Effect of Cu F Co-doping on the Properties of AgSnO2 Contact

Abstract
:1. Introduction
2. Models, Calculation Method, and Experiment
2.1. Models and Calculation Method
2.2. Preparation of Samples
3. Simulation Analysis
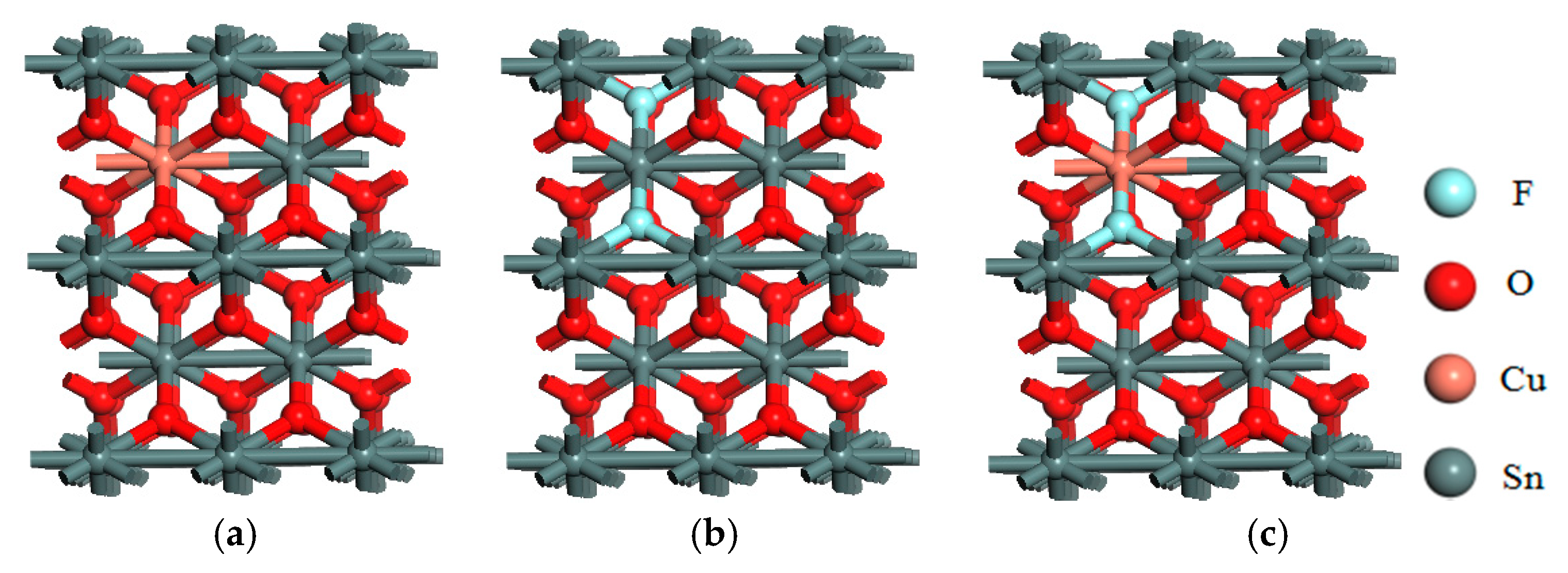
3.1. Crystal Structure and Stability
3.2. Electronic Structure
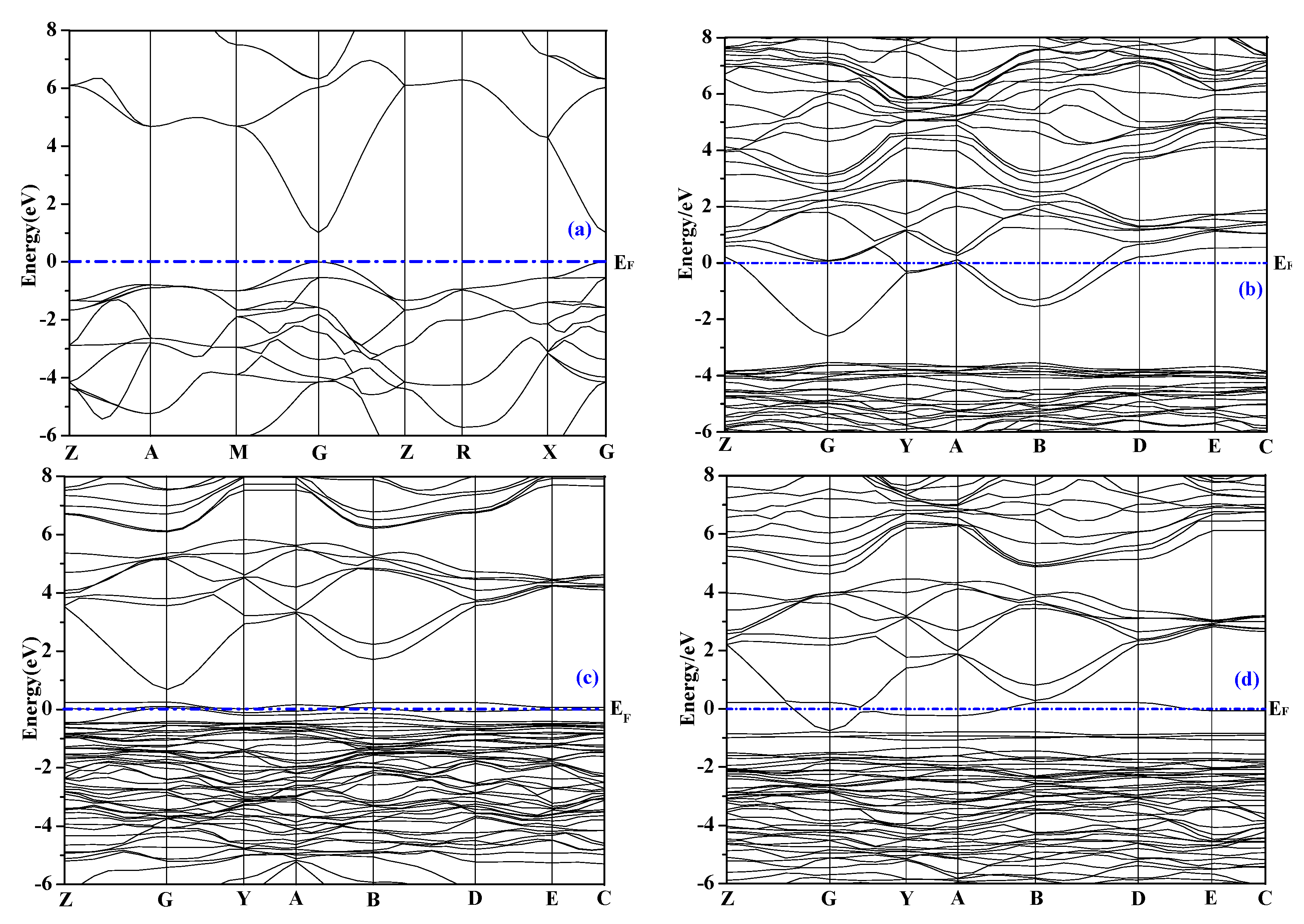
3.2.1. Energy Band Structure
3.2.2. Electron Effective Mass and Ionizing Energy of Donor
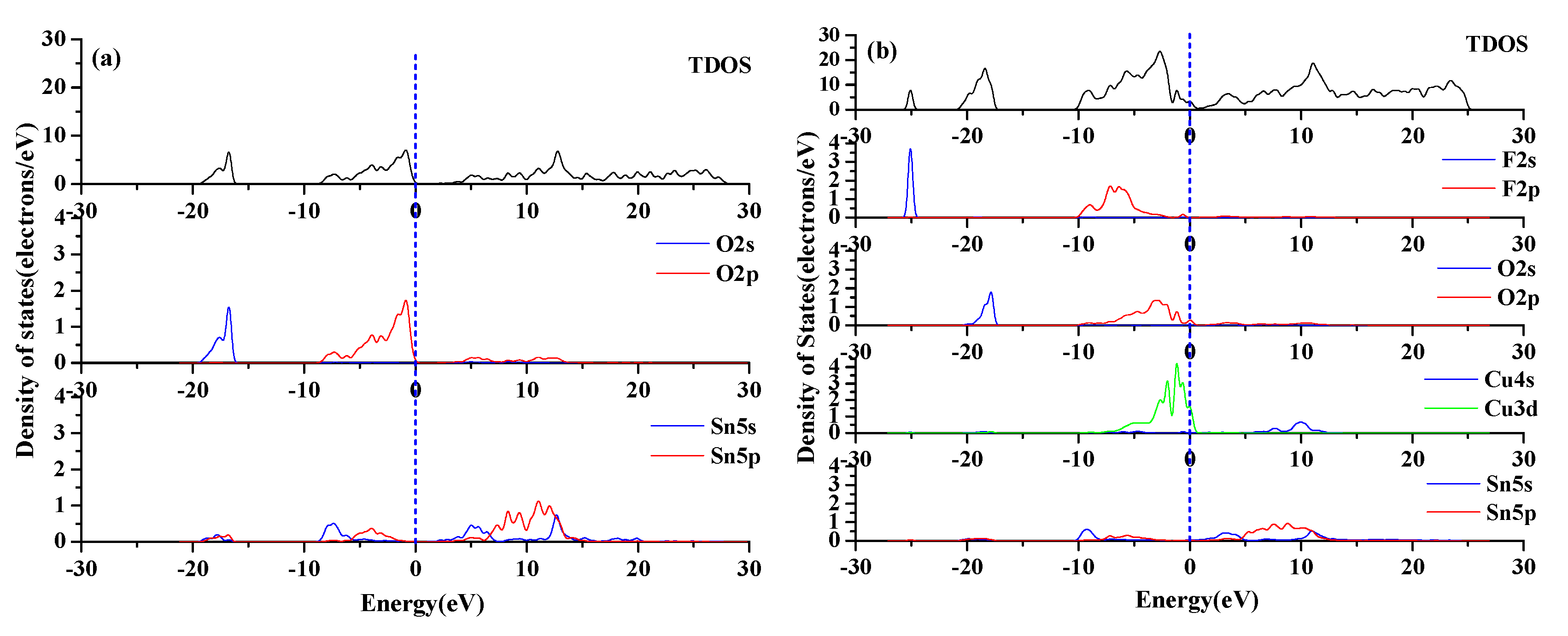
3.2.3. Density of States
3.3. Mechanical Properties
(C11 + C33 − 2C13) > 0, [2(C11 − C12) + C33 + 4C13] > 0
3.4. Debye Temperature
4. Experiment
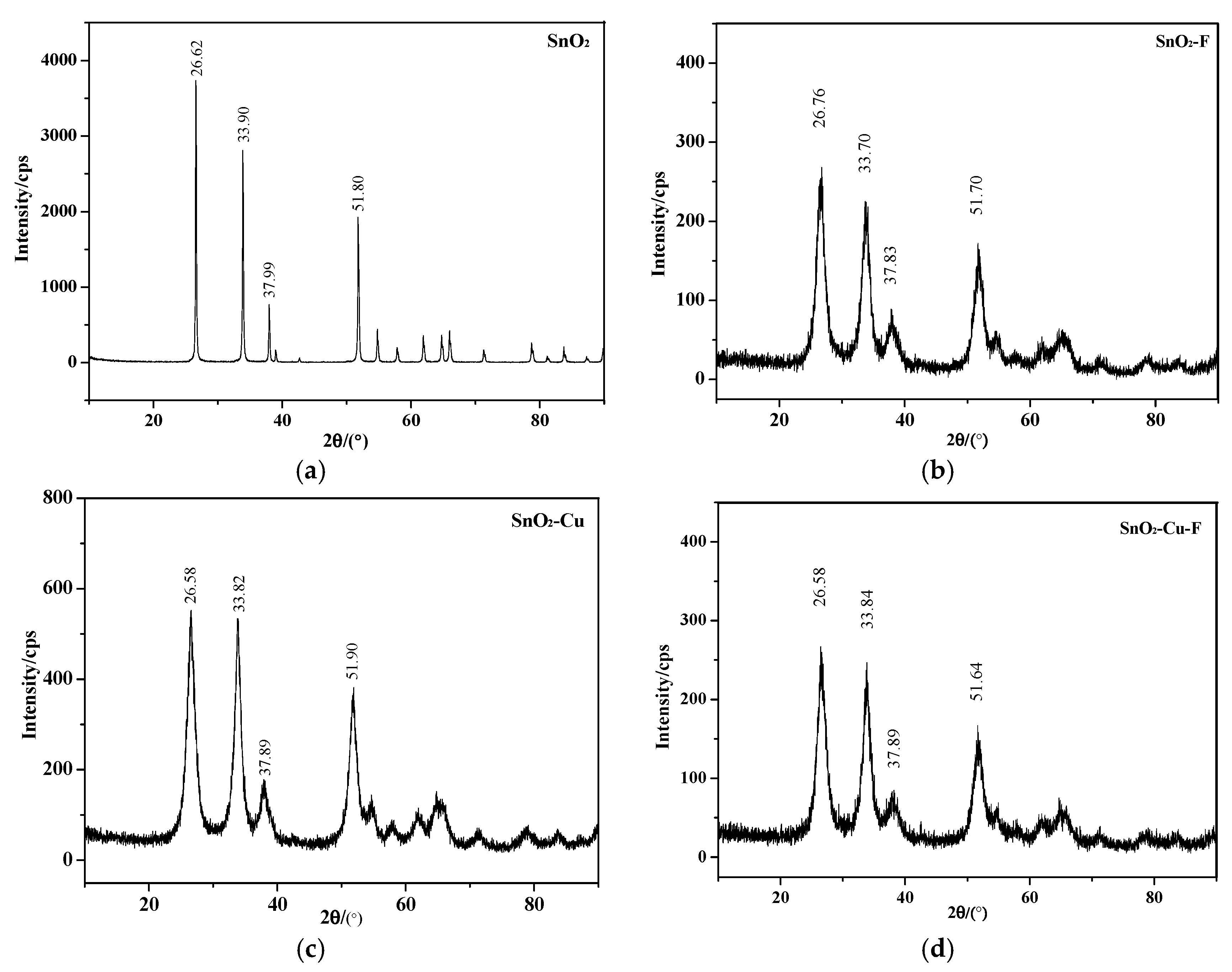
4.1. X-Ray Diffraction Experiment
4.2. Wettability Test
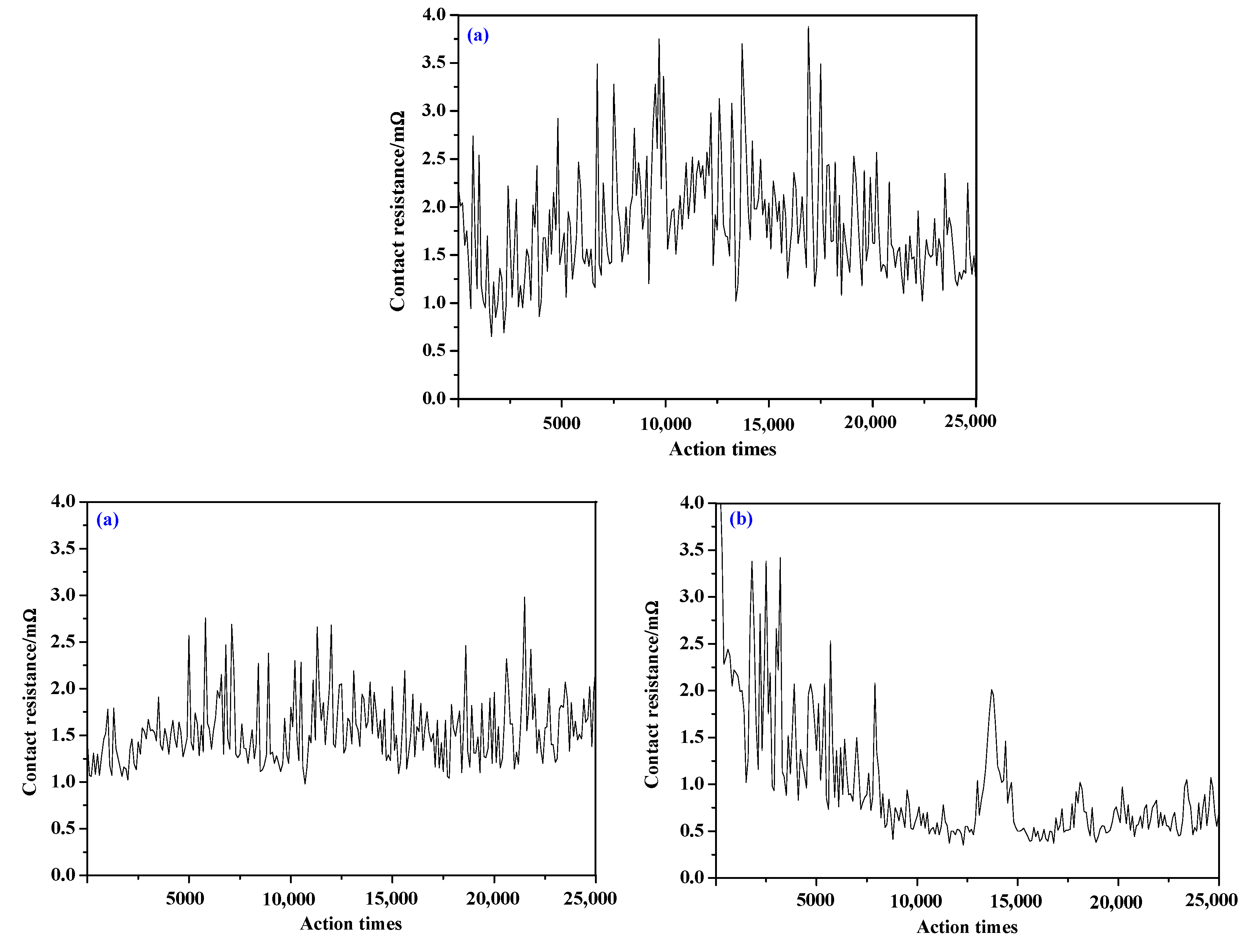
4.3. Hardness, Conductivity, and Electrical Contact Simulation Experiment
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Qiao, X.; Shen, Q.; Chen, L.; He, Q.; Fan, X.; Yang, H. Research progress in AgSnO2 electrical contact materials. Mater. Rep. 2013, 27, 1–6. [Google Scholar]
- Swingler, J.; Sumption, A. Arc erosion of AgSnO2 electrical contacts at different stages of a break operation. Rare Met. 2010, 29, 248–254. [Google Scholar] [CrossRef]
- Wang, J.; Wang, Y.; Yang, Z. Effect of CuO additive on the wettability and interface behavior of silver/Tin Oxide. Rare Met. Mater. Eng. 2005, 34, 405–408. [Google Scholar]
- Xu, J.; Huang, S.; Wang, Z.; Lu, D.-X.; Yuan, T.-S. Simulative calculation of electronic structure of F-doped SnO2. Acta Phys. Sin. 2007, 56, 7195–7200. [Google Scholar]
- Bolzan, A.A.; Fong, C.; Kennedy, B.J.; Howard, C.J. Structural studies of rutile-type metal dioxides. Acta Crystallogr. 2010, 53, 373–380. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Wang, D.; Zhang, Y.; Su, L.; Chen, S.; Wang, X.; Sun, P.; Yi, C. Electronic structure and optical properties of Cu-O co-doped AlN. Acta Phys. Sin. 2018, 67, 210–217. [Google Scholar]
- Ding, C.; Li, W.; Liu, J.; Wang, L.-L.; Cai, Y.; Pan, P.-F. First principle study of electronic structure of Sb,S Co-doped SnO2. Acta Phys. Sin. 2018, 67, 141–147. [Google Scholar]
- Dolbec, R.; Khakani, M.A.E.; Serventi, A.M.; Trudeau, M.; Saint-Jacques, R.G. Microstructure and Physical Properties of Nanostructured tin Oxide Thin Films Grown by Mans of Pulsed Laser Deposition. Thin Solid Films 2002, 419, 230–236. [Google Scholar] [CrossRef]
- Kittel, C. Introduction to Solid State Physics, 8th ed.; Chemical Industry Press: Beijing, China, 2004; p. 241. (In Chinese) [Google Scholar]
- Hou, Q.; Wu, Y.; Zhao, C. Study on the effect of In-2N co-doping at preferential locality on the photoelectric function of ZnO(GGA+U). Acta Phys. Sin. 2014, 63, 342–348. [Google Scholar]
- Finger, L.W. Physical properties of crystals, their representation by tensors and matrices. Phys. Today 1985, 36, 536. [Google Scholar] [CrossRef]
- Hill, R. The elastic behaviour of crystalline aggregate. Proc. Phys. Soc. 2002, 65, 349–354. [Google Scholar] [CrossRef]
- Voigt, W.; Fernandez, E.P.; Hsia, S.L. Transformation of testosterone into 17β-hydroxy-5α-androstan-3-one by microsomal preparations of human skin. J. Biol. Chem. 1970, 245, 5594–5599. [Google Scholar] [PubMed]
- Reuss, A.; Angrew, Z. Optimal bounds and microgeometries for elastic two-phase composities. Math. Mech. Solids 1929, 9, 45–48. [Google Scholar]
- Pugh, S.F. XCII. Relations between the elastic moduli and the plastic properties of polycrystalline pure metals. Philos. Mag. Ser. 2009, 45, 823–843. [Google Scholar] [CrossRef]
- Liu, S.; Zhan, Y.; Wu, J.; Wei, X. Insight into structural, mechanical, electronic and thermodynamic properties of intermetallic phases in Zr–Sn system from first-principles calculations. J. Phys. Chem. Solids 2015, 86, 177–185. [Google Scholar] [CrossRef]
- Anderson, O.L. A simplified method for calculating the debye temperature from elastic contants. J. Phys. Chem. Solids 1963, 24, 909–917. [Google Scholar] [CrossRef]
- Wang, H.; Wang, J.; Du, J.; Meng, F. Influence of rare earth on the wetting ability of AgSnO2 contact material. Rare Met. Mater. Eng. 2014, 43, 1846–1849. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | SnO2 | SnO2-F | SnO2-Cu | SnO2-Cu-F |
|---|---|---|---|---|
| a/Å | 4.737 | 4.963 | 4.861 | 4.892 |
| b/Å | 4.737 | 4.970 | 4.845 | 4.909 |
| c/Å | 3.186 | 3.354 | 3.250 | 3.266 |
| V/Å3 | 286.0 | 330.8 | 306.2 | 313.6 |
| Ef/eV | - | −4.39 | −1.97 | −8.89 |
| Doping Model | C11/GPa | C12/GPa | C13/GPa | C33/GPa | C44/GPa | C66/GPa |
|---|---|---|---|---|---|---|
| SnO2 | 204.4 | 131.2 | 114.3 | 357.0 | 86.90 | 177.6 |
| SnO2-F | 175.8 | 110.3 | 118.3 | 169.8 | 66.26 | 65.62 |
| SnO2-Cu | 169.1 | 126.5 | 116.4 | 175.1 | 50.25 | 49.75 |
| SnO2-Cu-F | 182.5 | 108.9 | 119.7 | 177.2 | 66.10 | 69.98 |
| Doping Model | B | G | E | γ | B/G | H |
|---|---|---|---|---|---|---|
| SnO2 | 161.5 | 96.07 | 219.3 | 0.2517 | 1.681 | 14.50 |
| SnO2-F | 143.3 | 65.76 | 171.1 | 0.3010 | 2.179 | 8.723 |
| SnO2-Cu | 149.2 | 53.89 | 144.3 | 0.3388 | 2.768 | 5.792 |
| SnO2-Cu-F | 146.9 | 70.45 | 182.2 | 0.2933 | 2.085 | 9.706 |
| Doping Model | ρ | Vt | Vl | Vm | ΘD |
|---|---|---|---|---|---|
| SnO2-F | 4.24 | 3661 | 7381 | 4399 | 484.5 |
| SnO2-Cu | 4.02 | 3938 | 7415 | 4110 | 452.4 |
| SnO2-Cu-F | 4.04 | 4176 | 7721 | 4660 | 513.2 |
| Doping Model | SnO2 | SnO2-F | SnO2-Cu | SnO2-Cu-F |
|---|---|---|---|---|
| Wetting angle (θ/°) | 99.65 | 65.45 | 2.15 | 1.15 |
| Contact | Hardness (HV) | Conductivity (mS⋅m−1) | Contact Resistance (mΩ) | Average Arc Energy (mJ) | Average Arc Duration (ms) |
|---|---|---|---|---|---|
| AgSnO2 | 117.1 | 26.44 | 1.814 | 215.3 | 3.34 |
| AgSnO2-Cu | 76.12 | 28.87 | 1.502 | 184.2 | 2.51 |
| AgSnO2-Cu-F | 82.03 | 31.20 | 1.048 | 190.6 | 2.76 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.-q.; Liu, Z.; Chen, L.; Yu, S.-m.; Zhu, Y.-c. Effect of Cu F Co-doping on the Properties of AgSnO2 Contact. Materials 2019, 12, 2315. https://doi.org/10.3390/ma12142315
Wang J-q, Liu Z, Chen L, Yu S-m, Zhu Y-c. Effect of Cu F Co-doping on the Properties of AgSnO2 Contact. Materials. 2019; 12(14):2315. https://doi.org/10.3390/ma12142315
Chicago/Turabian StyleWang, Jing-qin, Zhou Liu, Ling Chen, Shuang-miao Yu, and Yan-cai Zhu. 2019. "Effect of Cu F Co-doping on the Properties of AgSnO2 Contact" Materials 12, no. 14: 2315. https://doi.org/10.3390/ma12142315
APA StyleWang, J. -q., Liu, Z., Chen, L., Yu, S. -m., & Zhu, Y. -c. (2019). Effect of Cu F Co-doping on the Properties of AgSnO2 Contact. Materials, 12(14), 2315. https://doi.org/10.3390/ma12142315