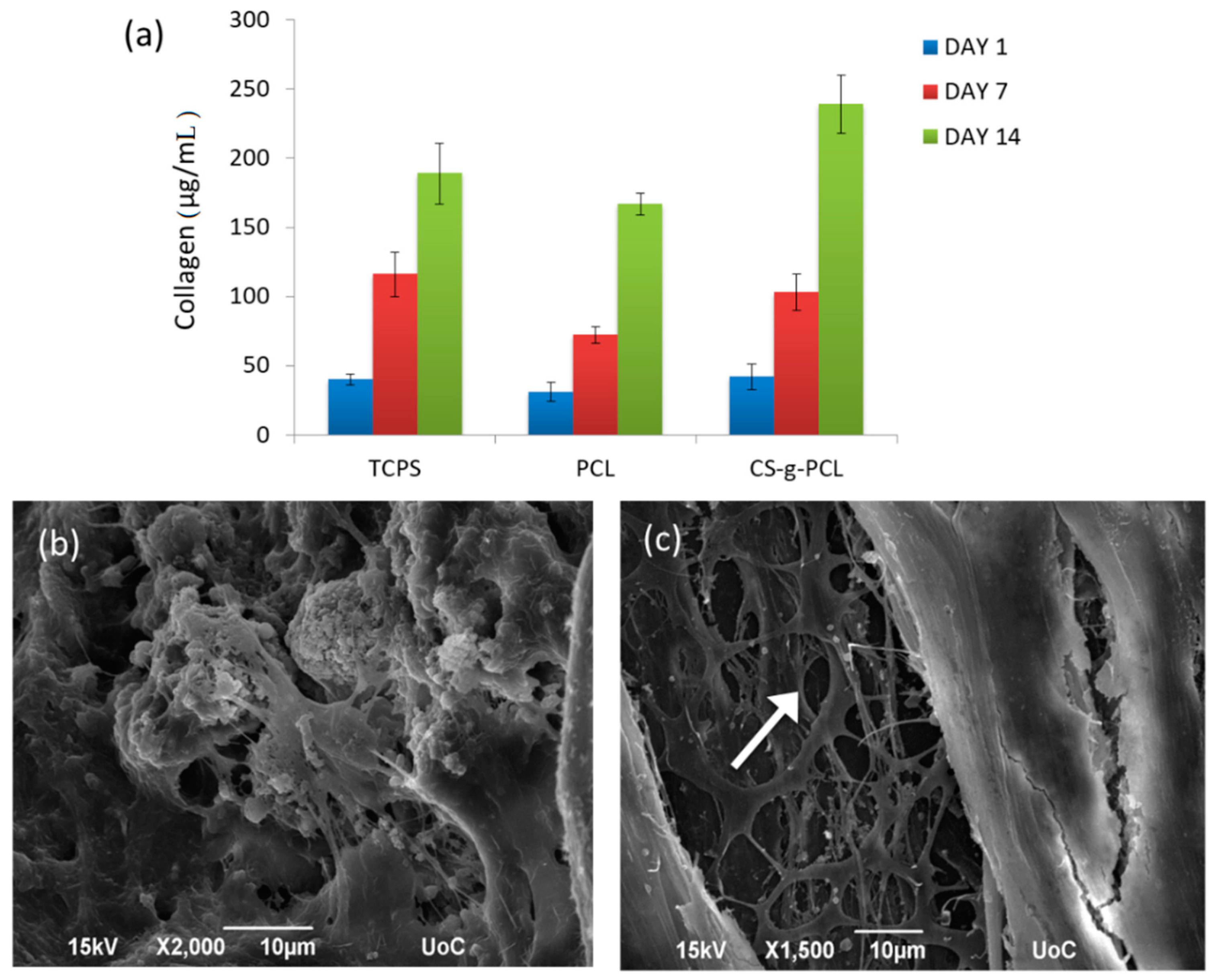
3.7. In Vitro Performance
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Roger, V.L.; Go, A.S.; Lloyd-Jones, D.M.; Benjamin, E.J.; Berry, J.D.; Borden, W.B.; Bravata, D.M.; Dai, S.; Ford, E.S.; Fox, C.S.; et al. Heart disease and stroke statistics—2012 update: A report from the American Heart Association. Circulation 2012, 125, e2–e220. [Google Scholar] [PubMed]
- Topol, E.J. Current status and future prospects for acute myocardial infarction therapy. Circulation 2003, 108, III6–III13. [Google Scholar] [CrossRef] [PubMed]
- Atkins, B.Z.; Hueman, M.T.; Meuchel, J.M.; Cottman, M.J.; Hutcheson, K.A.; Taylor, D.A. Myogenic cell transplantation improves in vivo regional performance in infarcted rabbit myocardium. J. Heart Lung Transplant. 1999, 18, 1173–1180. [Google Scholar] [CrossRef]
- Bursac, N.; Papadaki, M.; Cohen, R.J.; Schoen, F.J.; Eisenberg, S.R.; Carrier, R.; Vunjak-Novakovic, G.; Freed, L.E. Cardiac muscle tissue engineering: Toward an in vitro model for electrophysiological studies. Am. J. Physiol. 1999, 277, H433–H444. [Google Scholar] [CrossRef] [PubMed]
- Li, R.K.; Yau, T.M.; Weisel, R.D.; Mickle, D.A.; Sakai, T.; Choi, A.; Jia, Z.Q. Construction of a bioengineered cardiac graft. J. Thorac. Cardiovasc. Surg. 2000, 119, 368–375. [Google Scholar] [CrossRef] [Green Version]
- Tomita, S.; Li, R.K.; Weisel, R.D.; Mickle, D.A.; Kim, E.J.; Sakai, T.; Jia, Z.Q. Autologous transplantation of bone marrow cells improves damaged heart function. Circulation 1999, 100, II247–II256. [Google Scholar] [CrossRef]
- Davis, M.E.; Hsieh, P.C.; Grodzinsky, A.J.; Lee, R.T. Custom design of the cardiac microenvironment with biomaterials. Circ. Res. 2005, 97, 8–15. [Google Scholar] [CrossRef]
- Jawad, H.; Lyon, A.R.; Harding, S.E.; Ali, N.N.; Boccaccini, A.R. Myocardial tissue engineering. Br. Med. Bull. 2008, 87, 31–47. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Zheng, L.; Li, C.; Zhang, D.; Xiao, Y.; Guan, G.; Zhu, W. A novel and simple procedure to synthesize chitosan-graft-polycaprolactone in an ionic liquid. Carbohydr. Polym. 2013, 94, 505–510. [Google Scholar] [CrossRef]
- Honma, T.; Senda, T.; Inoue, Y. Thermal properties and crystallization behaviour of blends of poly(ε-caprolactone) with chitin and chitosan. Polym. Int. 2003, 52, 1839–1846. [Google Scholar] [CrossRef]
- Kumar, M.N.; Muzzarelli, R.A.; Muzzarelli, C.; Sashiwa, H.; Domb, A.J. Chitosan chemistry and pharmaceutical perspectives. Chem. Rev. 2004, 104, 6017–6084. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, M.; Obara, K.; Nakamura, S.; Fujita, M.; Masuoka, K.; Kanatani, Y.; Takase, B.; Hattori, H.; Morimoto, Y.; Ishihara, M.; et al. Chitosan hydrogel as a drug delivery carrier to control angiogenesis. J. Artif. Organs 2006, 9, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Tian, M.; Yang, Z.; Kuwahara, K.; Nimni, M.E.; Wan, C.X.; Han, B. Delivery of demineralized bone matrix powder using a thermogelling chitosan carrier. Acta Biomater. 2012, 8, 753–762. [Google Scholar] [CrossRef] [PubMed]
- Risbud, M.V.; Hardikar, A.A.; Bhat, S.V.; Bhonde, R.R. pH-sensitive freeze-dried chitosan-polyvinyl pyrrolidone hydrogels as controlled release system for antibiotic delivery. J. Control. Release 2000, 68, 23–30. [Google Scholar] [CrossRef]
- Chi, N.H.; Yang, M.C.; Chung, T.W.; Chou, N.K.; Wang, S.S. Cardiac repair using chitosan-hyaluronan/silk fibroin patches in a rat heart model with myocardial infarction. Carbohydr. Polym. 2013, 92, 591–597. [Google Scholar] [CrossRef] [PubMed]
- Kurdi, M.; Chidiac, R.; Hoemann, C.; Zouein, F.; Zgheib, C.; Booz, G.W. Hydrogels as a platform for stem cell delivery to the heart. Congest. Heart Fail. 2010, 16, 132–135. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.K.; Juenet, M.; Meddahi-Pelle, A.; Letourneur, D. Polysaccharide-based strategies for heart tissue engineering. Carbohydr. Polym. 2015, 116, 267–277. [Google Scholar] [CrossRef]
- Madihally, S.V.; Matthew, H.W.T. Porous chitosan scaffolds for tissue engineering. Biomaterials 1999, 20, 1133–1142. [Google Scholar] [CrossRef]
- Van der Giessen, W.J.; Lincoff, A.M.; Schwartz, R.S.; van Beusekom, H.M.; Serruys, P.W.; Holmes, D.R., Jr.; Ellis, S.G.; Topol, E.J. Marked inflammatory sequelae to implantation of biodegradable and nonbiodegradable polymers in porcine coronary arteries. Circulation 1996, 94, 1690–1697. [Google Scholar] [CrossRef]
- Htay, A.S.; Teoh, S.H.; Hutmacher, D.W. Development of perforated microthin poly(epsilon-caprolactone) films as matrices for membrane tissue engineering. J. Biomater. Sci. Polym. E 2004, 15, 683–700. [Google Scholar] [CrossRef]
- Aliabadi, H.M.; Mahmud, A.; Sharifabadi, A.D.; Lavasanifar, A. Micelles of methoxy poly (ethylene oxide)-b-poly(epsilon-caprolactone) as vehicles for the solubilization and controlled delivery of Cyclosporine A. J. Control Release 2005, 104, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Pok, S.; Myers, J.D.; Madihally, S.V.; Jacot, J.G. A multilayered scaffold of a chitosan and gelatin hydrogel supported by a PCL core for cardiac tissue engineering. Acta Biomater. 2013, 9, 5630–5642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinha, V.R.; Bansal, K.; Kaushik, R.; Kumria, R.; Trehan, A. Poly-epsilon-caprolactone microspheres and nanospheres: An overview. Int. J. Pharm. 2004, 278, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Cruz, D.M.G.; Coutinho, D.F.; Mano, J.F.; Ribelles, J.L.G.; Sanchez, M.S. Physical interactions in macroporous scaffolds based on poly(epsilon-caprolactone)/chitosan semi-interpenetrating polymer networks. Polymer 2009, 50, 2058–2064. [Google Scholar] [CrossRef]
- Sarasam, A.; Madihally, S.V. Characterization of chitosan-polycaprolactone blends for tissue engineering applications. Biomaterials 2005, 26, 5500–5508. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.P.; Pandey, J.K.; Rutot, D.; Degee, P.; Dubois, P. Biodegradation of poly(epsilon-caprolactone)/starch blends and composites in composting and culture environments: The effect of compatibilization on the inherent biodegradability of the host polymer. Carbohydr. Res. 2003, 338, 1759–1769. [Google Scholar] [CrossRef]
- Vazqueztorres, H.; Cruzramos, C.A. Blends of Cellulosic Esters with Poly(Caprolactone)—Characterization by Dsc, Dma, and Waxs. J. Appl. Polym. Sci. 1994, 54, 1141–1159. [Google Scholar] [CrossRef]
- Yang, A.L.; Wu, R.J.; Zhu, P.F. Thermal analysis and miscibility of chitin/polycaprolactone blends. J. Appl. Polym. Sci. 2001, 81, 3117–3123. [Google Scholar] [CrossRef]
- Garcia Cruz, D.M.; Gomez Ribelles, J.L.; Salmeron Sanchez, M. Blending polysaccharides with biodegradable polymers. I. Properties of chitosan/polycaprolactone blends. J. Biomed. Mater. Res. Part B Appl. Biomater. 2008, 85, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Cai, G.Q.; Jiang, H.L.; Chen, Z.J.; Tu, K.H.; Wang, L.Q.; Zhu, K.J. Synthesis, characterization and self-assemble behavior of chitosan-O-poly(epsilon-caprolactone). Eur. Polym. J. 2009, 45, 1674–1680. [Google Scholar] [CrossRef]
- Chen, H.L.; Huang, J.; Yu, J.H.; Liu, S.Y.; Gu, P. Electrospun chitosan-graft-poly (epsilon-caprolactone)/poly (epsilon-caprolactone) cationic nanofibrous mats as potential scaffolds for skin tissue engineering. Int. J. Biol. Macromol. 2011, 48, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Neves, S.C.; Moreira Teixeira, L.S.; Moroni, L.; Reis, R.L.; Van Blitterswijk, C.A.; Alves, N.M.; Karperien, M.; Mano, J.F. Chitosan/poly(epsilon-caprolactone) blend scaffolds for cartilage repair. Biomaterials 2011, 32, 1068–1079. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Wu, H.; Cao, X.Y.; Dalai, S. Compressive mechanical properties and biodegradability of porous poly(caprolactone)/chitosan scaffolds. Polym. Degrad. Stabil. 2008, 93, 1736–1741. [Google Scholar] [CrossRef]
- Chatzinikolaidou, M.; Kaliva, M.; Batsali, A.; Pontikoglou, C.; Vamvakaki, M. Wharton’s Jelly Mesenchymal Stem Cell Response on Chitosan-graft-poly (epsilon -caprolactone) Copolymer for Myocardium Tissue Engineering. Curr. Pharm. Des. 2014, 20, 2030–2039. [Google Scholar] [CrossRef] [PubMed]
- Doerner, M.F.; Nix, W.D. A method for interpreting the data from depth-sensing indentation instruments. J. Mater. Res. 2011, 1, 601–609. [Google Scholar] [CrossRef]
- Hertz, H. On the contact of solid, elastic bodies. J. Für Die Reine Und Angew. Math. 1881, 92, 156–171. [Google Scholar]
- Oliver, W.C.; Pharr, G.M. An Improved Technique for Determining Hardness and Elastic-Modulus Using Load and Displacement Sensing Indentation Experiments. J. Mater. Res. 1992, 7, 1564–1583. [Google Scholar] [CrossRef]
- Sneddon, I.N. The relation between load and penetration in the axisymmetric boussinesq problem for a punch of arbitrary profile. Int. J. Eng. Sci. 1965, 3, 47–57. [Google Scholar] [CrossRef]
- Skarmoutsou, A.; Lolas, G.; Charitidis, C.A.; Chatzinikolaidou, M.; Vamvakaki, M.; Farsari, M. Nanomechanical properties of hybrid coatings for bone tissue engineering. J. Mech. Behav. Biomed. Mater. 2013, 25, 48–62. [Google Scholar] [CrossRef]
- Chatzinikolaidou, M.; Rekstyte, S.; Danilevicius, P.; Pontikoglou, C.; Papadaki, H.; Farsari, M.; Vamvakaki, M. Adhesion and growth of human bone marrow mesenchymal stem cells on precise-geometry 3D organic-inorganic composite scaffolds for bone repair. Mater. Sci. Eng. C Mater. Biol. Appl. 2015, 48, 301–309. [Google Scholar] [CrossRef]
- Chatzinikolaidou, M.; Pontikoglou, C.; Terzaki, K.; Kaliva, M.; Kalyva, A.; Papadaki, E.; Vamvakaki, M.; Farsari, M. Recombinant human bone morphogenetic protein 2 (rhBMP-2) immobilized on laser-fabricated 3D scaffolds enhance osteogenesis. Colloids Surf. B Biointerfaces 2017, 149, 233–242. [Google Scholar] [CrossRef]
- Duan, K.R.; Chen, H.L.; Huang, J.; Yu, J.H.; Liu, S.Y.; Wang, D.X.; Li, Y.P. One-step synthesis of amino-reserved chitosan-graft-polycaprolactone as a promising substance of biomaterial. Carbohydr. Polym. 2010, 80, 498–503. [Google Scholar] [CrossRef]
- Marchessault, R.H.; Pearson, F.G.; Liang, C.Y. Infrared spectra of crystalline polysaccharides. VI. Effect of orientation on the tilting spectra of chitin films. Biochim. Biophys. Acta 1960, 45, 499–507. [Google Scholar] [CrossRef]
- De Britto, D.; Campana-Filho, S.P. Kinetics of the thermal degradation of chitosan. Thermochim. Acta 2007, 465, 73–82. [Google Scholar] [CrossRef]
- Georgieva, V.; Zvezdova, D.; Vlaev, L. Non-isothermal kinetics of thermal degradation of chitosan. Chem. Cent. J. 2012, 6, 81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kweon, H.; Yoo, M.K.; Park, I.K.; Kim, T.H.; Lee, H.C.; Lee, H.S.; Oh, J.S.; Akaike, T.; Cho, C.S. A novel degradable polycaprolactone networks for tissue engineering. Biomaterials 2003, 24, 801–808. [Google Scholar] [CrossRef]
- Feng, H.; Dong, C.M. Preparation and characterization of chitosan-graft-poly (epsilon-caprolactone) with an organic catalyst. J. Polym. Sci. Polym. Chem. 2006, 44, 5353–5361. [Google Scholar] [CrossRef]
- Chen, E.C.; Wu, T.M. Isothermal crystallization kinetics and thermal behavior of poly(epsilon-caprolactone)/multi-walled carbon nanotube composites. Polym. Degrad. Stabil. 2007, 92, 1009–1015. [Google Scholar] [CrossRef]
- Jaworska, M.; Sakurai, K.; Gaudon, P.; Guibal, E. Influence of chitosan characteristics on polymer properties. I: Crystallographic properties. Polym. Int. 2003, 52, 198–205. [Google Scholar] [CrossRef]
- Wang, S.F.; Shen, L.; Zhang, W.D.; Tong, Y.J. Preparation and mechanical properties of chitosan/carbon nanotubes composites. Biomacromolecules 2005, 6, 3067–3072. [Google Scholar] [CrossRef] [PubMed]
- Li, X.D.; Gao, H.S.; Scrivens, W.A.; Fei, D.L.; Xu, Y.; Sutton, M.A.; Reynolds, A.P.; Myrick, M.L. Nanomechanical characterization of single-walled carbon nanotube reinforced epoxy composited. Nanotechnology 2004, 15, 1416–1423. [Google Scholar] [CrossRef]
- Liu, L.; Li, Y.; Liu, H.; Fang, Y. Synthesis and characterization of chitosan-graft-polycaprolactone copolymers. Eur. Polym. J. 2004, 40, 2739–2744. [Google Scholar] [CrossRef]
- Chen, Q.-Z.; Bismarck, A.; Hansen, U.; Junaid, S.; Tran, M.Q.; Harding, S.E.; Ali, N.N.; Boccaccini, A.R. Characterisation of a soft elastomer poly (glycerol sebacate) designed to match the mechanical properties of myocardial tissue. Biomaterials 2008, 29, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Roeder, B.A.; Kokini, K.; Sturgis, J.E.; Robinson, J.P.; Voytik-Harbin, S.L. Tensile mechanical properties of three-dimensional type I collagen extracellular matrices with varied microstructure. J. Biomech. Eng. 2002, 124, 214–222. [Google Scholar] [CrossRef]
- Webb, A.R.; Yang, J.; Ameer, G.A. Biodegradable polyester elastomers in tissue engineering. Expert Opin. Biol. Ther. 2004, 4, 801–812. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Shite, J.; Takaoka, H.; Shinke, T.; Imuro, Y.; Ozawa, T.; Otake, H.; Matsumoto, D.; Ogasawara, D.; Paredes, O.L.; et al. Myocardial stiffness is an important determinant of the plasma brain natriuretic peptide concentration in patients with both diastolic and systolic heart failure. Eur. Heart J. 2006, 27, 832–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oyen, M.L.; Cook, R.F. Load-displacement during sharp indentation of viscous-elastic-plastic materials. J. Mater. Res. 2003, 18, 5630–5642. [Google Scholar] [CrossRef]
- Dong, Y.M.; Ruan, Y.H.; Wang, H.W.; Zhao, Y.G.; Bi, D.X. Studies on glass transition temperature of chitosan with four techniques. J. Appl. Polym. Sci. 2004, 93, 1553–1558. [Google Scholar] [CrossRef]
- Nielsen, L.E.; Landel, R.F. Mechanical Properties of Polymers and Composites, 2nd ed.; Marcel Dekker: New York, NY, USA, 1994. [Google Scholar]
- Dee, K.C.; Puleo, D.A.; Bizios, R. An Introduction to Tissue-Biomaterial Interactions; John Wiley & Sons: Hoboken, NJ, USA, 2002; ISBN 978-0-471-25394-5. [Google Scholar]
- Shalaby, S.W. Biomedical Polymers; Hanser Publishers: New York, NY, USA, 1994. [Google Scholar]
- Lu, L.; Peter, S.J.; Lyman, M.D.; Lai, H.L.; Leite, S.M.; Tamada, J.A.; Uyama, S.; Vacanti, J.P.; Langer, R.; Mikos, A.G. In vitro and in vivo degradation of porous poly (dl-lactic-co-glycolic acid) foams. Biomaterials 2000, 21, 1837–1845. [Google Scholar] [CrossRef]
- Odelius, K.; Hoglund, A.; Kumar, S.; Hakkarainen, M.; Ghosh, A.K.; Bhatnagar, N.; Albertsson, A.C. Porosity and pore size regulate the degradation product profile of polylactide. Biomacromolecules 2011, 12, 1250–1258. [Google Scholar] [CrossRef]
- Hofmann, D.; Entrialgo-Castano, M.; Kratz, K.; Lendlein, A. Knowledge-based approach towards hydrolytic degradation of polymer-based biomaterials. Adv. Mater. 2009, 21, 3237–3245. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.C.; Jiang, Y.; Zhang, Y.; Ye, Z.Y.; Tan, W.S.; Lang, M.D. Effect of porosity on long-term degradation of poly (epsilon-caprolactone) scaffolds and their cellular response. Polym. Degrad. Stabil. 2013, 98, 209–218. [Google Scholar] [CrossRef]
- Shor, L.; Guceri, S.; Wen, X.; Gandhi, M.; Sun, W. Fabrication of three-dimensional polycaprolactone/hydroxyapatite tissue scaffolds and osteoblast-scaffold interactions in vitro. Biomaterials 2007, 28, 5291–5297. [Google Scholar] [CrossRef] [PubMed]
- Thuaksuban, N.; Nuntanaranont, T.; Pattanachot, W.; Suttapreyasri, S.; Cheung, L.K. Biodegradable polycaprolactone-chitosan three-dimensional scaffolds fabricated by melt stretching and multilayer deposition for bone tissue engineering: Assessment of the physical properties and cellular response. Biomed. Mater. 2011, 6, 015009. [Google Scholar] [CrossRef] [PubMed]
- Sheehy, E.J.; Mesallati, T.; Vinardell, T.; Kelly, D.J. Engineering cartilage or endochondral bone: A comparison of different naturally derived hydrogels. Acta Biomater. 2015, 13, 245–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Georgopoulou, A.; Kaliva, M.; Vamvakaki, M.; Chatzinikolaidou, M. Osteogenic Potential of Pre-Osteoblastic Cells on a Chitosan-graft-Polycaprolactone Copolymer. Materials 2018, 11, 490. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.A.; Sampaio, L.C.; Gobin, A. Building new hearts: A review of trends in cardiac tissue engineering. Am. J. Transplant. 2014, 14, 2448–2459. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.; Budina, E.; Stoppel, W.L.; Sullivan, K.E.; Emani, S.; Emani, S.M.; Black, L.D., 3rd. Cardiac extracellular matrix-fibrin hybrid scaffolds with tunable properties for cardiovascular tissue engineering. Acta Biomater. 2015, 14, 84–95. [Google Scholar] [CrossRef] [PubMed]
- Oberwallner, B.; Brodarac, A.; Choi, Y.H.; Saric, T.; Anic, P.; Morawietz, L.; Stamm, C. Preparation of cardiac extracellular matrix scaffolds by decellularization of human myocardium. J. Biomed. Mater. Res. Part A 2014, 102, 3263–3272. [Google Scholar] [CrossRef]
- Gishto, A.; Farrell, K.; Kothapalli, C.R. Tuning composition and architecture of biomimetic scaffolds for enhanced matrix synthesis by murine cardiomyocytes. J. Biomed. Mater. Res. Part A 2015, 103, 693–708. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Days | PCL | CS-g-PCL | ||
|---|---|---|---|---|
| Weight (g) | % Weight Loss (%) | Weight (g) | % Weight Loss (%) | |
| 0 | 0.135 ± 0.003 | 0 | 0.082 ± 0.002 | 0 |
| 7 | 0.120 ± 0.002 | 11 ± 2 | 0.073 ± 0.002 | 11 ± 4 |
| 14 | 0.114 ± 0.002 | 16 ± 2 | 0.067 ± 0.002 | 18 ± 4 |
| 21 | 0.110 ± 0.001 | 19 ± 2 | 0.053 ± 0.002 | 35 ± 4 |
| Sample | Elastic Modulus (MPa) | Hardness (MPa) |
|---|---|---|
| PCL | 443 ± 44 | 13 ± 1.5 |
| CS | 615 ± 55 | 54 ± 5 |
| CS-g-PCL | 550 ± 88 | 45 ± 4 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Charitidis, C.A.; Dragatogiannis, D.A.; Milioni, E.; Kaliva, M.; Vamvakaki, M.; Chatzinikolaidou, M. Synthesis, Nanomechanical Characterization and Biocompatibility of a Chitosan-Graft-Poly(ε-caprolactone) Copolymer for Soft Tissue Regeneration. Materials 2019, 12, 150. https://doi.org/10.3390/ma12010150
Charitidis CA, Dragatogiannis DA, Milioni E, Kaliva M, Vamvakaki M, Chatzinikolaidou M. Synthesis, Nanomechanical Characterization and Biocompatibility of a Chitosan-Graft-Poly(ε-caprolactone) Copolymer for Soft Tissue Regeneration. Materials. 2019; 12(1):150. https://doi.org/10.3390/ma12010150
Chicago/Turabian StyleCharitidis, Costas A., Dimitrios A. Dragatogiannis, Eleni Milioni, Maria Kaliva, Maria Vamvakaki, and Maria Chatzinikolaidou. 2019. "Synthesis, Nanomechanical Characterization and Biocompatibility of a Chitosan-Graft-Poly(ε-caprolactone) Copolymer for Soft Tissue Regeneration" Materials 12, no. 1: 150. https://doi.org/10.3390/ma12010150
APA StyleCharitidis, C. A., Dragatogiannis, D. A., Milioni, E., Kaliva, M., Vamvakaki, M., & Chatzinikolaidou, M. (2019). Synthesis, Nanomechanical Characterization and Biocompatibility of a Chitosan-Graft-Poly(ε-caprolactone) Copolymer for Soft Tissue Regeneration. Materials, 12(1), 150. https://doi.org/10.3390/ma12010150