Nitrogen-Doped Superporous Activated Carbons as Electrocatalysts for the Oxygen Reduction Reaction

 ,
,  ,
,  and
and 
Abstract
:1. Introduction
- (i)
- Carbonization of a nitrogen-containing precursor (such as pyridine, melamine, and polyaniline), which can be followed by an activation process (either physical or chemical).
- (ii)
- Hydrothermal carbonization of nitrogen containing-compounds (glucosamine, cyanuric acid, etc.).
- (iii)
- Templating approaches using a nitrogen-containing precursor followed by a thermal treatment.
- (iv)
- Post-thermal treatments of a material previously synthesized with a nitrogen-containing reactant in gas or liquid phase.
2. Materials and Methods
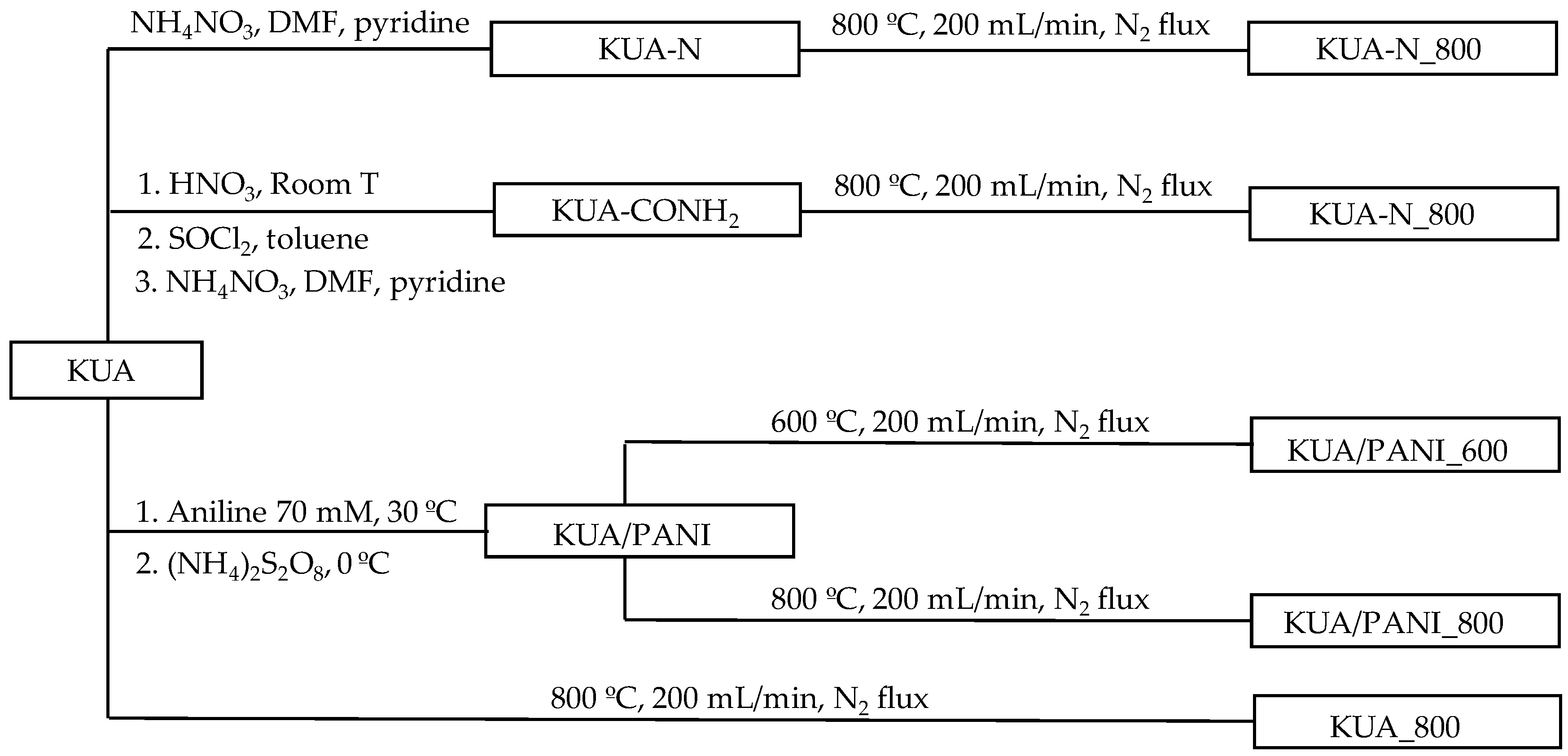
2.1. Synthesis of Activated Carbons
2.1.1. Pristine Activated Carbon
2.1.2. Chemical Functionalization of Activated Carbon at Mild Conditions
2.1.3. Preparation of Polyaniline/Activated Carbon Composite
2.1.4. Post-Thermal Treatments
2.2. Porous Texture and Surface Chemistry Characterization
2.3. Electrochemical Activity towards ORR
3. Results and Discussion
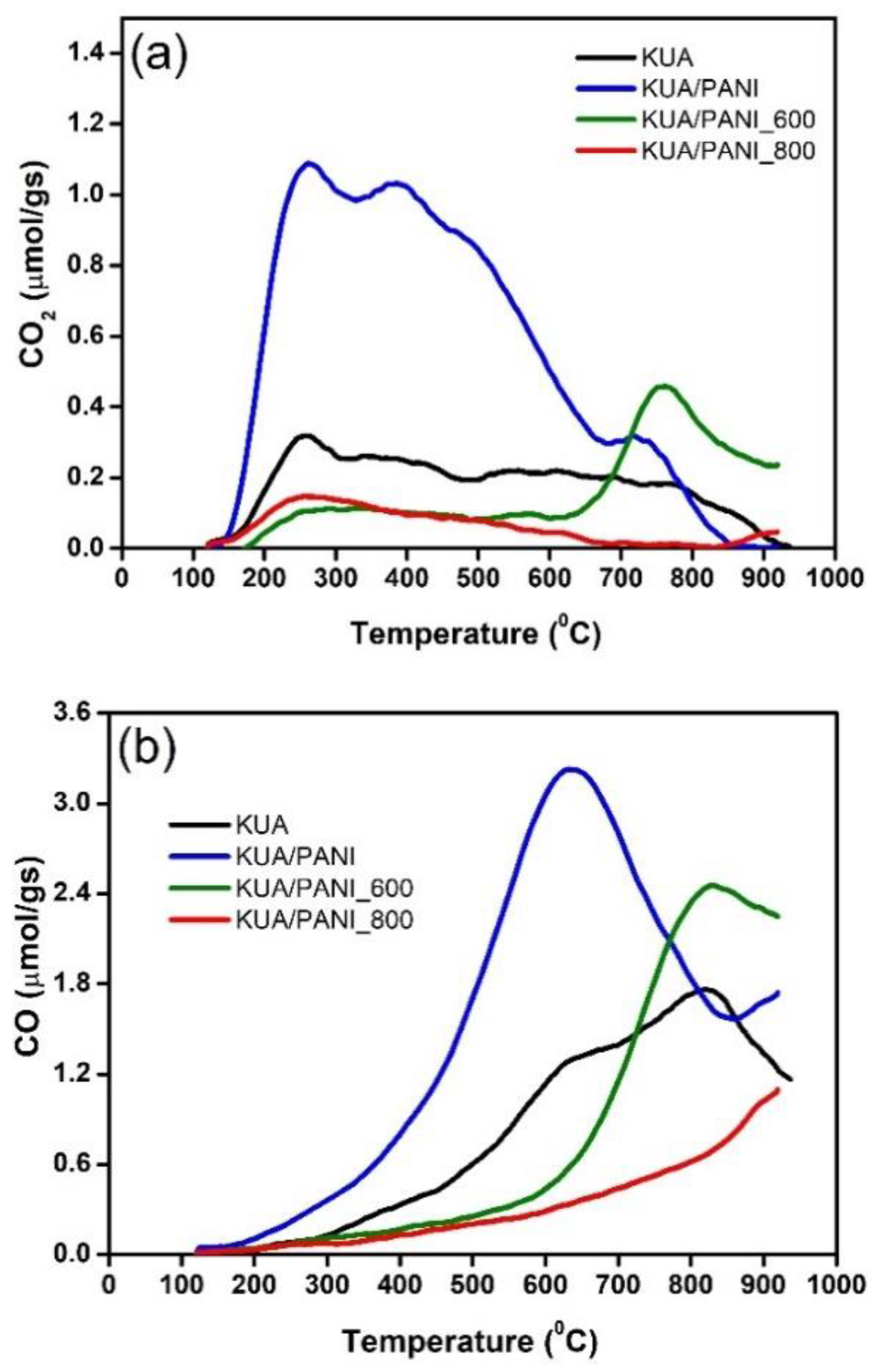
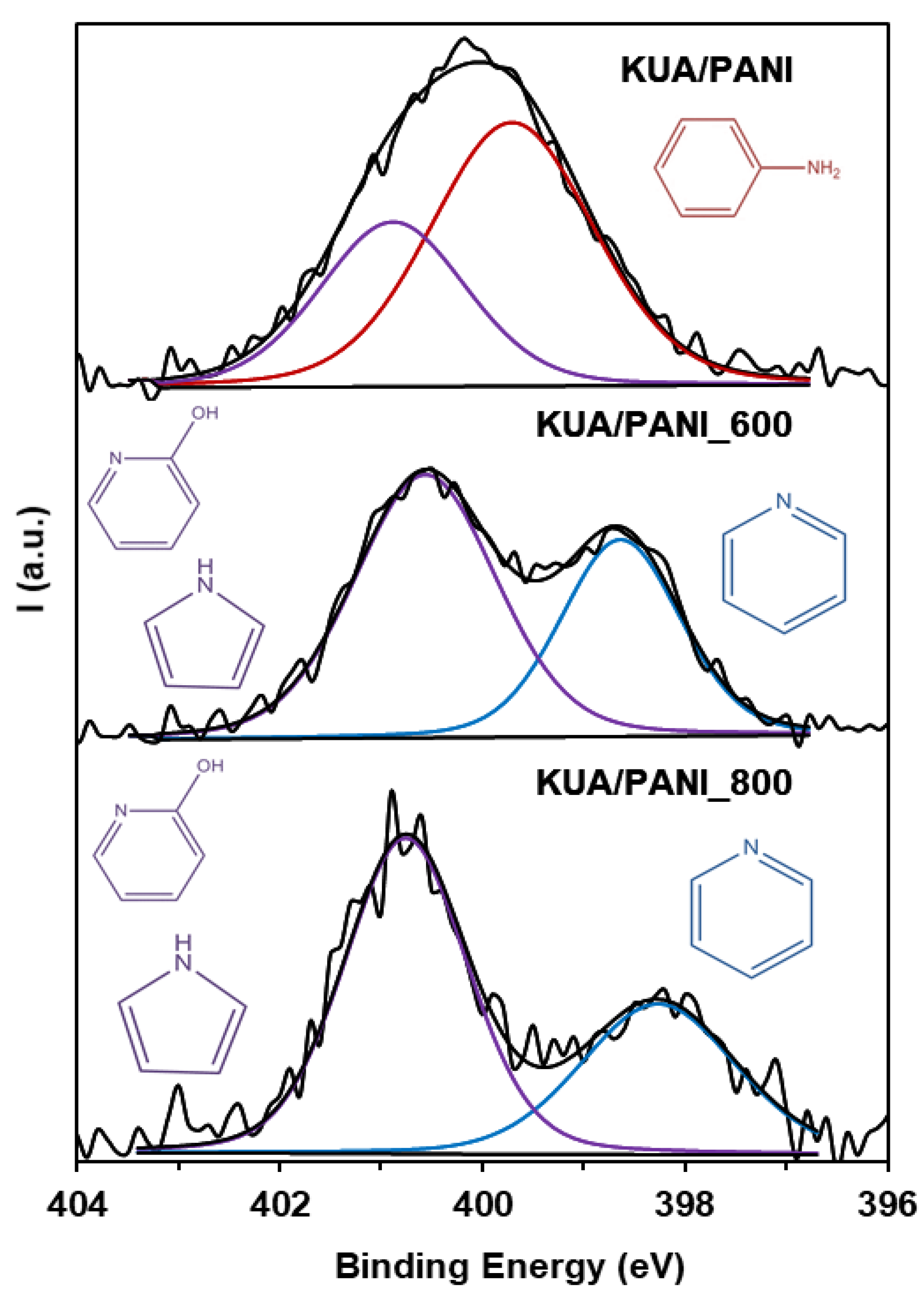
3.1. Surface Chemistry Characterization
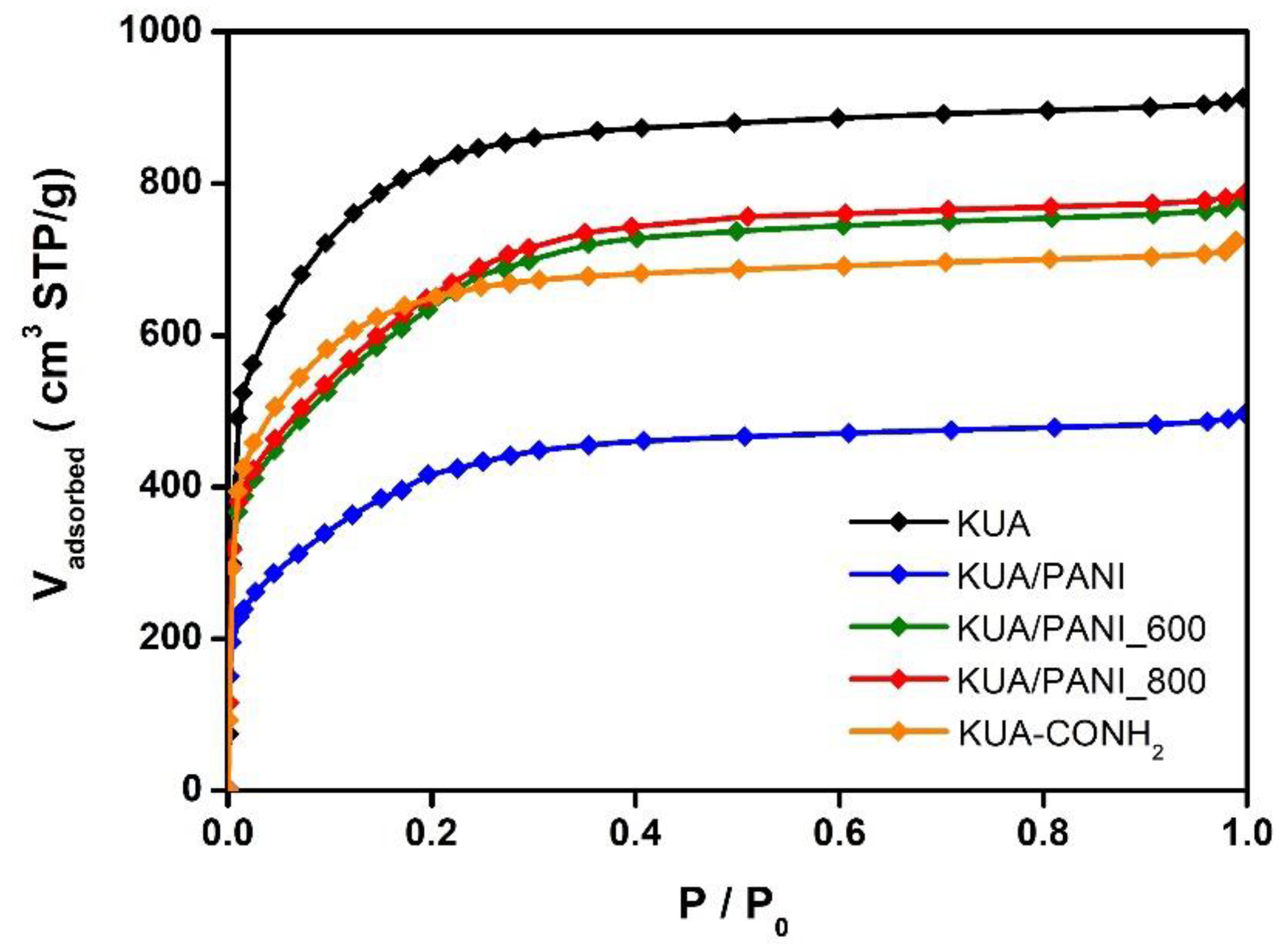
3.2. Porous Texture Characterization
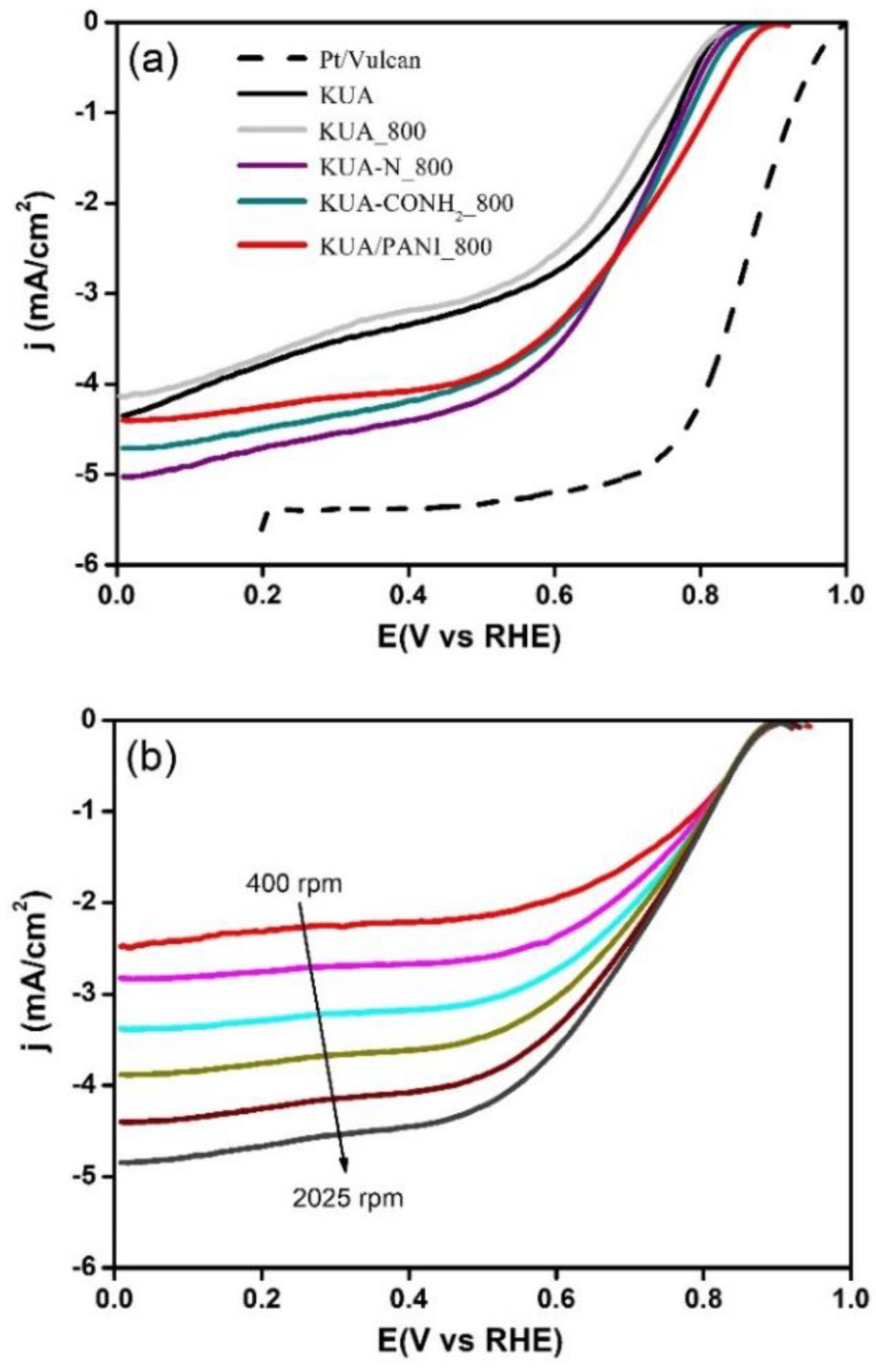
3.3. Electroactivity Towards ORR
3.3.1. Pristine Activated Carbon
3.3.2. N-Doped Activated Carbons at Mild Conditions
3.3.3. Polyaniline/Activated Carbon Composite
3.3.4. Heat-Treated Activated Carbons
3.3.5. Selectivity to Water Formation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Shao, M.; Chang, Q.; Dodelet, J.P.; Chenitz, R. Recent Advances in Electrocatalysts for Oxygen Reduction Reaction. Chem. Rev. 2016, 116, 3594–3657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bezerra, C.W.B.; Zhang, L.; Liu, H.; Lee, K.; Marques, A.L.B.; Marques, E.P.; Wang, H.; Zhang, J. A review of heat-treatment effects on activity and stability of PEM fuel cell catalysts for oxygen reduction reaction. J. Power Sources 2007, 173, 891–908. [Google Scholar] [CrossRef]
- Bezerra, C.W.B.; Zhang, L.; Lee, K.; Liu, H.; Marques, A.L.B.; Marques, E.P.; Wang, H.; Zhang, J. A review of Fe-N/C and Co-N/C catalysts for the oxygen reduction reaction. Electrochim. Acta 2008, 53, 4937–4951. [Google Scholar] [CrossRef]
- Yu, X.; Ye, S. Recent advances in activity and durability enhancement of Pt/C catalytic cathode in PEMFC. Part II: Degradation mechanism and durability enhancement of carbon supported platinum catalyst. J. Power Sources 2007, 172, 145–154. [Google Scholar] [CrossRef]
- Borup, R.; Meyers, J.; Pivovar, B.; Kim, Y.S.; Mukundan, R.; Garland, N.; Myers, D.; Wilson, M.; Garzon, F.; Wood, D.; et al. Scientific aspects of polymer electrolyte fuel cell durability and degradation. Chem. Rev. 2007, 107, 3904–3951. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Jia, Y.; Yao, X. Defects on carbons for electrocatalytic oxygen reduction. Chem. Soc. Rev. 2018, 47, 7628–7658. [Google Scholar] [CrossRef]
- Morozan, A.; Jousselme, B.; Palacin, S. Low-platinum and platinum-free catalysts for the oxygen reduction reaction at fuel cell cathodes. Energy Environ. Sci. 2011, 4, 1238–1254. [Google Scholar] [CrossRef]
- Ozaki, J.; Kimura, N.; Anahara, T.; Oya, A. Preparation and oxygen reduction activity of BN-doped carbons. Carbon N. Y. 2007, 45, 1847–1853. [Google Scholar] [CrossRef]
- Samanta, A.; Ohsaka, T.; Mondal, S.; Noh, S.H.; Raj, C.R.; Okajima, T. Emerging new generation electrocatalysts for the oxygen reduction reaction. J. Mater. Chem. A 2016, 4, 11156–11178. [Google Scholar]
- Kundu, S.; Nagaiah, T.C.; Xia, W.; Wang, Y.; Van Dommele, S.; Bitter, J.H.; Santa, M.; Grundmeier, G.; Bron, M.; Schuhmann, W.; et al. Electrocatalytic activity and stability of nitrogen-containing carbon nanotubes in the oxygen reduction reaction. J. Phys. Chem. C 2009, 113, 14302–14310. [Google Scholar] [CrossRef]
- Wu, G.; Santandreu, A.; Kellogg, W.; Gupta, S.; Ogoke, O.; Zhang, H.; Wang, H.L.; Dai, L. Carbon nanocomposite catalysts for oxygen reduction and evolution reactions: From nitrogen doping to transition-metal addition. Nano Energy 2016, 29, 83–110. [Google Scholar] [CrossRef]
- Wu, K.H.; Wang, D.W.; Su, D.S.; Gentle, I.R. A Discussion on the Activity Origin in Metal-Free Nitrogen-Doped Carbons for Oxygen Reduction Reaction and their Mechanisms. ChemSusChem 2015, 8, 2772–2788. [Google Scholar] [CrossRef]
- Gabe, A.; Ruiz-Rosas, R.; González-Gaitán, C.; Morallón, E.; Cazorla-Amorós, D. Modeling of oxygen reduction reaction in porous carbon materials in alkaline medium. Effect of microporosity. J. Power Sources 2019, 412, 451–464. [Google Scholar] [CrossRef]
- Deng, Y.; Xie, Y.; Zou, K.; Ji, X. Review on recent advances in nitrogen-doped carbons: Preparations and applications in supercapacitors. J. Mater. Chem. A 2015, 4, 1144–1173. [Google Scholar] [CrossRef]
- Gabe, A.; Mostazo-López, M.J.; Salinas-Torres, D.; Morallón, E.; Cazorla-Amorós, D. Synthesis of Conducting Polymer/Carbon Material Composites and Their Application in Electrical Energy Storage. In Hybrid Polymer Composite Materials: Processing; Woodhead Publishing: Duxford, UK, 2017; pp. 173–209. [Google Scholar]
- Wu, G.; More, K.L.; Johnston, C.M.; Zelenay, P. High-Performance Electrocatalysts for Oxygen Reduction Derived from Polyaniline, Iron, and Cobalt. Science 2012, 332, 443–447. [Google Scholar] [CrossRef]
- Liang, H.W.; Wei, W.; Wu, Z.S.; Feng, X.; Müllen, K. Mesoporous metal-nitrogen-doped carbon electrocatalysts for highly efficient oxygen reduction reaction. J. Am. Chem. Soc. 2013, 135, 16002–16005. [Google Scholar] [CrossRef]
- Gavrilov, N.; Pašti, I.A.; Mitrić, M.; Travas-Sejdić, J.; Ćirić-Marjanović, G.; Mentus, S.V. Electrocatalysis of oxygen reduction reaction on polyaniline-derived nitrogen-doped carbon nanoparticle surfaces in alkaline media. J. Power Sources 2012, 220, 306–316. [Google Scholar] [CrossRef]
- Quilez-Bermejo, J.; González-Gaitán, C.; Morallón, E.; Cazorla-Amorós, D. Effect of carbonization conditions of polyaniline on its catalytic activity towards ORR. Some insights about the nature of the active sites. Carbon 2017, 119, 62–71. [Google Scholar] [CrossRef]
- Quílez-Bermejo, J.; Morallón, E.; Cazorla-Amorós, D. Oxygen-reduction catalysis of N-doped carbons prepared: Via heat treatment of polyaniline at over 1100 °C. Chem. Commun. 2018, 54, 4441–4444. [Google Scholar] [CrossRef]
- Silva, R.; Voiry, D.; Chhowalla, M.; Asefa, T. Efficient metal-free electrocatalysts for oxygen reduction: Polyaniline-derived N- and O-doped mesoporous carbons. J. Am. Chem. Soc. 2013, 135, 7823–7826. [Google Scholar] [CrossRef]
- Zhang, J.; Zhao, Z.; Xia, Z.; Dai, L. A metal-free bifunctional electrocatalyst for oxygen reduction and oxygen evolution reactions. Nat. Nanotechnol. 2015, 10, 444. [Google Scholar] [CrossRef]
- Zhou, F.; Wang, G.; Huang, F.; Zhang, Y.; Pan, M. Polyaniline derived N- and O-enriched high surface area hierarchical porous carbons as an efficient metal-free electrocatalyst for oxygen reduction. Electrochim. Acta 2017, 257, 73–81. [Google Scholar] [CrossRef]
- Zhao, A.; Masa, J.; Muhler, M.; Schuhmann, W.; Xia, W. N-doped carbon synthesized from N-containing polymers as metal-free catalysts for the oxygen reduction under alkaline conditions. Electrochim. Acta 2013, 98, 139–145. [Google Scholar] [CrossRef]
- Lai, L.; Potts, J.R.; Zhan, D.; Wang, L.; Poh, C.K.; Tang, C.; Gong, H.; Shen, Z.; Lin, J.; Ruoff, R.S. Exploration of the active center structure of nitrogen-doped graphene-based catalysts for oxygen reduction reaction. Energy Environ. Sci. 2012, 5, 7936–7942. [Google Scholar] [CrossRef]
- Salinas-Torres, D.; Shiraishi, S.; Morallón, E.; Cazorla-Amorós, D. Improvement of carbon materials performance by nitrogen functional groups in electrochemical capacitors in organic electrolyte at severe conditions. Carbon 2015, 82, 205–213. [Google Scholar] [CrossRef]
- Mostazo-López, M.J.; Ruiz-Rosas, R.; Morallón, E.; Cazorla-Amorós, D. Generation of nitrogen functionalities on activated carbons by amidation reactions and Hofmann rearrangement: Chemical and electrochemical characterization. Carbon 2015, 91, 252–265. [Google Scholar] [CrossRef]
- Mostazo-López, M.J.; Ruiz-Rosas, R.; Morallón, E.; Cazorla-Amorós, D. Nitrogen doped superporous carbon prepared by a mild method. Enhancement of supercapacitor performance. Int. J. Hydrogen Energy 2016, 41, 19691–19701. [Google Scholar] [CrossRef] [Green Version]
- Mostazo-López, M.J.; Ruiz-Rosas, R.; Shiraishi, S.; Morallón, E.; Cazorla-Amorós, D. Nitrogen Doped Activated Carbons Prepared at Mild Conditions as Electrodes for Supercapacitors in Organic Electrolyte. In Proceedings of the 7th International Conference on Carbon for Energy Storage and Environment Protection, Lyon, France, 23–26 October 2017. [Google Scholar]
- Lozano-Castelló, D.; Lillo-Ródenas, M.A.; Cazorla-Amorós, D.; Linares-Solano, A. Preparation of activated carbons from Spanish anthracite: I. Activation by KOH. Carbon 2001, 39, 741–749. [Google Scholar] [CrossRef]
- Bleda-Martínez, M.J.; Morallón, E.; Cazorla-Amorós, D. Polyaniline/porous carbon electrodes by chemical polymerisation: Effect of carbon surface chemistry. Electrochim. Acta 2007, 52, 4962–4968. [Google Scholar] [CrossRef] [Green Version]
- Cazorla-Amorós, D.; Alcañiz-Monge, J.; De La Casa-Lillo, M.A.; Linares-Solano, A. CO2 as an adsorptive to characterize carbon molecular sieves and activated carbons. Langmuir 1998, 14, 4589–4596. [Google Scholar] [CrossRef]
- Jagiello, J.; Olivier, J.P. 2D-NLDFT adsorption models for carbon slit-shaped pores with surface energetical heterogeneity and geometrical corrugation. Carbon 2013, 55, 70–80. [Google Scholar] [CrossRef]
- Bard, A.J.; Faulkner, L.R. Electrochemical Methods. Fundamentals and Applications, 2nd ed.; John Wiley & Sons: New York, NY, USA, 2000; ISBN 978-0-471-04372-0. [Google Scholar]
- Salinas-Torres, D.; Sieben, J.M.; Lozano-Castello, D.; Morallón, E.; Burghammer, M.; Riekel, C.; Cazorla-Amorós, D. Characterization of activated carbon fiber/polyaniline materials by position-resolved microbeam small-angle X-ray scattering. Carbon 2012, 50, 1051–1056. [Google Scholar] [CrossRef]
- Figueiredo, J.L.; Pereira, M.F.R.; Freitas, M.M.A.; Órfão, J.J.M. Modification of the surface chemistry of activated carbons. Carbon 1999, 37, 1379–1389. [Google Scholar] [CrossRef]
- Raymundo-Piñero, E.; Cazorla-Amorós, D.; Linares-Solano, A.; Find, J.; Wild, U.; Schlögl, R. Structural characterization of N-containing activated carbon fibers prepared from a low softening point petroleum pitch and a melamine resin. Carbon 2002, 40, 597–608. [Google Scholar] [CrossRef]
- Jansen, R.J.J.; van Bekkum, H. XPS of nitrogen-containing functional groups on activated carbon. Carbon 1995, 33, 1021–1027. [Google Scholar] [CrossRef]
- Yamada, Y.; Kim, J.; Matsuo, S.; Sato, S. Nitrogen-containing graphene analyzed by X-ray photoelectron spectroscopy. Carbon 2014, 70, 59–74. [Google Scholar] [CrossRef]
- Kuroki, S.; Hosaka, Y.; Yamauchi, C. A solid-state NMR study of the carbonization of polyaniline. Carbon 2013, 55, 160–167. [Google Scholar] [CrossRef]
- Rozlívková, Z.; Trchová, M.; Exnerová, M.; Stejskal, J. The carbonization of granular polyaniline to produce nitrogen-containing carbon. Synth. Met. 2011, 161, 1122–1129. [Google Scholar] [CrossRef]
- Latham, K.G.; Dose, W.M.; Allen, J.A.; Donne, S.W. Nitrogen doped heat treated and activated hydrothermal carbon: NEXAFS examination of the carbon surface at different temperatures. Carbon 2018, 128, 179–190. [Google Scholar] [CrossRef]
- Gara, M.; Compton, R.G. Activity of carbon electrodes towards oxygen reduction in acid: A comparative study. New J. Chem. 2011, 35, 2647–2652. [Google Scholar] [CrossRef]
- Waki, K.; Wong, R.A.; Oktaviano, H.S.; Fujio, T.; Nagai, T.; Kimoto, K.; Yamada, K. Non-nitrogen doped and non-metal oxygen reduction electrocatalysts based on carbon nanotubes: Mechanism and origin of ORR activity. Energy Environ. Sci. 2014, 7, 1950–1958. [Google Scholar] [CrossRef]
- Subramanian, N.P.; Li, X.; Nallathambi, V.; Kumaraguru, S.P.; Colon-Mercado, H.; Wu, G.; Lee, J.W.; Popov, B.N. Nitrogen-modified carbon-based catalysts for oxygen reduction reaction in polymer electrolyte membrane fuel cells. J. Power Sources 2009, 188, 38–44. [Google Scholar] [CrossRef]
- Matter, P.H.; Ozkan, U.S. Non-metal catalysts for dioxygen reduction in an acidic electrolyte. Catal. Lett. 2006, 109, 115–123. [Google Scholar] [CrossRef]
- Matter, P.H.; Zhang, L.; Ozkan, U.S. The role of nanostructure in nitrogen-containing carbon catalysts for the oxygen reduction reaction. J. Catal. 2006, 239, 83–96. [Google Scholar] [CrossRef]
- Chu, X.; Kinoshita, K. Surface modification of carbons for enhanced electrochemical activity. Mater. Sci. Eng. B 1997, 49, 53–60. [Google Scholar] [CrossRef]
- Rao, C.V.; Cabrera, C.R.; Ishikawa, Y. In search of the active site in nitrogen-doped carbon nanotube electrodes for the oxygen reduction reaction. J. Phys. Chem. Lett. 2010, 1, 2622–2627. [Google Scholar] [CrossRef]
- González-Gaitán, C.; Ruiz-Rosas, R.; Morallón, E.; Cazorla-Amorós, D. Functionalization of carbon nanotubes using aminobenzene acids and electrochemical methods. Electroactivity for the oxygen reduction reaction. Int. J. Hydrogen Energy 2015, 40, 11242–11253. [Google Scholar] [CrossRef]
- Tuci, G.; Zafferoni, C.; Rossin, A.; Luconi, L.; Milella, A.; Ceppatelli, M.; Innocenti, M.; Liu, Y.; Pham-Huu, C.; Giambastiani, G. Chemical functionalization of N-doped carbon nanotubes: A powerful approach to cast light on the electrochemical role of specific N-functionalities in the oxygen reduction reaction. Catal. Sci. Technol. 2016, 6, 6226–6236. [Google Scholar] [CrossRef]
- Ikeda, T.; Boero, M.; Huang, S.F.; Terakura, K.; Oshima, M.; Ozaki, J.I.; Hang, S.F.; Terakura, K.; Oshima, M.; Ozaki, J.I. Carbon Alloy Catalysts: Active Sites for Oxygen Reduction Reaction. J. Phys. Chem. C 2008, 112, 14706–14709. [Google Scholar] [CrossRef]
- Chung, J.H.; Kwon, H.C.; Woo, S.I.; Choi, C.H.; Chung, M.W. Nitrogen-doped graphene/carbon nanotube self-assembly for efficient oxygen reduction reaction in acid media. Appl. Catal. B Environ. 2013, 144, 760–766. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | CO2 (µmol/g) | CO (µmol/g) | O (µmol/g) | OXPS (at.%) | NXPS (at.%) |
|---|---|---|---|---|---|
| KUA | 450 | 1970 | 2870 | 8.8 | 0.3 |
| KUA_800 | 170 | 620 | 960 | 2.3 | - |
| KUA/PANI | 1320 | 3560 | 6200 | 11.4 | 6.0 |
| KUA/PANI_600 | 380 | 1860 | 2620 | 4.5 | 4.7 |
| KUA/PANI_800 | 210 | 740 | 1050 | 6.7 | 2.0 |
| KUA-CONH2 | 1140 | 2370 | 4650 | 10.2 | 4.2 |
| KUA-CONH2_800 | 140 | 570 | 830 | 4.5 | 2.1 |
| KUA-N | 450 | 1750 | 2640 | 7.5 | 3.7 |
| KUA-N_800 | 130 | 505 | 720 | 3.0 | 1.7 |
| Sample | Binding Energy (eV) | Functional Group | N (at.%) | Percentage of N Species |
|---|---|---|---|---|
| KUA/PANI | 400.8 ± 0.2 | Positive N | 2.2 | 36 |
| 399.7 ± 0.2 | Amines | 3.8 | 64 | |
| KUA/PANI_600 | 400.6 ± 0.2 | Pyrrole, pyridone | 2.9 | 61 |
| 398.6 ± 0.2 | Pyridine, Imine | 1.8 | 39 | |
| KUA/PANI_800 | 400.8 ± 0.2 | Pyrrole, pyridone | 1.4 | 70 |
| 398.3 ± 0.2 | Pyridine, imine | 0.5 | 30 | |
| KUA-N | 401.9 ± 0.2 | Quaternary | 0.4 | 10 |
| 400.7 ± 0.2 | Pyrrole,pyridone | 0.9 | 25 | |
| 399.8 ± 0.2 | Amide, Lactam, Amine, Imide | 1.3 | 35 | |
| 398.7 ± 0.2 | Pyridine, Imine | 1.1 | 30 | |
| KUA-N_800 | 402.7 ± 0.2 | Oxidized N | 0.2 | 14 |
| 400.8 ± 0.2 | Pyrrole, pyridone | 0.9 | 51 | |
| 398.7 ± 0.2 | Pyridine, imine | 0.6 | 35 | |
| KUA-CONH2 | 400.7 ± 0.2 | Pyrrole, pyridone | 0.7 | 19 |
| 399.8 ± 0.2 | Amide, lactam, amine, imide | 1.9 | 50 | |
| 398.8 ± 0.2 | Pyridine, imine | 1.2 | 31 | |
| KUA-CONH2_800 | 402.5 ± 0.2 | Oxidized N | 0.2 | 11 |
| 400.8 ± 0.2 | Pyrrole, pyridone | 0.9 | 52 | |
| 398.7 ± 0.2 | Pyridine, imine | 0.6 | 37 |
| Sample | SBET (m2/g) | VDRN2 (cm3/g) | VDRCO2 (cm3/g) |
|---|---|---|---|
| KUA | 3080 | 1.19 | 0.57 |
| KUA_800 | 2680 | 1.05 | 0.49 |
| KUA/PANI | 1590 | 0.54 | 0.37 |
| KUA/PANI_600 | 2420 | 0.81 | 0.49 |
| KUA/PANI_800 | 2470 | 0.84 | 0.56 |
| KUA-COOH | 2770 | 1.06 | 0.49 |
| KUA-CONH2 | 2390 | 0.97 | 0.45 |
| KUA-CONH2_500 | 2630 | 1.02 | 0.41 |
| KUA-CONH2_800 | 2630 | 1.0 | 0.43 |
| KUA-N | 2960 | 1.18 | 0.52 |
| KUA-N_500 | 2800 | 1.11 | 0.49 |
| KUA-N_800 | 2770 | 1.09 | 0.48 |
| Sample | Eonset (V versus RHE) | n (at 0.5 V) |
|---|---|---|
| KUA | 0.82 | 2.5 |
| KUA_800 | 0.82 | 2.7 |
| KUA/PANI | 0.80 | 2.4 |
| KUA/PANI_600 | 0.82 | 2.7 |
| KUA/PANI_800 | 0.88 | 3.4 |
| KUA-CONH2 | 0.79 | 2.6 |
| KUA-CONH2_800 | 0.85 | 3.4 |
| KUA-N | 0.81 | 2.8 |
| KUA-N_800 | 0.84 | 3.1 |
| Pt/Vulcan | 0.98 | 3.9 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mostazo-López, M.J.; Salinas-Torres, D.; Ruiz-Rosas, R.; Morallón, E.; Cazorla-Amorós, D. Nitrogen-Doped Superporous Activated Carbons as Electrocatalysts for the Oxygen Reduction Reaction. Materials 2019, 12, 1346. https://doi.org/10.3390/ma12081346
Mostazo-López MJ, Salinas-Torres D, Ruiz-Rosas R, Morallón E, Cazorla-Amorós D. Nitrogen-Doped Superporous Activated Carbons as Electrocatalysts for the Oxygen Reduction Reaction. Materials. 2019; 12(8):1346. https://doi.org/10.3390/ma12081346
Chicago/Turabian StyleMostazo-López, María José, David Salinas-Torres, Ramiro Ruiz-Rosas, Emilia Morallón, and Diego Cazorla-Amorós. 2019. "Nitrogen-Doped Superporous Activated Carbons as Electrocatalysts for the Oxygen Reduction Reaction" Materials 12, no. 8: 1346. https://doi.org/10.3390/ma12081346
APA StyleMostazo-López, M. J., Salinas-Torres, D., Ruiz-Rosas, R., Morallón, E., & Cazorla-Amorós, D. (2019). Nitrogen-Doped Superporous Activated Carbons as Electrocatalysts for the Oxygen Reduction Reaction. Materials, 12(8), 1346. https://doi.org/10.3390/ma12081346