Revealing High Oxygen Evolution Catalytic Activity of Fluorine-Doped Carbon in Alkaline Media


Abstract
:
{kind=link}
{kind=link}
{kind=link}
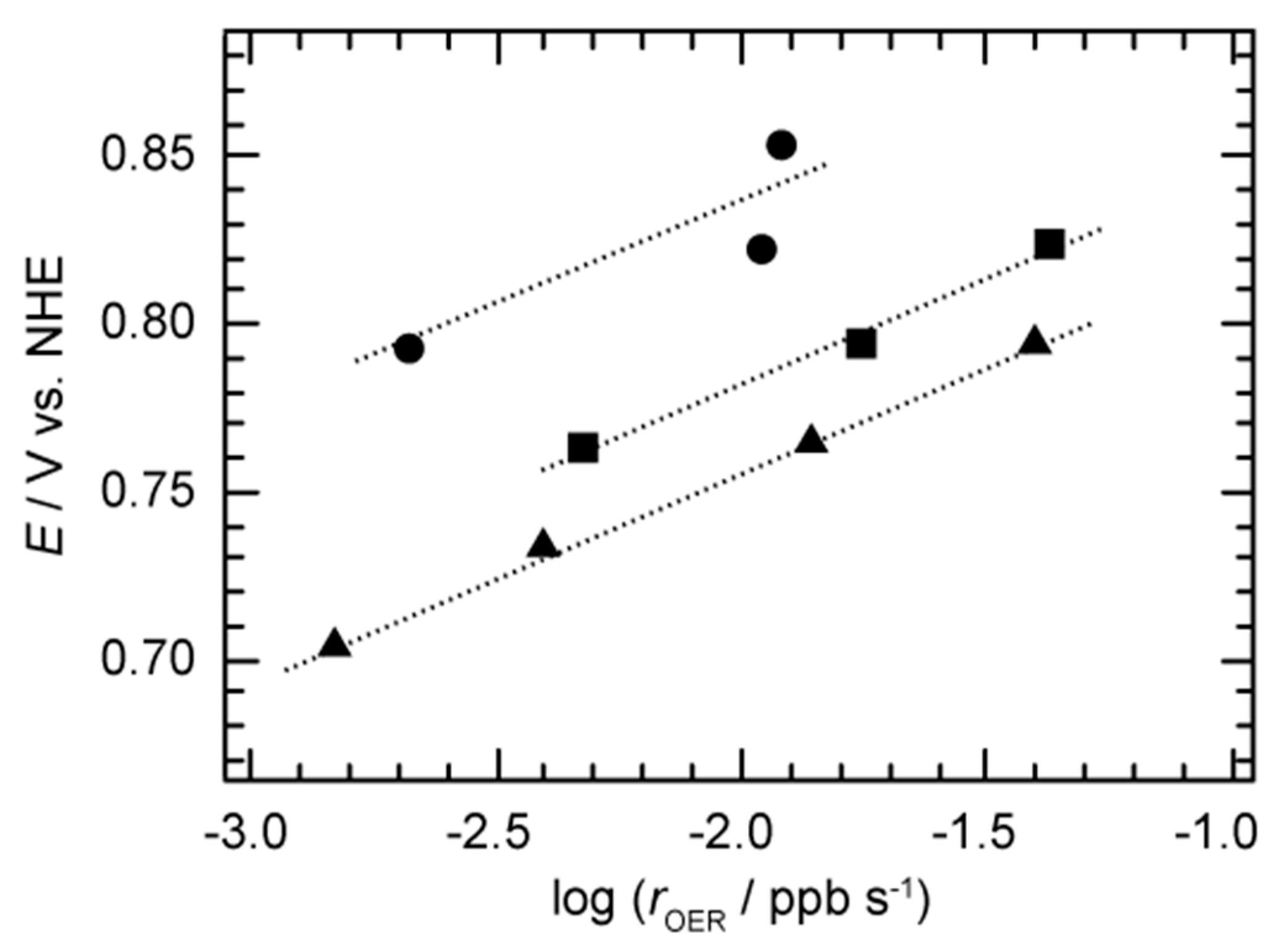{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Sample Preparation
2.2. Electrochemical Characterization
2.3. Theoretical Calculation
3. Results and Discussion
3.1. Evaluation of pH-Dependent Tafel Slopes from Electrochemical Measurement and O2 Sensor
3.2. Kinetic Model and Theoretical Calculation for F–Carbon
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Suen, N.-T.; Hung, S.-F.; Quan, Q.; Zhang, N.; Xu, Y.-J.; Chen, H.M. Electrocatalysis for the oxygen evolution reaction: Recent development and future perspectives. Chem. Soc. Rev. 2017, 46, 337–365. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, Y.; Sato, E. Electrocatalytic properties of transition metal oxides for oxygen evolution reaction. Mater. Chem. Phys. 1986, 14, 397–426. [Google Scholar] [CrossRef]
- Reier, T.; Oezaslan, M.; Strasser, P. Electrocatalytic Oxygen Evolution Reaction (OER) on Ru, Ir, and Pt Catalysts: A Comparative Study of Nanoparticles and Bulk Materials. ACS Catal. 2012, 2, 1765–1772. [Google Scholar] [CrossRef]
- Kim, J.; Zhou, R.; Murakoshi, K.; Yasuda, S. Advantage of semi-ionic bonding in fluorine-doped carbon materials for the oxygen evolution reaction in alkaline media. RSC Adv. 2018, 8, 14152–14156. [Google Scholar] [CrossRef]
- Xiao, Z.; Huang, X.; Xu, L.; Yan, D.; Huo, J.; Wang, S. Edge-selectively phosphorus-doped few-layer graphene as an efficient metal-free electrocatalyst for the oxygen evolution reaction. Chem. Commun. 2016, 52, 13008–13011. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Zhang, J.; Jiang, S.P. Are metal-free pristine carbon nanotubes electrocatalytically active? Chem. Commun. 2015, 51, 13764–13767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, G.L.; Zhao, M.Q.; Yu, D.; Kong, X.Y.; Huang, J.Q.; Zhang, Q.; Wei, F. Nitrogen-Doped Graphene/Carbon Nanotube Hybrids: In Situ Formation on Bifunctional Catalysts and Their Superior Electrocatalytic Activity for Oxygen Evolution/Reduction Reaction. Small 2014, 10, 2251–2259. [Google Scholar] [CrossRef]
- Zhao, Y.; Nakamura, R.; Kamiya, K.; Nakanishi, S.; Hashimoto, K. Nitrogen-doped carbon nanomaterials as non-metal electrocatalysts for water oxidation. Nat. Commun. 2013, 4, 1–7. [Google Scholar] [CrossRef]
- Staud, N.; Sokol, H.; Ross, P.N. The Corrosion of Carbon Black Anodes in Alkaline Electrolyte. IV. Current Efficiencies for Oxygen Evolution from Metal Oxide-Impregnated Graphitized Furnace Blacks. J. Electrochem. Soc. 1989, 136, 3570–3576. [Google Scholar] [CrossRef]
- Yi, Y.; Weinberg, G.; Prenzel, M.; Greiner, M.; Heumann, S.; Becker, S.; Schlögl, R. Electrochemical corrosion of a glassy carbon electrode. Catal. Today 2017, 295, 32–40. [Google Scholar] [CrossRef]
- McCrory, C.C.L.; Jung, S.; Peters, J.C.; Jaramillo, T.F. Benchmarking Heterogeneous Electrocatalysts for the Oxygen Evolution Reaction. J. Am. Chem. Soc. 2013, 135, 16977–16987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suntivich, J.; May, K.J.; Gasteiger, H.A.; Goodenough, J.B.; Shao-Horn, Y. A perovskite oxide optimized for oxygen evolution catalysis from molecular orbital principles. Science 2011, 334, 1383–1385. [Google Scholar] [CrossRef] [PubMed]
- Görlin, M.; Chernev, P.; De Araújo, J.F.; Reier, T.; Dresp, S.; Paul, B.; Krähnert, R.; Dau, H.; Strasser, P. Oxygen Evolution Reaction Dynamics, Faradaic Charge Efficiency, and the Active Metal Redox States of Ni–Fe Oxide Water Splitting Electrocatalysts. J. Am. Chem. Soc. 2016, 138, 5603–5614. [Google Scholar] [CrossRef] [PubMed]
- Berkes, B.B.; Jozwiuk, A.; Vračar, M.; Sommer, H.; Brezesinski, T.; Janek, J. Online Continuous Flow Differential Electrochemical Mass Spectrometry with a Realistic Battery Setup for High-Precision, Long-Term Cycling Tests. Anal. Chem. 2015, 87, 5878–5883. [Google Scholar] [CrossRef]
- Kanan, M.W.; Nocera, D.G. In Situ Formation of an Oxygen-Evolving Catalyst in Neutral Water Containing Phosphate and Co2+. Science 2008, 321, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Itoh, K.; Hagiwara, R.; Fukunaga, T.; Ito, Y. On the so-called “semi-ionic” C–F bond character in fluorine–GIC. Carbon 2004, 42, 3243–3249. [Google Scholar] [CrossRef]
- Corrigan, D.A. The Catalysis of the Oxygen Evolution Reaction by Iron Impurities in Thin Film Nickel Oxide Electrodes. J. Electrochem. Soc. 1987, 134, 377–384. [Google Scholar] [CrossRef]
- Trotochaud, L.; Young, S.L.; Ranney, J.K.; Boetther, S.W. Nickel-Iron Oxyhydroxide Oxygen Evolution Electrocatalysis: The Role of Intentional and Incidental Iron Incorporation. J. Am. Chem. Soc. 2014, 136, 6744–6753. [Google Scholar] [CrossRef]
- Bockris, J.O.M. Kinetics of Activation Controlled Consecutive Electrochemical Reactions: Anodic Evolution of Oxygen. J. Chem. Phys. 1956, 24, 817–827. [Google Scholar] [CrossRef]
- Surendranath, Y.; Kanan, M.W.; Nocera, D.G. Mechanistic Studies of the Oxygen Evolution Reaction by a Cobalt-Phosphate Catalyst at Neutral pH. J. Am. Chem. Soc. 2010, 132, 16501–16509. [Google Scholar] [CrossRef]
- Shinagawa, T.; Garcia-Esparza, A.T.; Takanabe, K. Insight on Tafel slopes from a microkinetic analysis of aqueous electrocatalysis for energy conversion. Sci. Rep. 2015, 5, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Iwakura, C.; Fukuda, K.; Tamura, H. The anodic evolution of oxygen on platinum oxide electrode in alkaline solutions. Electrochim. Acta 1976, 21, 501–508. [Google Scholar] [CrossRef]
- Bockris, J.O.M.; Otagawa, T. The Electrocatalysis of Oxygen Evolution on Perovskites. J. Electrochem. Soc. 1984, 131, 290–302. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.; Fukushima, T.; Zhou, R.; Murakoshi, K. Revealing High Oxygen Evolution Catalytic Activity of Fluorine-Doped Carbon in Alkaline Media. Materials 2019, 12, 211. https://doi.org/10.3390/ma12020211
Kim J, Fukushima T, Zhou R, Murakoshi K. Revealing High Oxygen Evolution Catalytic Activity of Fluorine-Doped Carbon in Alkaline Media. Materials. 2019; 12(2):211. https://doi.org/10.3390/ma12020211
Chicago/Turabian StyleKim, Jeheon, Tomohiro Fukushima, Ruifeng Zhou, and Kei Murakoshi. 2019. "Revealing High Oxygen Evolution Catalytic Activity of Fluorine-Doped Carbon in Alkaline Media" Materials 12, no. 2: 211. https://doi.org/10.3390/ma12020211
APA StyleKim, J., Fukushima, T., Zhou, R., & Murakoshi, K. (2019). Revealing High Oxygen Evolution Catalytic Activity of Fluorine-Doped Carbon in Alkaline Media. Materials, 12(2), 211. https://doi.org/10.3390/ma12020211