In Vitro Corrosion Behavior of Biodegradable Iron Foams with Polymeric Coating


 ,
,  , ,
, ,
Abstract
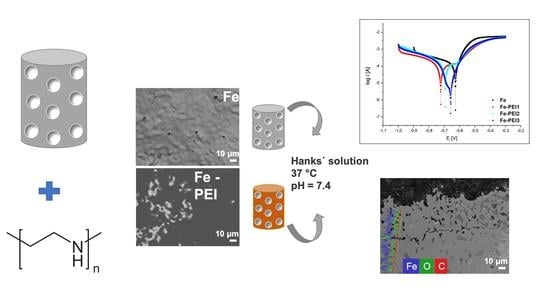
:
1. Introduction
2. Materials and Methods
2.1. Material Preparation
2.2. PEI Coating Preparation
2.3. Microstructure and Surface Characterization
2.4. Electrochemical Corrosion Testing
2.5. Immersion Test
3. Results and Discussion
3.1. Material Characterization
3.1.1. Morphology of the Sintered Iron Foam
3.1.2. Characterization of the Polymer Coating
3.2. Degradation Study
3.2.1. Potentiodynamic Polarization Tests
3.2.2. Static Degradation Tests
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wu, G.; Ibrahim, J.M. Surface design of biodegradable magnesium alloys—A review. Surf. Coat. Technol. 2013, 233, 2–12. [Google Scholar] [CrossRef]
- Yun, Y.; Dong, Z.; Lee, N.; Liu, Y.; Xue, D.; Guo, X.; Kuhlmann, J.; Doepke, A.; Halsall, H.B.; Heineman, W.; et al. Revolutionizing biodegradable metals. Mater. Today 2009, 12, 22–32. [Google Scholar] [CrossRef]
- Han, H.S.; Loffredo, S.; Jun, I.; Edwards, J.; Kim, Y.C.; Seok, H.K.; Witte, F.; Mantovani, D.; Glyn-Jones, S. Current status and outlook on the clinical translation of biodegradable metals. Mater. Today 2019, 23, 57–71. [Google Scholar] [CrossRef]
- Li, H.; Zheng, Y.; Qin, L. Progress of biodegradable metals. Prog. Nat. Sci. Mater. Int. 2014, 24, 414–422. [Google Scholar] [CrossRef] [Green Version]
- Tan, L.; Yu, X.; Wan, P.; Yang, K. Biodegradable materials for bone repairs: a review. J. Mater. Sci. Technol. 2013, 6, 503–513. [Google Scholar] [CrossRef]
- Yusop, A.H.; Bakir, A.A.; Shaharom, N.A.; Abdul Kadir, M.R.; Hermawan, H. Porous Biodegradable Metals for Hard Tissue Scaffolds: A Review. Int. J. Biomater. 2012, 2012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seitz, J.M.; Durisin, M.; Goldman, J.; Drelich, J.W. Recent Advances in Biodegradable Metals for Medical Sutures: A Critical Review. Adv. Healthc. Mater. 2015, 4, 1915–1936. [Google Scholar] [CrossRef]
- Mahyudin, F.; Widhiyanto, L.; Hermawan, H. Biomaterials in orthopaedics. In Biomaterials and Medical Devices; Springer: Cham, Switzerland, 2016; pp. 161–181. [Google Scholar]
- Kraus, T.; Moszner, F.; Fischerauer, S.; Fiedler, M.; Martinelli, E.; Eichler, J.; Witte, F.; Willbold, E.; Schinhammer, M.; Meischel, M.; et al. Biodegradable Fe-based alloys for use in osteosynthesis: Outcome of an in vivo study after 52 weeks. Acta Biomater. 2014, 10, 3346–3353. [Google Scholar] [CrossRef]
- Schinhammer, M.; Steiger, P.; Moszner, F.; Löffler, J.F.; Uggowitzer, P.J. Degradation performance of biodegradable FeMnC(Pd) alloys. Mater. Sci. Eng. C 2013, 33, 1882–1893. [Google Scholar] [CrossRef]
- Huang, T.; Cheng, J.; Zheng, Y.F. In vitro degradation and biocompatibility of Fe–Pd and Fe–Pt composites fabricated by spark plasma sintering. Mater. Sci. Eng. C 2014, 35, 43–53. [Google Scholar] [CrossRef]
- Schinhammer, M.; Hänzi, A.C.; Löffler, J.F.; Uggowitzer, P.J. Design strategy for biodegradable Fe-based alloys for medical applications. Acta Biomater. 2010, 6, 1705–1713. [Google Scholar] [CrossRef] [PubMed]
- Chou, D.T.; Wells, D.; Hong, D.; Lee, B.; Kuhn, H.; Kumta, P.N. Novel processing of iron-manganese alloy-based biomaterials by inkjet 3-D printing. Acta Biomater. 2013, 9, 8593–8603. [Google Scholar] [CrossRef] [PubMed]
- Oriňaková, R.; Oriňak, A.; Markušová, L.; Labbanczová, E.; Kupková, M.; Hrubovčáková, M.; Fedorková, A. Biodegradable Open Cell Iron Foams for Potential Skeletal Application. Powder Metall. Prog. 2012, 12, 219–223. [Google Scholar]
- Hermawan, H.; Alamdari, H.; Mantovani, D.; Dubé, D. Iron–manganese: New class of metallic degradable biomaterials prepared by powder metallurgy. Powder Metall. 2008, 51, 38–45. [Google Scholar] [CrossRef]
- Hong, D.; Chou, D.T.; Velikokhatnyi, O.I.; Roy, A.; Lee, B.; Swink, I.; Issaev, I.; Kuhn, H.A.; Kumta, P.N. Binder-jetting 3D printing and alloy development of new biodegradable Fe-Mn-Ca/Mg alloys. Acta Biomater. 2016, 45, 375–386. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.; Huang, T.; Zheng, Y.F. Microstructure, mechanical property, biodegradation behavior, and biocompatibility of biodegradable Fe-Fe2O3 composites. J. Biomed. Mater. Res. Part A 2014, 102, 2277–2287. [Google Scholar] [CrossRef]
- Bakhsheshi-Rad, H.R.; Ismail, A.F.; Aziz, M.; Hadisi, Z.; Omidi, M.; Chen, X. Antibacterial activity and corrosion resistance of Ta2O5 thin film and electrospun PCL/MgO-Ag nanofiber coatings on biodegradable Mg alloy implants. Ceram. Int. 2019, 45, 11883–11892. [Google Scholar] [CrossRef]
- Bakhsheshi-Rad, H.R.; Akbari, M.; Ismail, A.F.; Aziz, M.; Hadisi, Z.; Pagan, E.; Daroonparvar, M.; Chen, X. Coating biodegradable magnesium alloys with electrospun poly-L-lactic acid-åkermanite-doxycycline nanofibers for enhanced biocompatibility, antibacterial activity, and corrosion resistance. Surf. Coat. Technol. 2019, 377, 124898. [Google Scholar] [CrossRef]
- Hufenbach, J.; Wendrock, H.; Kochta, F.; Kühn, U.; Gebert, A. Novel biodegradable Fe-Mn-C-S alloy with superior mechanical and corrosion properties. Mater. Lett. 2017, 186, 330–333. [Google Scholar] [CrossRef]
- Cheng, J.; Zheng, Y.F. In vitro study on newly designed biodegradable Fe-X composites (X = W, CNT) prepared by spark plasma sintering. J. Biomed. Mater. Res. Part B Appl. Biomater. 2013, 4, 485–497. [Google Scholar] [CrossRef]
- Zhang, Q.; Cao, P. Degradable porous Fe-35wt.%Mn produced via powder sintering from NH4HCO3 porogen. Mater. Chem. Phys. 2015, 163, 394–401. [Google Scholar] [CrossRef]
- Gorejová, R.; Haverová, L.; Oriňaková, R.; Oriňak, A.; Oriňak, M. Recent advancements in Fe-based biodegradable materials for bone repair. J. Mater. Sci. 2019, 54, 1913–1947. [Google Scholar] [CrossRef]
- Čapek, J.; Vojtěch, D.; Oborná, A. Microstructural and mechanical properties of biodegradable iron foam prepared by powder metallurgy. Mater. Des. 2015, 83, 468–482. [Google Scholar] [CrossRef]
- Li, Y.; Jahr, H.; Lietaert, K.; Pavanram, P.; Yilmaz, A.; Fockaert, L.; Leeflang, M.A.; Pouran, B.; Gonzalez-Garcia, Y.; Weinans, H.; et al. Additively manufactured biodegradable porous iron. Acta Biomater. 2018, 77, 380–393. [Google Scholar] [CrossRef]
- Posada, V.M.; Orozco, C.; Ramirez Patino, J.F.; Fernandez-Morales, P. Human Bone Inspired Design of an Mg Alloy-Based Foam. Mater. Sci. Forum 2018, 933, 291–296. [Google Scholar] [CrossRef]
- Daud, N.M.; Sing, N.B.; Yusop, A.H.; Majid, F.A.A.; Hermawan, H. Degradation and in vitro cell-material interaction studies on hydroxyapatite-coated biodegradable porous iron for hard tissue scaffolds. J. Orthop. Transl. 2014, 2, 177–184. [Google Scholar]
- Oriňaková, R.; Oriňak, A.; Kupková, M.; Hrubovčáková, M.; Markušová-Bučková, L.; Giretová, M.; Medvecký, L.; Dobročka, E.; Petruš, O.; Kaľavský, F. In vitro degradation and cytotoxicity evaluation of iron biomaterials with hydroxyapatite film. Int. J. Electrochem. Sci. 2015, 10, 8158–8174. [Google Scholar]
- Wang, S.; Xu, Y.; Zhou, J.; Li, H.; Chang, J.; Huan, Z. In vitro degradation and surface bioactivity of iron-matrix composites containing silicate-based bioceramic. Bioact. Mater. 2017, 2, 10–18. [Google Scholar] [CrossRef]
- Haverová, L.; Oriňaková, R.; Oriňak, A.; Gorejova, R.; Baláž, M.; Vanýsek, P.; Kupkova, M.; Hrubovčáková, M.; Mudroň, P.; Radoňák, J.; et al. An In Vitro Corrosion Study of Open Cell Iron Structures with PEG Coating for Bone Replacement Applications. Metals 2018, 8, 499. [Google Scholar] [CrossRef] [Green Version]
- Yusop, A.H.M.; Daud, N.M.; Nur, H.; Kadir, M.R.A.; Hermawan, H. Controlling the degradation kinetics of porous iron by poly(lactic-co-glycolic acid) infiltration for use as temporary medical implants. Sci. Rep. 2015, 5, 11194. [Google Scholar] [CrossRef]
- Oriňaková, R.; Gorejová, R.; Macko, J.; Oriňak, A.; Kupková, M.; Hrubovčáková, M.; Ševc, J.; Smith, R.M. Evaluation of in vitro biocompatibility of open cell iron structures with PEG coating. Appl. Surf. Sci. 2019, 475, 515–518. [Google Scholar] [CrossRef]
- Vancha, A.R.; Govindaraju, S.; Parsa, K.V.L.; Jasti, M.; González-García, M.; Ballestero, R.P. Use of polyethyleneimine polymer in cell culture as attachment factor and lipofection enhancer. BMC Biotechnol. 2004, 4, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, F.; Hu, S.; Lu, Y.; Yang, H.; Zhao, Y.; Li, L. Effects of coatings of polyethyleneimine and thyme essential oil combined with chitosan on sliced fresh channa argus during refrigerated storage. J. Food Process Eng. 2015, 38, 225–233. [Google Scholar] [CrossRef]
- Xia, T.; Kovochich, M.; Liong, M.; Meng, H.; Kabehie, S.; George, S.; Zink, J.I.; Nel, A.E. Polyethyleneimine coating enhances the cellular uptake of mesoporous silica nanoparticles and allows safe delivery of siRNA and DNA constructs. ACS Nano 2009, 3, 3273–3286. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.A.; Park, T.E.; Singh, B.; Maharjan, S.; Firdous, J.; Cho, M.H.; Kang, S.K.; Yun, C.H.; Choi, Y.J.; Cho, C.S. Major degradable polycations as carriers for DNA and siRNA. J. Control. Release 2014, 193, 74–89. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zheng, L.; Cai, H.; Sun, W.; Shen, M.; Zhang, G.; Shi, X. Polyethyleneimine-mediated synthesis of folic acid-targeted iron oxide nanoparticles for invivo tumor MR imaging. Biomaterials 2013, 34, 8382–8392. [Google Scholar] [CrossRef]
- Yao, X.; Zhou, N.; Wan, L.; Su, X.; Sun, Z.; Mizuguchi, H.; Yoshioka, Y.; Nakagawa, S.; Zhao, R.C.; Gao, J.Q. Polyethyleneimine-coating enhances adenoviral transduction of mesenchymal stem cells. Biochem. Biophys. Res. Commun. 2014, 447, 383–387. [Google Scholar] [CrossRef]
- Dong, P.; Hao, W.; Wang, X.; Wang, T. Fabrication and biocompatibility of polyethyleneimine/heparin self-assembly coating on NiTi alloy. Thin Solid Films 2008, 516, 5168–5171. [Google Scholar] [CrossRef]
- Bergstrand, A.; Rahmani-Monfared, G.; Östlund, Å.; Nydén, M.; Holmberg, K. Comparison of PEI-PEG and PLL-PEG copolymer coatings on the prevention of protein fouling. J. Biomed. Mater. Res. Part A 2009, 88, 608–615. [Google Scholar] [CrossRef]
- Oriňaková, R.; Oriňak, A.; Bučková, L.M.; Giretová, M.; Medvecký, L.; Labbanczová, E.; Kupková, M.; Hrubovčáková, M.; Kovaľ, K. Iron based degradable foam structures for potential orthopedic applications. Int. J. Electrochem. Sci. 2013, 8, 12451–12465. [Google Scholar]
- Yin, C.Y.; Aroua, M.K.; Daud, W.M.A.W. Metal-polyethyleneimine-activated carbon interaction parameter at equilibrium adsorption capacity. J. Appl. Sci. 2010, 10, 1192–1195. [Google Scholar] [CrossRef] [Green Version]
- Dhiman, M.; Chalke, B.; Polshettiwar, V. Efficient Synthesis of Monodisperse Metal (Rh, Ru, Pd) Nanoparticles Supported on Fibrous Nanosilica (KCC-1) for Catalysis. ACS Sustain. Chem. Eng. 2015, 3, 3224–3230. [Google Scholar] [CrossRef]
- Li, J.; Tang, W.; Yang, H.; Dong, Z.; Huang, J.; Li, S.; Wang, J.; Jin, J.; Ma, J. Enhanced-electrocatalytic activity of Ni 1−x Fe x alloy supported on polyethyleneimine functionalized MoS 2 nanosheets for hydrazine oxidation. RSC Adv. 2014, 4, 1988–1995. [Google Scholar] [CrossRef]
- Singh, S.; Thomas, V.; Martyshkin, D.; Kozlovskaya, V.; Kharlampieva, E.; Catledge, S.A. Spatially controlled fabrication of a bright fluorescent nanodiamond-array with enhanced far-red Si-V luminescence. Nanotechnology 2014, 25, 045302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Liu, J.; Yao, W.; Cen, W.; Wang, H.; Weng, X.; Wu, Z. The effects of surface acidity on CO2 adsorption over amine functionalized protonated titanate nanotubes. RSC Adv. 2013, 3, 18803–18810. [Google Scholar] [CrossRef]
- Sanchez-Cortes, S.; Berenguel, R.M.; Madejón, A.; Pérez-Méndez, M. Adsorption of polyethyleneimine on silver nanoparticles and its interaction with a plasmid DNA: A surface-enhanced Raman scattering study. Biomacromolecules 2002, 3, 655–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rezaei, F.; Jones, C.W. Stability of Supported Amine Adsorbents to SO2 and NOx in Postcombustion CO2 Capture. 1. Single-Component Adsorption. Ind. Eng. Chem. Res. 2013, 52, 12192–12201. [Google Scholar] [CrossRef]
- Chaufer, B.; Rabiller-Baudry, M.; Bouguen, A.; Labbé, J.P.; Quémerais, A. Spectroscopic characterization of zirconia coated by polymers with amine groups. Langmuir 2000, 16, 1852–1860. [Google Scholar] [CrossRef]
- Kupková, M.; Hrubovčáková, M.; Kupka, M.; Oriňáková, R.; Turoňová, A.M. Corrosion behaviour of powder metallurgy biomaterials from phosphated carbonyl-iron powders. Int. J. Electrochem. Sci. 2015, 10, 671–681. [Google Scholar]
- Zhang, E.; Chen, H.; Shen, F. Biocorrosion properties and blood and cell compatibility of pure iron as a biodegradable biomaterial. J. Mater. Sci. Mater. Med. 2010, 21, 2151–2163. [Google Scholar] [CrossRef]
- Chen, H.; Zhang, E.; Yang, K. Microstructure, corrosion properties and bio-compatibility of calcium zinc phosphate coating on pure iron for biomedical application. Mater. Sci. Eng. C 2014, 34, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Kupková, M.; Hrubovčáková, M.; Kupka, M.; Oriňaková, R.; Turoňová, A.M. Sintering behaviour, graded microstructure and corrosion performance of sintered Fe-Mn biomaterials. Int. J. Electrochem. Sci. 2015, 10, 9256–9268. [Google Scholar]
- Wei, J.; Zhou, B.; Wan, T.; Liu, K.; Gong, S.; Wu, J.; Xu, S. Effect of Sulfate and Chloride ions on pitting corrosion behavior of 2Cr12MoV Steel at pH 6 and 90 °C. Int. J. Electrochem. Sci. 2018, 13, 11596–11606. [Google Scholar] [CrossRef]
- Kislenko, V.N.; Oliynyk, L.P. Complex formation of polyethyleneimine with copper(II), nickel(II), and cobalt(II) ions. J. Polym. Sci. Part A Polym. Chem. 2002, 40, 914–922. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fe-PEI1 | Fe-PEI2 | Fe-PEI3 | |
|---|---|---|---|
| Average PEI weight (mg) | 15.9 | 41.4 | 53.6 |
| Average PEI content (wt %) | 1.9 | 5.0 | 6.6 |
| SBET (g m −2) | |||
|---|---|---|---|
| Fe | Fe-PEI1 | Fe-PEI2 | Fe-PEI3 |
| 1.19 | 0.92 | 0.61 | 0.04 |
| Sample | Ecorr (V) | icorr (A) | jcorr (μA cm−2) | PR (Ω cm−2) | CR (mm y−1) |
|---|---|---|---|---|---|
| Fe | −0.627 | 11.91 × 10−5 | 1.18 × 10−2 | 0.017 | 0.045 |
| Fe-PEI1 | −0.722 | 37.11 × 10−5 | 4.52 × 10−2 | 0.041 | 0.172 |
| Fe-PEI2 | −0.687 | 102.95 × 10−5 | 3.10 × 10−2 | 0.039 | 0.118 |
| Fe-PEI3 | −0.658 | 5.39 × 10−5 | 15.49 × 10−2 | 4.460 | 0.590 |
| CR [mm y −1] | |||
|---|---|---|---|
| Week of Immersion | 4 | 8 | 12 |
| Fe | 0.004 ± 0.0015 | 0.005 ± 0.0030 | 0.005 ± 0.0034 |
| Fe-PEI1 | 0.024 ± 0.0052 | 0.006 ± 0.0008 | 0.015 ± 0.0047 |
| Fe-PEI2 | 0.148 ± 0.0420 | 0.037 ± 0.0113 | 0.021 ± 0.0209 |
| Fe-PEI3 | 0.697 ± 0.0398 | 1.547 ± 0.0793 | 0.199 ± 0.0109 |
| pH ± 0.2 | ||||
|---|---|---|---|---|
| Week of Immersion | 0 | 4 | 8 | 12 |
| Fe | 7.40 | 7.46 | 7.75 | 8.06 |
| Fe-PEI1 | 7.40 | 7.43 | 7.06 | 8.08 |
| Fe-PEI2 | 7.40 | 7.69 | 7.53 | 8.23 |
| Fe-PEI3 | 7.40 | 7.48 | 8.25 | 8.73 |
| Total Iron Concentration (mg mL −1) | |||
|---|---|---|---|
| Week of Immersion | 4 | 8 | 12 |
| Fe | 0.0243 | 0.0265 | 0.0316 |
| Fe-PEI1 | 0.0151 | 0.0278 | 0.0284 |
| Fe-PEI2 | 0.0184 | 0.0224 | 0.0251 |
| Fe-PEI3 | 0.0139 | 0.0283 | 0.0337 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gorejová, R.; Oriňaková, R.; Orságová Králová, Z.; Baláž, M.; Kupková, M.; Hrubovčáková, M.; Haverová, L.; Džupon, M.; Oriňak, A.; Kaľavský, F.; et al. In Vitro Corrosion Behavior of Biodegradable Iron Foams with Polymeric Coating. Materials 2020, 13, 184. https://doi.org/10.3390/ma13010184
Gorejová R, Oriňaková R, Orságová Králová Z, Baláž M, Kupková M, Hrubovčáková M, Haverová L, Džupon M, Oriňak A, Kaľavský F, et al. In Vitro Corrosion Behavior of Biodegradable Iron Foams with Polymeric Coating. Materials. 2020; 13(1):184. https://doi.org/10.3390/ma13010184
Chicago/Turabian StyleGorejová, Radka, Renáta Oriňaková, Zuzana Orságová Králová, Matej Baláž, Miriam Kupková, Monika Hrubovčáková, Lucia Haverová, Miroslav Džupon, Andrej Oriňak, František Kaľavský, and et al. 2020. "In Vitro Corrosion Behavior of Biodegradable Iron Foams with Polymeric Coating" Materials 13, no. 1: 184. https://doi.org/10.3390/ma13010184
APA StyleGorejová, R., Oriňaková, R., Orságová Králová, Z., Baláž, M., Kupková, M., Hrubovčáková, M., Haverová, L., Džupon, M., Oriňak, A., Kaľavský, F., & Kovaľ, K. (2020). In Vitro Corrosion Behavior of Biodegradable Iron Foams with Polymeric Coating. Materials, 13(1), 184. https://doi.org/10.3390/ma13010184