Teicoplanin-Modified HPLC Column as a Source of Experimental Parameters for Prediction of the Anticonvulsant Activity of 1,2,4-Triazole-3-Thiones by the Regression Models

 , , , , and
, , , , and
Abstract
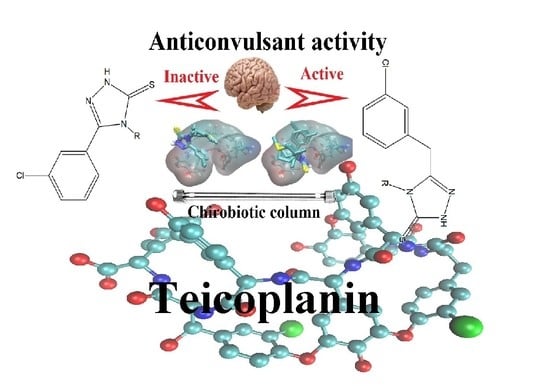
:
1. Introduction
2. Methods and Materials
2.1. Solutes and Reagents
2.2. HPLC Measurement
2.3. Differential Scanning Calorimetry (DSC) Experiments
2.4. The Methodology of Statistical Analysis
2.5. The Methodology of Molecular Modeling
2.6. Evaluation of Anticonvulsant Activity
3. Results and Discussion
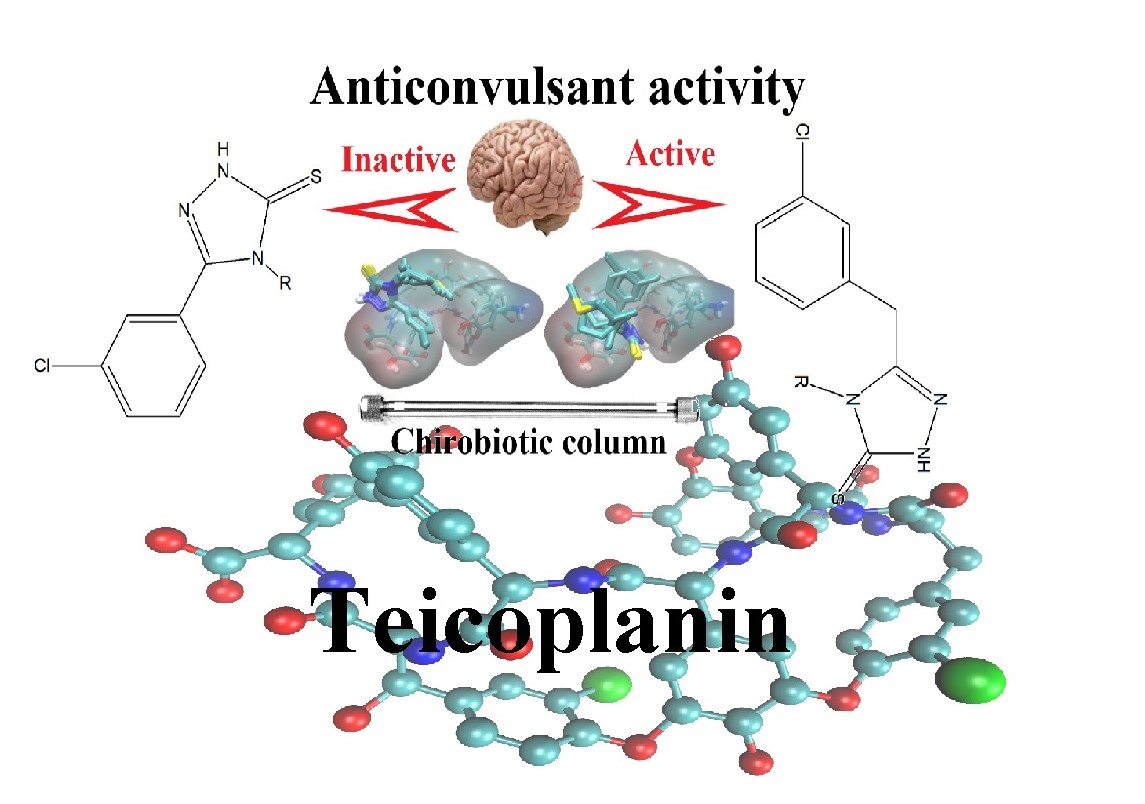
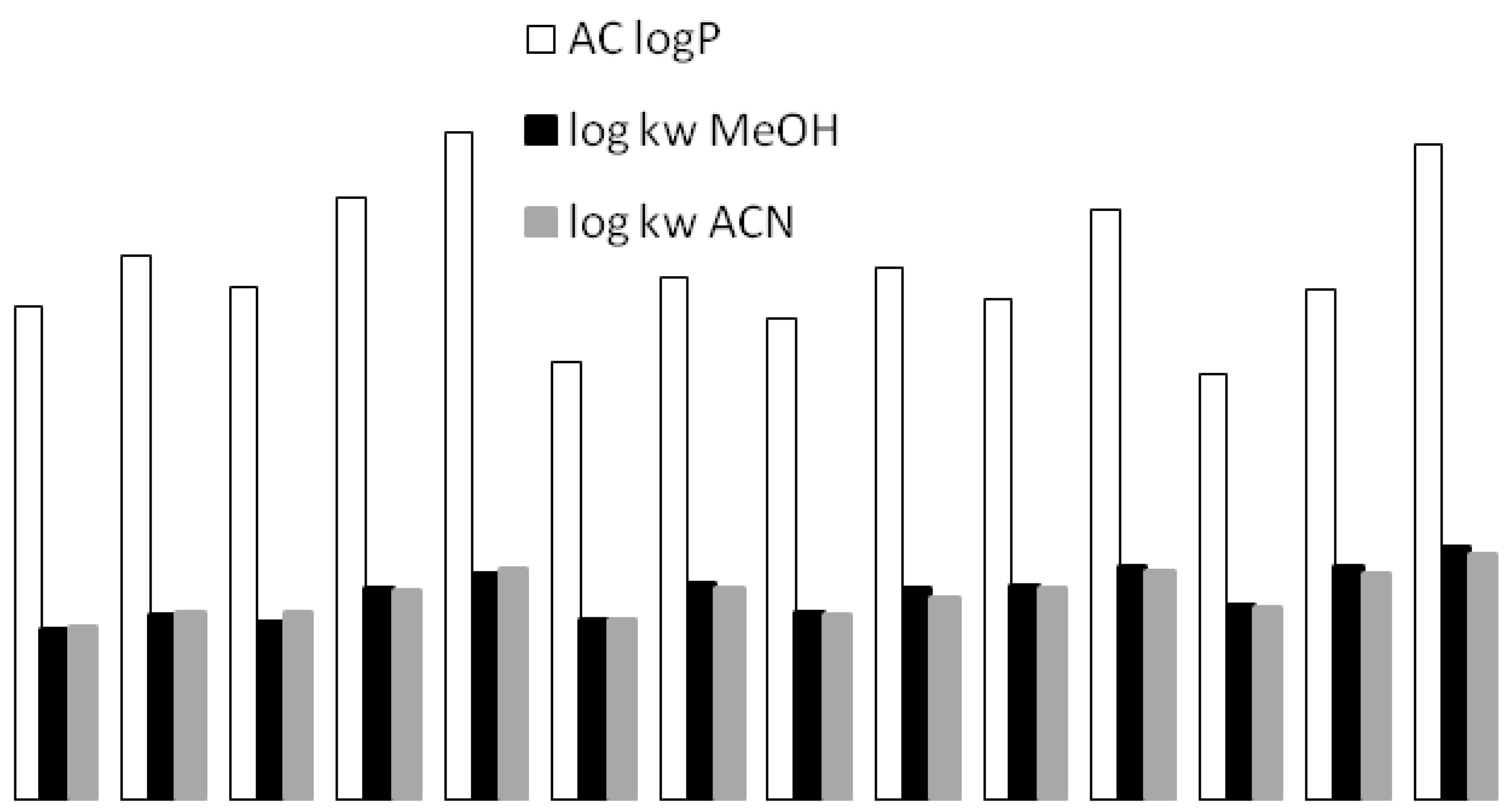
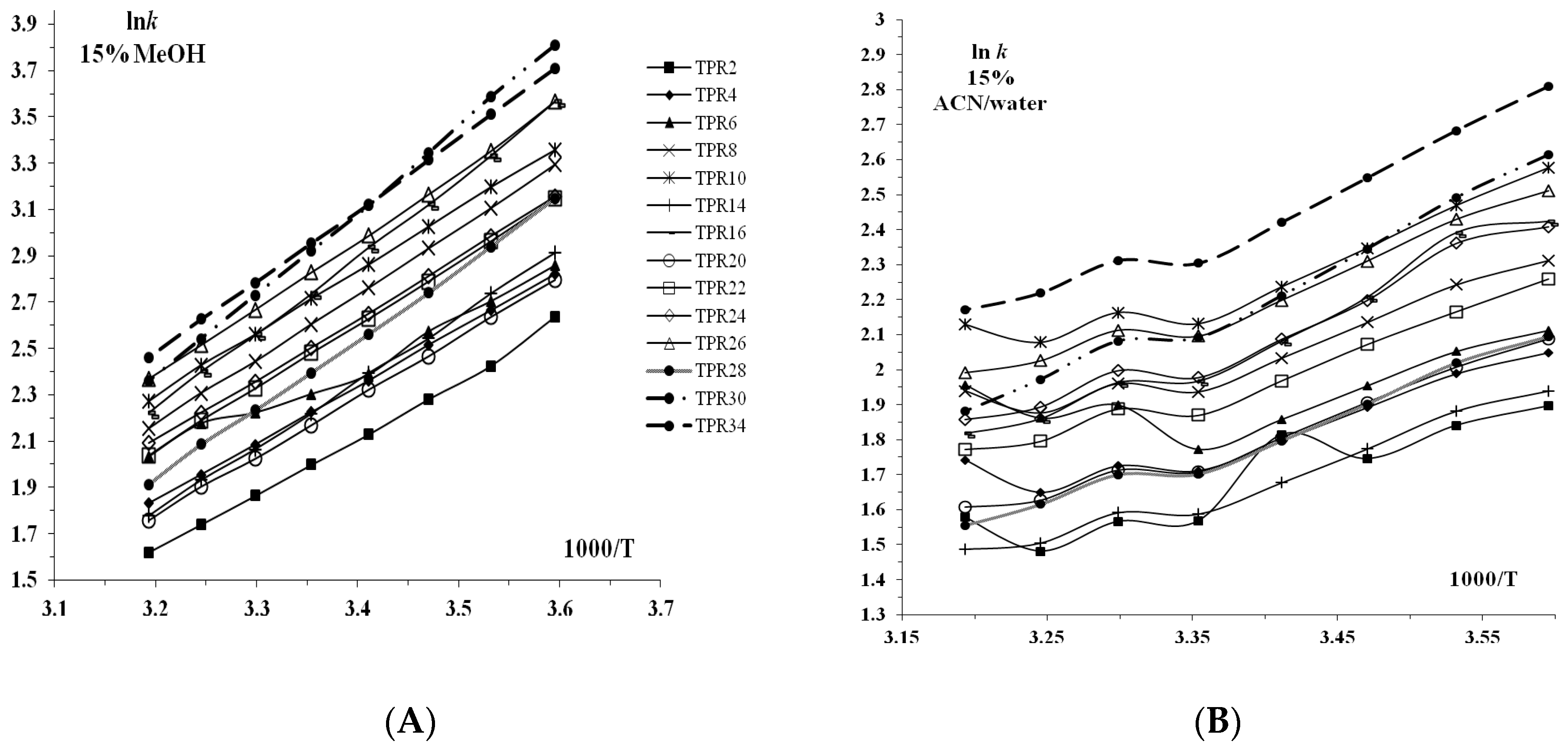
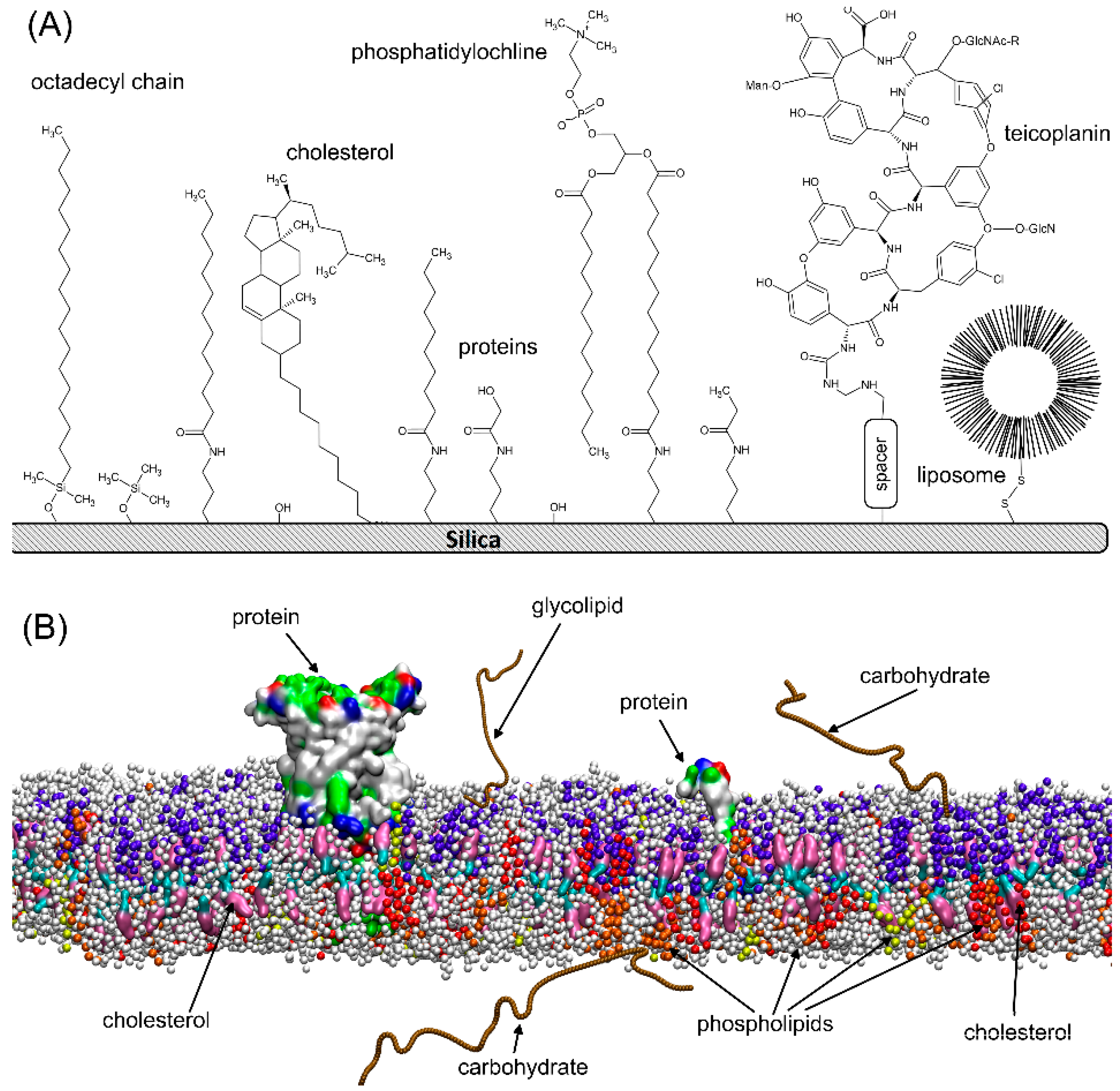
3.1. Retention Behavior on Teicoplanin Column
n = 14, R2 = 0.8976, F = 113.9818, se = 0.062873
n = 14, R2 = 0.9901, F = 1294.178, se = 0.019135
3.2. Thermodynamic Studies
3.3. The Phase Transition of Teicoplanin
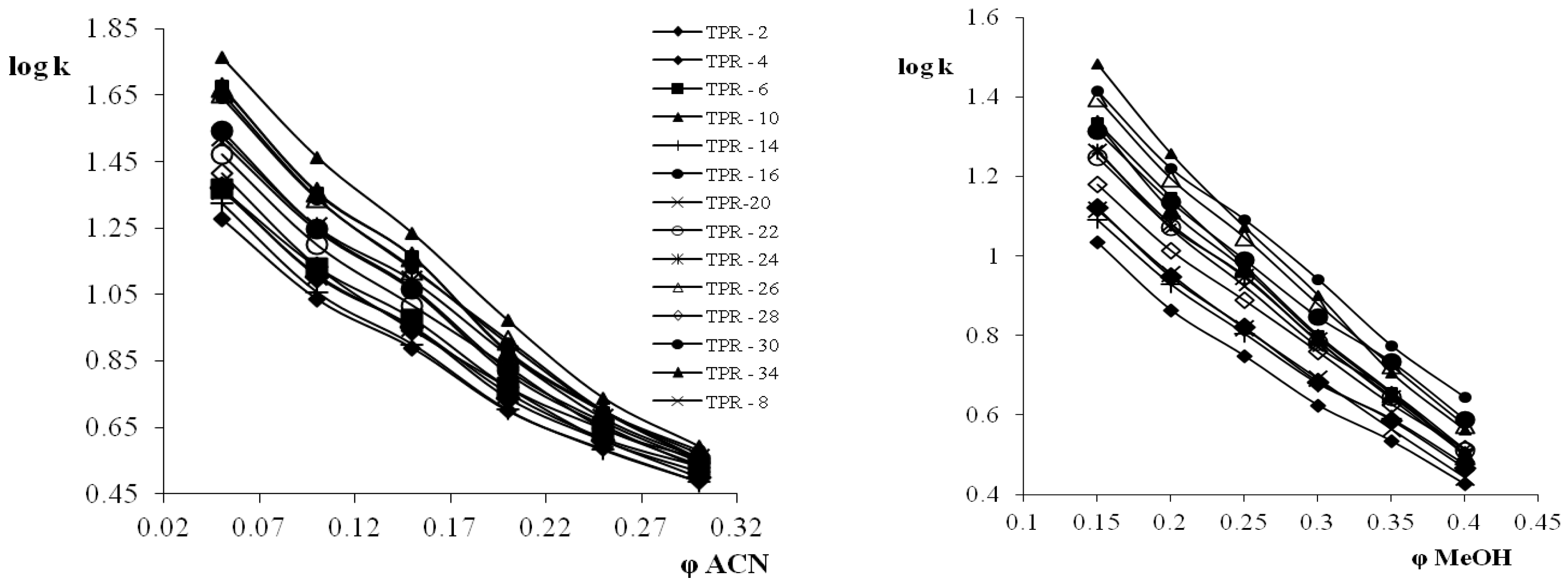
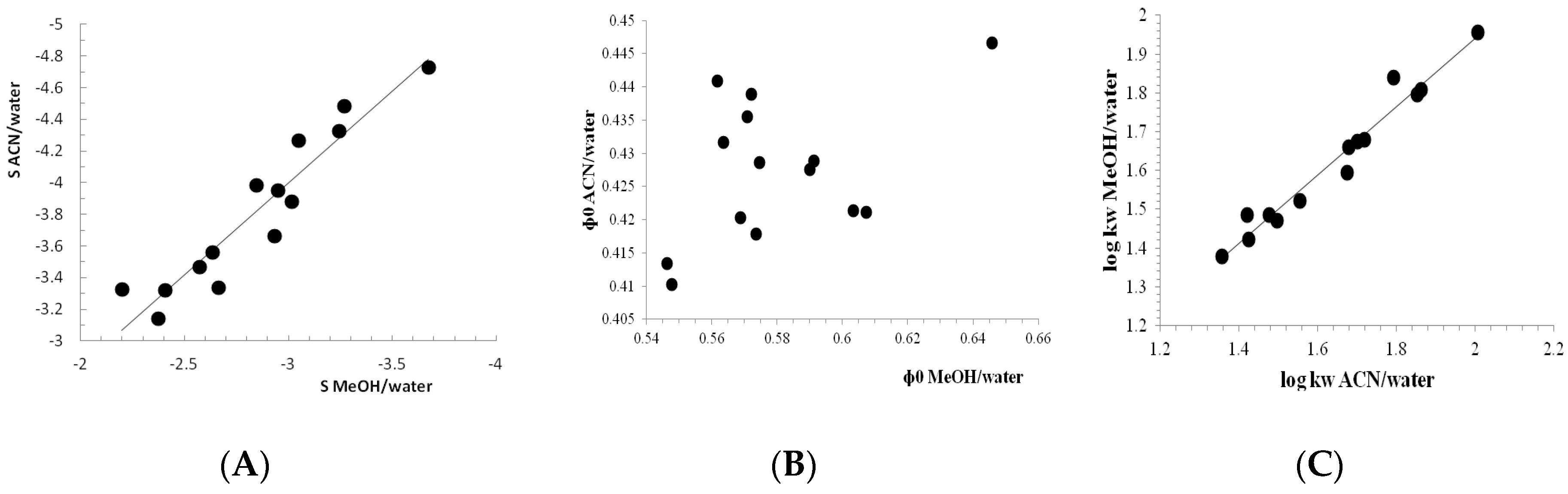
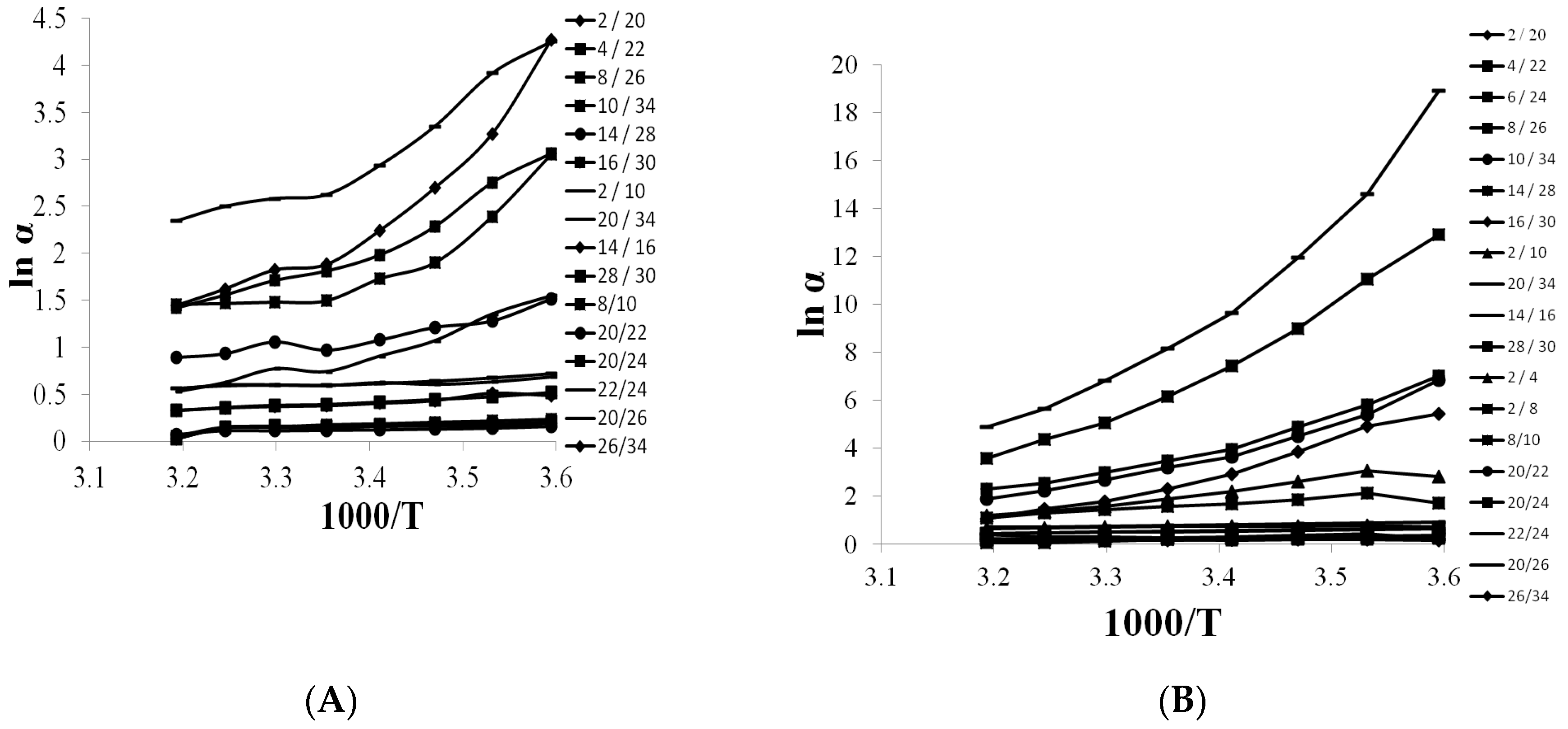
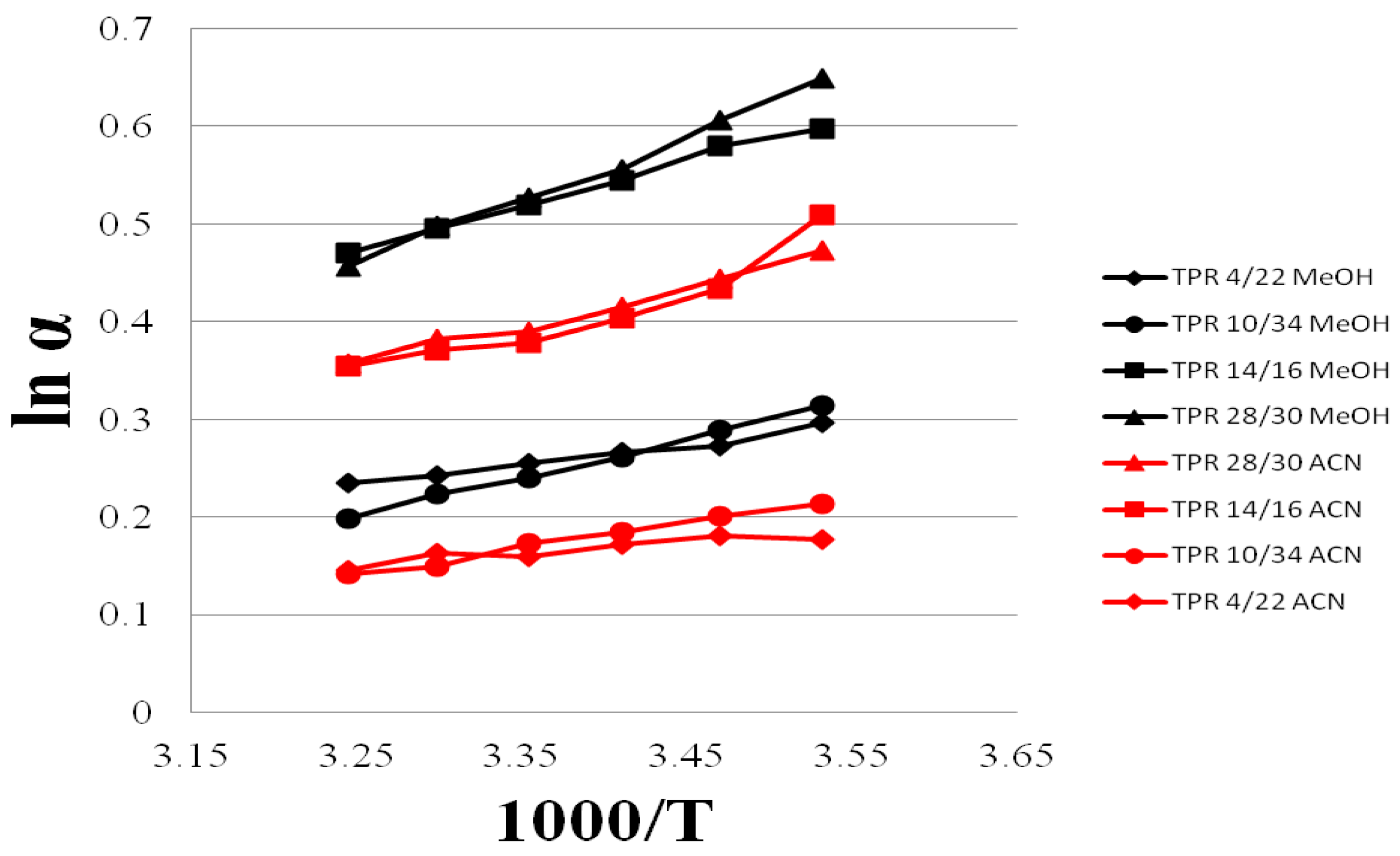
3.4. Selectivity of Teicpolanin Column towards Studied Triazoles
3.5. Cross-Correlation of Chromatographic and Thermodynamic Parameters with Theoretical Structural Descriptors
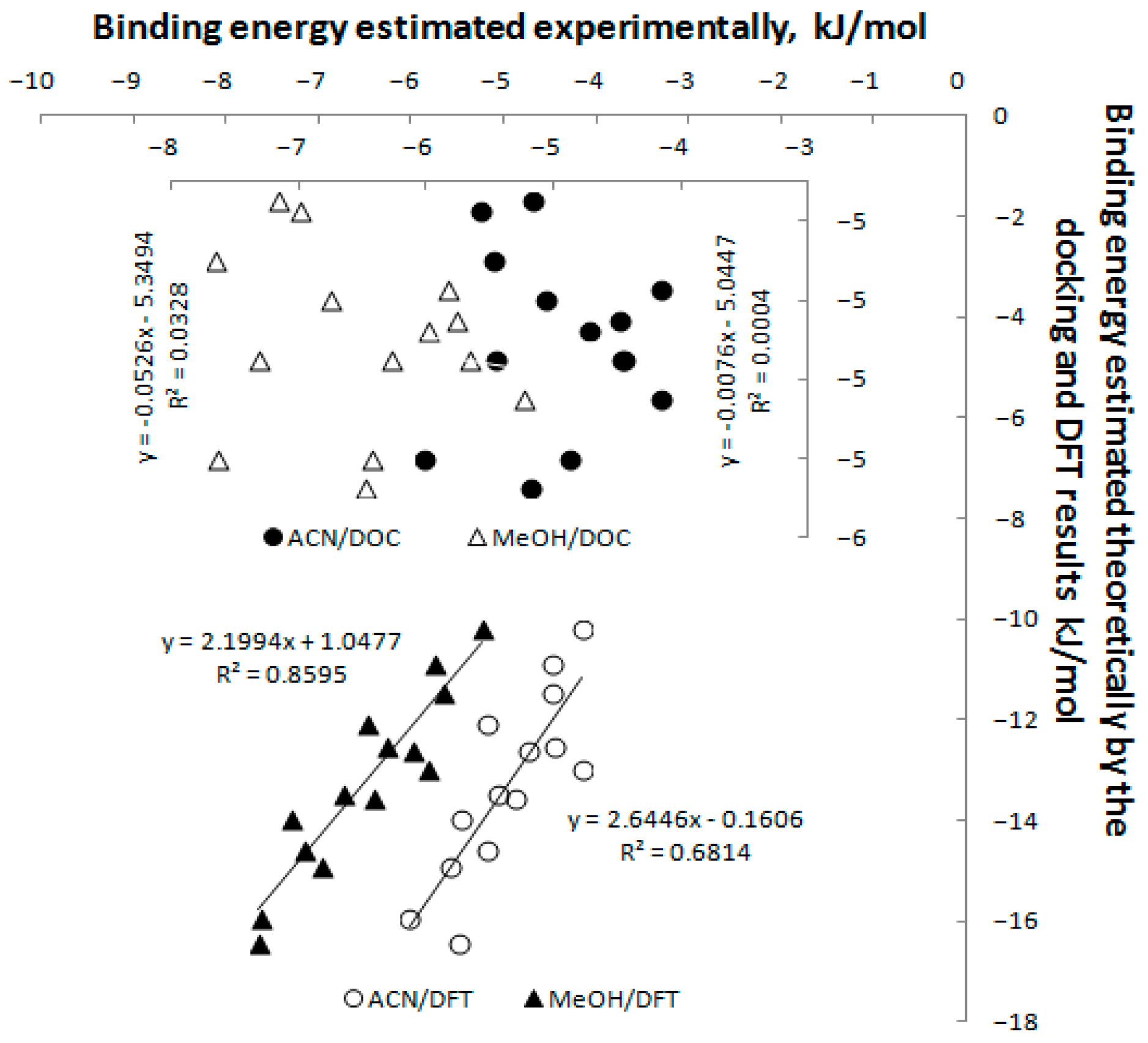
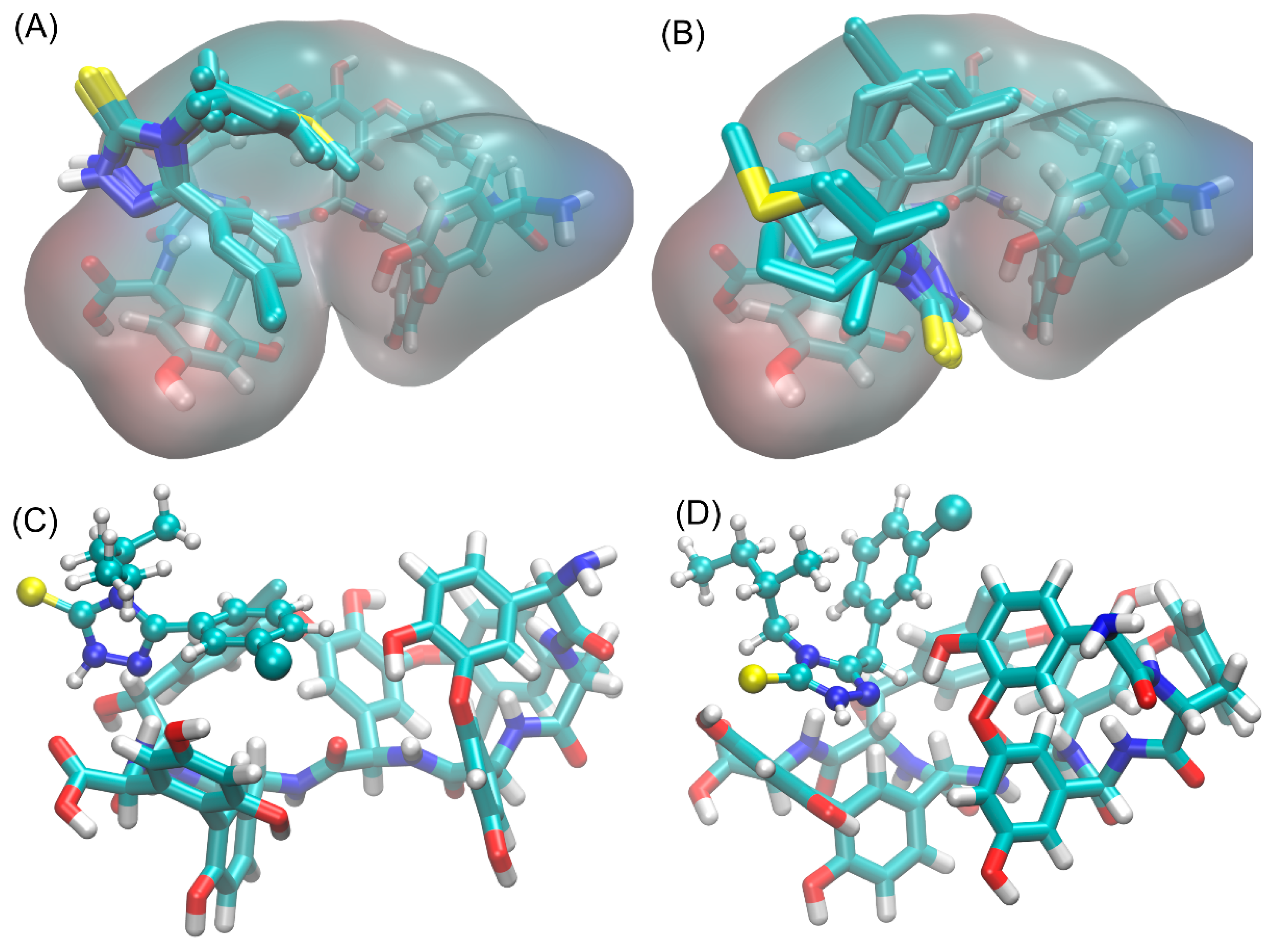
3.6. Study of Interactions of Teicoplanin with Investigated Triazoles by Docking Simulation
3.7. Statistical Analysis of Thermodynamic and Chromatographic Parameters for Activity Evaluation
3.7.1. Activity Division
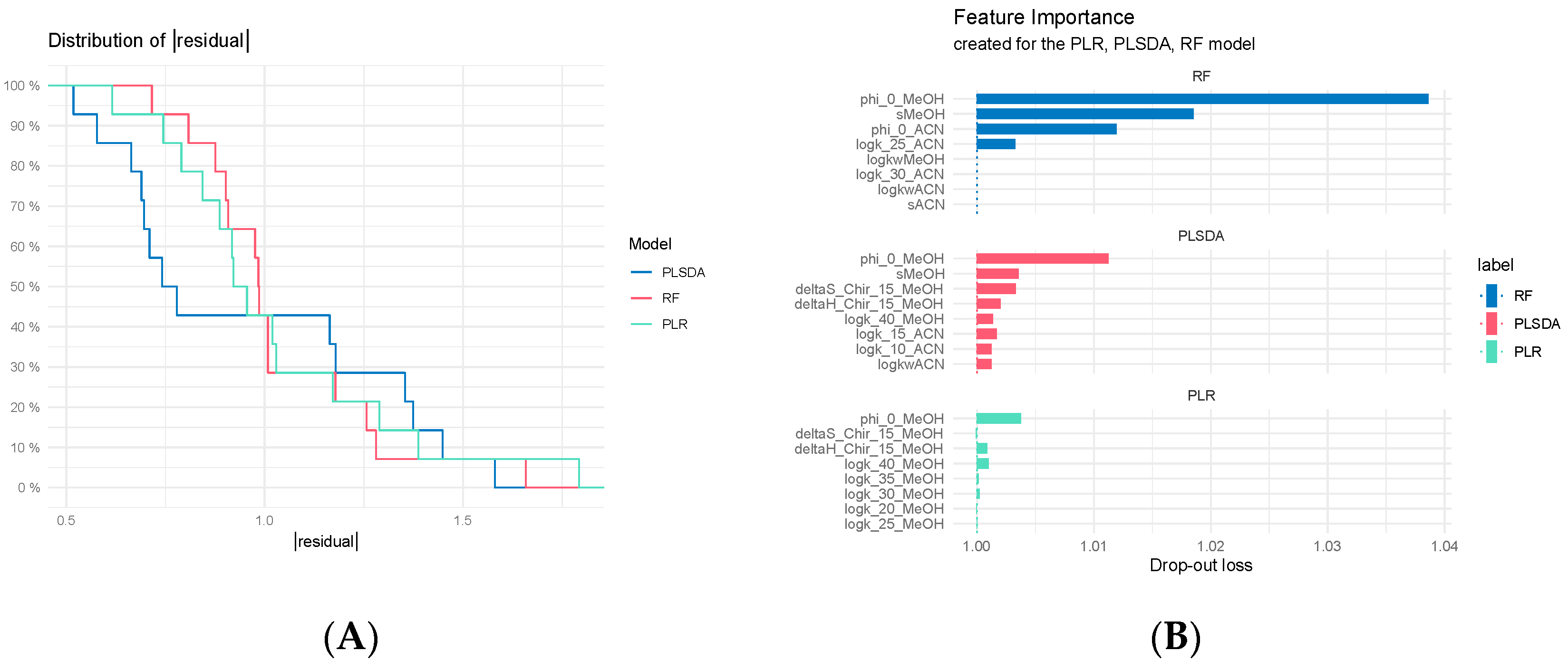
3.7.2. Comparison of Classification Models
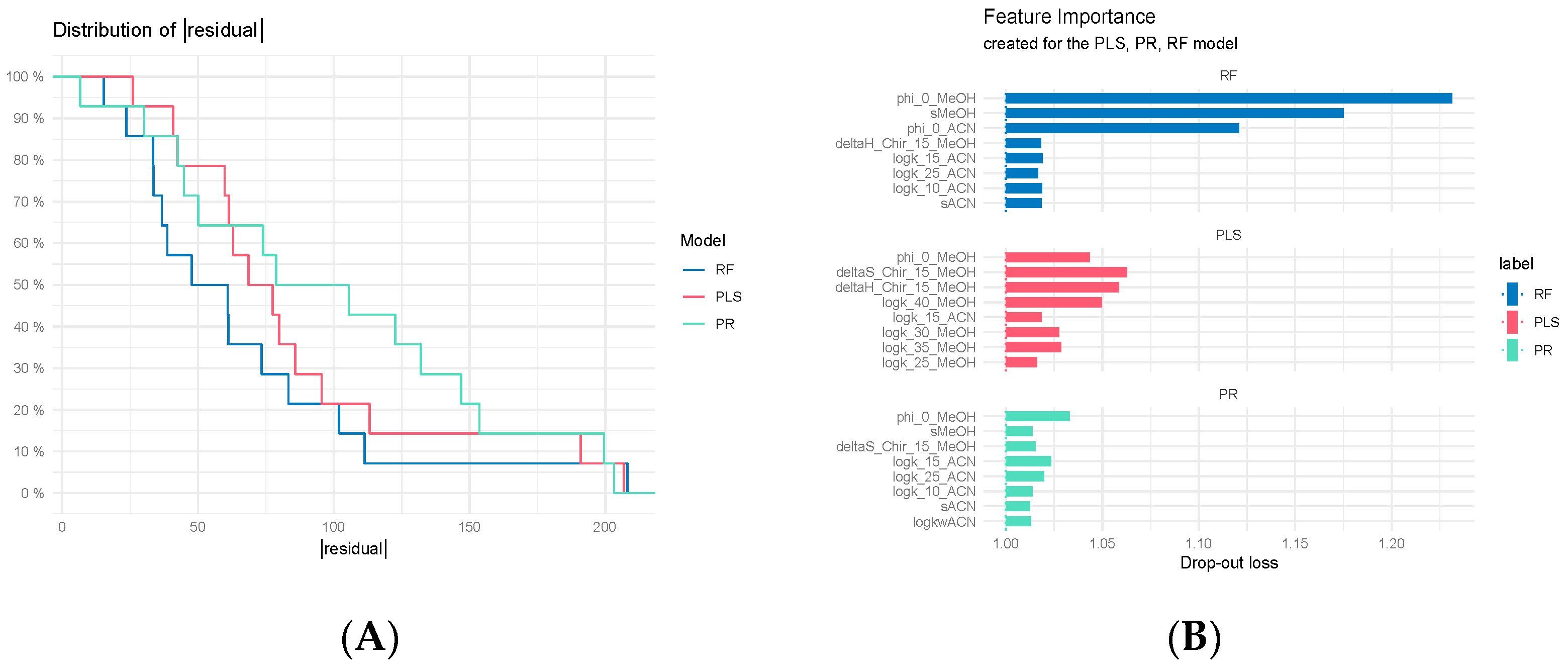
3.7.3. Regression Model Comparisons
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kirchmair, J. Drug Metabolism Prediction, 1st ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2014. [Google Scholar]
- Pliska, V.; Testa, B.; van de Waterbeemd, H. Lipophilicity in Drug Action and Toxicology; John Wiley & Sons: Weinheim, Germany, 1996. [Google Scholar]
- Kaliszan, R. Structure and Retention in Chromatography. A Chemometric Approach; Harwood Academic Publishers: Amsterdam, The Netherlands, 1997. [Google Scholar]
- Van de Waterbeemd, H.; Carter, R.E.; Grassy, G.; Kubinyi, H.; Martin, Y.C.; Tute, M.S.; Willett, P. Glossary of terms used in computational drug design (IUPAC Recommendations 1997). Pure Appl. Chem. 1997, 69, 1137. [Google Scholar] [CrossRef]
- Lambert, W.J. Modeling Oil-Water Partitioning and Membrane Permeation Using Reversed-Phase Chromatography. J. Chromatogr. A 1993, 656, 469. [Google Scholar] [CrossRef]
- Dorsey, J.G. Implications for Biological Partitioning Processes. J. Chromatogr. A 1993, 656, 485. [Google Scholar] [CrossRef]
- Kaliszan, R. Effect of Separation Conditions on Chromatographic Determination of Hydrophobicity of Acidic Xenobiotics. J. Chromatogr. B 1998, 717, 125. [Google Scholar] [CrossRef]
- Markuszewski, M.; Kaliszan, R. Quantitative Structure-Retention Relationships in Affinity High-Performance Liquid Chromatography. J. Chromatogr. B 2002, 768, 55. [Google Scholar] [CrossRef]
- Flieger, J.; Tatarczak-Michalewska, M.; Wujec, M.; Pitucha, M.; Świeboda, R. RP-HPLC analysis and in vitro identification of antimycobacterial activity of novel thiosemicarbazides and 1,2,4-triazole derivatives. J. Pharm. Biomed. Anal. 2015, 107, 501. [Google Scholar] [CrossRef]
- Ong, S.; Liu, H.; Pidgeon, C. Immobilized-Artificial Membrane Chromatography: Measurements of Membrane Partition Coefficient and Predicting Drug Membrane Permeability. J. Chromatogr. A 1996, 728, 113. [Google Scholar] [CrossRef]
- Yang, C.Y.; Cai, S.J.; Liu, H.; Pidgeon, C. Immobilized Artificial Membranes-Screens for Drug Membrane Interactions. Adv. Drug Deliv. Rev. 1997, 23, 229. [Google Scholar] [CrossRef]
- Lepont, C.; Poole, C.F. Retention Characteristics of an Immobilized Artificial Membrane Column in Reversed-Phase Liquid Chromatography. J. Chromatogr. A 2002, 946, 107. [Google Scholar] [CrossRef]
- Turnhofer, H.; Schnabel, J.; Betz, M.; Lipka, G.; Pidgeon, C.; Hauser, H. Cholesterol transfer protein located in the intestinal brush-border membrane. Partial purification and characterization. Biochim. Biophys. Acta 1991, 1064, 275. [Google Scholar] [CrossRef]
- Taillardat-Bertschinger, A.; Carrupt, P.A.; Barbato, F.; Testa, B. Immobilized Artificial Membrane HPLC in Drug Research. J. Med. Chem. 2003, 46, 1. [Google Scholar] [CrossRef] [PubMed]
- Flieger, J.; Trębacz, H.; Pizoń, M.; Kowalska, A.; Szczęsna, A.; Plech, T. High-performance liquid chromatography thermodynamic study of new potential antiepileptic compounds on a cholesterol column using isocratic elution with methanol/water and acetonitrile/water eluent systems. J. Sep. Sci. 2017, 40, 4176. [Google Scholar] [CrossRef] [PubMed]
- Flieger, J.; Pizon, M.; Plech., T. Chromatographic behavior of new antiepileptic active compounds on different reversed-phase materials. J. Chromatogr. A 2014, 1338, 188. [Google Scholar] [CrossRef] [PubMed]
- Hewen Li, H.; Zhao, T.; Sun, Z. Analytical techniques and methods for study of drug-lipid membrane interactions. Rev. Anal. Chem. 2018, 37, 20170012. [Google Scholar]
- Pereiraleite, C.; Nunes, C.; Reis, S. Interaction of Nonsteroidal Anti-Inflammatory Drugs with Membranes: In Vitro Assessment and Relevance for Their Biological Actions. Prog. Lipid Res. 2013, 52, 571–584. [Google Scholar] [CrossRef] [PubMed]
- Moravcova, D.; Planeta, J.; Wiedmer, S.K. Silica-Based Monolithic Capillary Columns Modified by Liposomes for Characterization of Analyte-Liposome Interactions by Capillary Liquid Chromatography. J. Chromatogr. A 2013, 1317, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Li, J.; Xu, L.; Shi, Z. Fast Immobilized Liposome Chromatography Based on Penetrable Silica Microspheres for Screening and Analysis of Permeable Compounds. J. Chromatogr. A 2012, 1233, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Fan, P.; Chen, M.; Hefesha, H.; Scriba, G.K.E.; Gabel, D.; Fahr, A. Drug-Membrane Interaction on Immobilized Liposome Chromatography Compared to Immobilized Artificial Membrane (IAM), Liposome/Water, and Octan-1-ol/Water Systems. Helv. Chim. Acta 2010, 93, 203–211. [Google Scholar] [CrossRef]
- Baldwin, T.D.; Kelly, J.W. Application of Enzyme Biotechnology; Plenum Press: New York, NY, USA, 1992. [Google Scholar]
- Pidgeon, C.; Ong, S.; Liu, H.; Qiu, X.; Pidgeon, M.; Dantzig, A.H.; Munroe, J.; Hornback, W.J.; Kasher, J.S.; Glunz, L.; et al. IAM Chromatography: An in Vitro Screen for Predicting Drug Membrane Permeability. J. Med. Chem. 1995, 38, 590. [Google Scholar] [CrossRef] [PubMed]
- Stewart, B.H.; Chan, H. Use of Immobilized Artificial Membrane Chromatography for Drug Transport Applications. J. Pharm. Sci. 1998, 87, 1471. [Google Scholar] [CrossRef] [PubMed]
- Schäfer-Korting, M. Drug Delivery; Springer: Berlin/Heidelberg, Germany, 2010; p. 197. [Google Scholar]
- Thuerauf, N.; Fromm, M.F. The role of the transporter P-glycoprotein for disposition and effects of centrally acting drugs and for the pathogenesis of CNS diseases. Eur. Arch. Psychiatry Clin. Neurosci. 2006, 256, 281. [Google Scholar] [CrossRef] [PubMed]
- Persidsky, Y.; Ramirez, S.H.; Haorah, J.; Kanmogne, G.D. Blood-brain barrier; structural components and function under physiologic and pathologic conditions. J. Neuroimmune Pharmacol. 2006, 1, 223. [Google Scholar] [CrossRef] [PubMed]
- Marchi, N.; Betto, G.; Fazio, V.; Fan, Q.; Ghosh, C.; Machado, A.; Janigro, D. Blood-brain barrier damage and brain penetration of antiepileptic drugs: Role of serum proteins and brain edema. Epilepsia 2009, 50, 664. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, T.S.; Kirshner, D.A.; Lau, E.Y.; Wong, S.E.; Nilmeier, J.P.; Lightstone, F.C. A Method to Predict Blood-Brain Barrier Permeability of Drug-Like Compounds Using Molecular Dynamics Simulations. Biophys. J. 2014, 107, 630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pignatello, R.; Musumeci, T.; Basile, L.; Carbone, C.; Puglisi, G. Biomembrane models and drug-biomembrane interaction studies: Involvement in drug design and development. J. Pharm. Bioall. Sci. 2011, 3, 4. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, D.W.; Tang, Y.; Chen, S.; Zhou, Y.; Bagwill, C.; Chen, J. Macrocyclic antibiotics as a new class of chiral selectors for liquid chromatography. Anal. Chem. 1994, 66, 1473. [Google Scholar] [CrossRef]
- Ward, T.J.; Farris, A.B. Chiral separations using the macrocyclic antibiotics: A review. J. Chromatogr. A 2001, 906, 73. [Google Scholar] [CrossRef]
- Ilisz, I.; Berkecz, R.; Péter, A. Retention mechanism of high-performance liquid chromatographic enantioseparation on macrocyclic glycopeptide-based chiral stationary phases. J. Chromatogr. A 2009, 1216, 1845. [Google Scholar] [CrossRef] [PubMed]
- Aburachid Cardoso, P.; Costa César, I. Chiral method development strategies for HPLC using macrocyclic glycopeptide-based stationary phases. Chromatographia 2018, 81, 841. [Google Scholar] [CrossRef]
- Moldoveanu, S.C.; David, V. Stationary Phases and Columns for Chiral Chromatography. HPLC Method in Chemical Analysis; Elsevier: Amsterdam, The Netherlands, 2017; pp. 363–376. [Google Scholar]
- Tang, M.; Zhang, J.; Zhuang, S.; Liu, W. Development of chiral stationary phases for high-performance liquid chromatographic separation. Trends Anal. Chem. 2012, 39, 180. [Google Scholar] [CrossRef]
- Kaproń, B.; Łuszczki, J.J.; Płazińska, A.; Siwek, A.; Karcz, T.; Gryboś, A.; Nowak, G.; Makuch-Kocka, A.; Walczak, K.; Langner, E.; et al. Development of the 1,2,4-triazole-based anticonvulsant drug candidates acting on the voltage-gated sodium channels. Insights from in-vivo, in-vitro, and in-silico studies. Eur. J. Pharm. Sci. 2019, 129, 42. [Google Scholar] [PubMed]
- Plech, T.; Łuszczki, J.J.; Wujec, M.; Flieger, J.; Pizoń, M. Synthesis, characterization and preliminary anticonvulsant evaluation of some 4-alkyl-1,2,4-triazoles. Eur. J. Med. Chem. 2013, 60, 208. [Google Scholar] [CrossRef] [PubMed]
- Plech, T.; Kaproń, B.; Łuszczki, J.J.; Paneth, A.; Siwek, A.; Kołaczkowski, M.; Żołnierek, M.; Nowak, G. Studies on the anticonvulsant activity of 4-alkyl-1,2,4-triazole-3-thiones and their effect on gabaergic system. Eur. J. Med. Chem. 2014, 86, 690. [Google Scholar] [CrossRef] [PubMed]
- Kaproń, B.; Łuszczki, J.J.; Siwek, A.; Karcz, T.; Nowak, G.; Zagaja, M.; Andres-Mach, M.; Stasiłowicz, M.; Cielecka-Piontek, J.; Kocki, J. Preclinical evaluation of 1,2,4-triazole-based compounds targeting voltage-gated sodium channels (VGSCs) as promising anticonvulsant drug candidates. Bioorg. Chem. 2020, 94, 103355. [Google Scholar] [CrossRef] [PubMed]
- Kaproń, B.; Łuszczki, J.; Paneth, A.; Wujec, M.; Siwek, A.; Karcz, T.; Mordyl, B.; Głuch-Lutwin, M.; Gryboś, A.; Nowak, G. Molecular mechanism of action and safety of 5-(3-chlorophenyl)-4-hexyl-2,4-dihydro-3H-1,2,4-triazole-3-thione—A novel anticonvulsant drug candidate. Int. J. Med. Sci. 2017, 14, 741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flieger, J.; Trębacz, H.; Pizoń, M.; Płazińska, A.; Płaziński, W.; Kowalska, A.; Szczęsna, A.; Plech, T. Thermodynamic study of new antiepileptic compounds by combining chromatography on the phosphatidylcholine biomimetic stationary phase and differential scanning calorimetry. J. Sep. Sci. 2019, 42, 2628. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. A Language and Environment for Statistical Computing. 2020. Available online: https://www.r-project.org/ (accessed on 2 May 2020).
- Kuhn, M. Caret: Classification and Regression Training. 2020. Available online: https://CRAN.R-project.org/package=caret (accessed on 2 May 2020).
- Biecek, P. DALEX: Explainers for Complex Predictive Models in R. J. Mach. Learn. Res. 2018, 19, 1. [Google Scholar]
- Kassambara, A. Ggpubr: ’Ggplot2’ Based Publication Ready Plots. Available online: https://CRAN.R-project.org/package=ggpubr (accessed on 2 May 2020).
- Liaw, A.; Wiener, M. Classification and Regression by random Forest. R. News 2002, 2, 18. [Google Scholar]
- Friedman, J.; Hastie, T.; Tibshirani, R. Regularization Paths for Generalized Linear Models via Coordinate Descent. J. Stat. Softw. 2010, 33, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noah, S.; Friedman, J.; Hastie, T.; Tibshirani, R. Regularization Paths for Cox’s Proportional Hazards Model via Coordinate Descent. J. Stat. Softw. 2011, 39, 1. [Google Scholar]
- Mevik, B.-H.; Wehrens, R.; Hovde Liland, K. Pls: Partial Least Squares and Principal Component Regression. Available online: https://CRAN.R-project.org/package=pls (accessed on 2 May 2020).
- Granitto, P.M.; Furlanello, C.; Biasioli, F.; Gasperi, F. Recursive Feature Elimination with Random Forest for PTR-MS Analysis of Agroindustrial Products. Chemom. Intell. Lab. 2006, 83, 83. [Google Scholar] [CrossRef]
- Kirkpatrick, S.; Gelatt, C.D.; Vecchi, M.P. Optimization by Simulated Annealing. Science 1983, 220, 671. [Google Scholar] [CrossRef] [PubMed]
- Browne, M.W. Cross-Validation Methods. J. Math. Psychol. 2000, 44, 108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tibshirani, R.; Bien, J.; Friedman, J.; Hastie, T.; Simon, N.; Taylor, J.; Tibshirani, R.J. Strong Rules for Discarding Predictors in Lasso-Type Problems: Strong Rules for Discarding Predictors. J. R. Stat. Soc. 2012, 74, 245. [Google Scholar] [CrossRef] [PubMed]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rappe, A.K.; Casewit, C.J.; Colwell, K.S.; Goddard, W.A.; Skiff, W.M. UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations. J. Am. Chem. Soc. 1992, 114, 10024. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hehre, W.J.; Radom, L.; Schleyer, P.R.; Pope, J. Ab Initio Molecular Orbital Theory; Wiley: New York, NY, USA, 1986; pp. 86–87. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision E.01; Gaussian, Inc.: Wallingford, UK, 2009. [Google Scholar]
- Soczewinski, E.; Wachtmeister, C.A. The relation between the composition of certain ternary two-phase solvent systems and RM values. J. Chromatogr. 1962, 7, 311. [Google Scholar] [CrossRef]
- Valko, K.; Sleger, P. New chromatographic hydrophobicity index (&) based on the slope and the intercept of the logk’ versus organic phase concentration plot. J. Chromatogr. A 1993, 631, 49. [Google Scholar]
- Barbaro, G.L.; Sapone, A.M.; Borea, A.; Variani, P.A.; Recanatini, M. Study of lipophilic character of serotonic ligand. J. Chromatogr. A 1996, 723, 135. [Google Scholar]
- Biagi, L.; Barbaro, A.M.; Sapone, A.; Recanatini, M. Determination lipophilicity by means of reversed phase thin-layer chromatography. Influence of the organic modifier on the slope of the thin-layer chromatographic equation. J. Chromatogr. A 1994, 669, 246. [Google Scholar] [CrossRef]
- Knox, H.; Pryde, A. Performance and selected applications of a new range of chemically bonded packing materials in high-performance liquid chromatography. J. Chromatogr. 1975, 112, 171. [Google Scholar] [CrossRef]
- Kazakevich, Y.V.; LoBrutto, R.; Chan, F.; Patel, T. Interpretation of the excess adsorption isotherms of organic eluent components on the surface of reversed-phase adsorbents: Effect on the analyte retention. J.Chromatogr. A 2001, 913, 75. [Google Scholar] [CrossRef]
- Hansch, C.; Leo, A.; Hoekman, D. Exploring QSAR—Hydrophobic, Electronic, and Steric Constants; American Chemical Society: Washington, DC, USA, 1995. [Google Scholar]
- Wildman, S.A.; Crippen, G.M. Prediction of physicochemical parameters by atomic contributions. J. Chem. Inf. Comput. Sci. 1999, 39, 868. [Google Scholar] [CrossRef]
- Chester, T.L.; Coym, J.W. Effect of phase ratio on van’t Hoff analysis in reversed-phase liquid chromatography, and phase-ratio independent estimation of transfer enthalpy. J. Chromatogr. A 2003, 1003, 101. [Google Scholar] [CrossRef]
- Dorsey, J.G.; Dill, K.A. The molecular mechanism of retention in reversed-phase liquid chromatography. Chem. Rev. 1989, 89, 331. [Google Scholar] [CrossRef]
- Sentell, K.B.; Dorsey, J.G. On the Calculation of the Stationary Phase Volume in Reversed Phase Chromatography. J. Liq. Chromatogr. 1988, 11, 1875. [Google Scholar] [CrossRef]
- Doyle, C.A.; Vickers, T.J.; Mann, C.K.; Dorsey, J.G. Characterization of C18-bonded liquid chromatographic stationary phases by Raman spectroscopy: The effect of temperature. J. Chromatogr. A 2000, 877, 41. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| General Formula (1) | General Formula (2) | ||
|---|---|---|---|
 |  | ||
| No. | R | No. | R |
| TPR-2 AC log P = 3.93 * 310.3 mg/kg bw ** |  | TPR-20 AC log P = 3.83 * (−) ** |  |
| TPR-4 AC log P = 4.33 * 221.7 mg/kg bw ** |  | TPR-22 AC log P = 4.23 * 130.4 mg/kg bw ** |  |
| TPR-6 AC log P = 4.08 * 295.1 mg/kg bw ** |  | TPR-24 AC log P = 3.98 * (−) ** |  |
| TPR-8 AC log P = 4.79 * (−) ** |  | TPR-26 AC log P = 4.69 * (−) ** |  |
| TPR-10 AC log P = 5.32 * (−) ** |  | TPR-34 AC log P = 5.22 * (−) ** |  |
| TPR-14 AC log Ps = 3.49 * 208.9 mg/kg bw ** |  | TPR-28 AC log P = 3.39 * 132.4 mg/kg bw ** |  |
| TPR-16 AC log P = 4.16 * 201.0 mg/kg bw ** |  | TPR-30 AC log P = 4.06 * 320.0 mg/kg bw ** |  |
| Symbol | MeOH | ACN | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| log kw | S | φ0 | R2 | F | se | log kw | S | φ0 | R2 | F | se | |
| TPR 2 | 1.3574 | −2.3728 | 0.5721 | 0.9902 | 404 | 0.0247 | 1.3794 | −3.1428 | 0.4389 | 0.9784 | 182 | 0.0488 |
| TPR 4 | 1.4772 | −2.5712 | 0.5745 | 0.9925 | 526 | 0.0234 | 1.4850 | −3.4647 | 0.4286 | 0.9796 | 192 | 0.0523 |
| TPR 6 | 1.4200 | −2.1986 | 0.6459 | 0.9804 | 200 | 0.0325 | 1.4859 | −3.3270 | 0.4466 | 0.9843 | 250 | 0.0440 |
| TPR 8 | 1.6778 | −2.9497 | 0.5688 | 0.9937 | 630 | 0.0246 | 1.6600 | −3.9492 | 0.4203 | 0.9863 | 289 | 0.0486 |
| TPR 10 | 1.7921 | −3.2712 | 0.5478 | 0.9956 | 913 | 0.0226 | 1.8393 | −4.4832 | 0.4103 | 0.9856 | 273 | 0.0567 |
| TPR 14 | 1.4238 | −2.4081 | 0.5912 | 0.9920 | 2519 | 0.0126 | 1.4227 | −3.3181 | 0.4288 | 0.9732 | 375 | 0.0419 |
| TPR 16 | 1.7177 | −2.8469 | 0.6034 | 0.9960 | 985 | 0.0190 | 1.6785 | −3.9831 | 0.4214 | 0.9818 | 216 | 0.0567 |
| TPR 20 | 1.4968 | −2.6651 | 0.5616 | 0.9974 | 1518 | 0.0143 | 1.4711 | −3.3363 | 0.4409 | 0.9765 | 166 | 0.0541 |
| TPR 22 | 1.6746 | −2.9338 | 0.5708 | 0.9983 | 2285 | 0.0128 | 1.5954 | −3.6636 | 0.4355 | 0.9815 | 213 | 0.0526 |
| TPR 24 | 1.6998 | −3.0168 | 0.5635 | 0.9984 | 2519 | 0.0126 | 1.6752 | −3.8815 | 0.4316 | 0.9895 | 375 | 0.0419 |
| TPR 26 | 1.8619 | −3.2460 | 0.5736 | 0.9979 | 1884 | 0.0156 | 1.8080 | −4.3276 | 0.4178 | 0.9858 | 278 | 0.0543 |
| TPR 28 | 1.5547 | −2.6348 | 0.5901 | 0.9979 | 1884 | 0.0156 | 1.5212 | −3.5573 | 0.4276 | 0.9858 | 278 | 0.0543 |
| TPR 30 | 1.8527 | −3.0514 | 0.6072 | 0.9972 | 1409 | 0.0170 | 1.7957 | −4.2644 | 0.4211 | 0.9851 | 264 | 0.0549 |
| TPR 34 | 2.0070 | −3.6746 | 0.5462 | 0.9966 | 1161 | 0.0226 | 1.9553 | −4.7297 | 0.4134 | 0.9915 | 466 | 0.0458 |
| Descriptive Statistics | ||||||||||||
| Mean | 1.644 | −2.846 | 0.580 | 0.995 | 1345.6 | 0.019 | 1.627 | −3.816 | 0.427 | 0.983 | 272.76 | 0.050 |
| Median | 1.676 | −2.890 | 0.573 | 0.996 | 1285.3 | 0.018 | 1.628 | −3.773 | 0.428 | 0.985 | 268.81 | 0.052 |
| Standard deviation | 0.188 | 0.388 | 0.025 | 0.005 | 755.2 | 0.006 | 0.169 | 0.476 | 0.010 | 0.005 | 81.19 | 0.005 |
| Skewness | 0.211 | −0.316 | 1.195 | −2.160 | 0.185 | 0.709 | 0.367 | −0.422 | 0.183 | −0.399 | 0.972 | −0.557 |
| Kurtosis | -0.873 | −0.088 | 1.931 | 5.447 | −1.240 | 0.022 | −0.916 | −0.968 | 0.714 | −0.132 | 0.723 | −1.171 |
| Coefficient of variation | 11.854 | 14.140 | 4.536 | 0.487 | 58.244 | 30.851 | 10.780 | 12.953 | 2.494 | 0.511 | 30.89 | 10.487 |
| MeOH | ACN | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| No TPR | ∆H° kJ/mol | ∆S°^ kJ/mol*K | ∆G° kJ/mol | R2 | F | Se | ∆H° kJ/mol | ∆S° kJ/mol*K | ∆G° kJ/mol | R2 | F | Se |
| 2 | −20.6090 | −0.0521 | −5.2303 | 0.9979 | 2882 | 0.017 | −8.3367 | −0.0142 | −4.1676 | 0.8175 | 27 | 0.072 |
| 4 | −20.5715 | −0.0502 | −5.7669 | 0.9999 | 50484 | 0.004 | −7.7317 | −0.0111 | −4.4872 | 0.8404 | 32 | 0.062 |
| 6 | −16.4897 | −0.0355 | −5.9824 | 0.9824 | 334 | 0.040 | −4.1104 | 0.0022 | −4.7408 | 0.3960 | 4 | 0.093 |
| 8 | −23.5131 | −0.0569 | −6.7458 | 0.9999 | 46120 | 0.005 | −8.8404 | −0.0129 | −5.0677 | 0.8828 | 45 | 0.059 |
| 10 | −22.4397 | −0.0524 | −6.9865 | 0.9996 | 16937 | 0.008 | −10.0121 | −0.0151 | −5.5932 | 0.8824 | 45 | 0.067 |
| 14 | −23.4591 | −0.0598 | −5.8228 | 0.9995 | 21550 | 0.007 | −9.8373 | −0.0194 | −4.1642 | 0.9670 | 131 | 0.045 |
| 16 | −27.5706 | −0.0693 | −7.1638 | 0.9995 | 12907 | 0.011 | −13.4231 | −0.0281 | −5.1833 | 0.9629 | 156 | 0.048 |
| 20 | −21.4193 | −0.0534 | −5.6540 | 0.9997 | 19402 | 0.007 | −10.2859 | −0.0198 | −4.4765 | 0.9668 | 175 | 0.035 |
| 22 | −22.7078 | −0.0552 | −6.4215 | 0.9999 | 44485 | 0.005 | −10.2560 | −0.0183 | −4.8815 | 0.9624 | 154 | 0.037 |
| 24 | −22.0695 | −0.0528 | −6.4815 | 0.9997 | 21550 | 0.007 | −11.9776 | −0.0231 | −5.1955 | 0.9562 | 131 | 0.047 |
| 26 | −24.5931 | −0.0586 | −7.3147 | 0.9994 | 10895 | 0.011 | −11.0007 | −0.0189 | −5.4615 | 0.9597 | 143 | 0.041 |
| 28 | −25.2271 | −0.0643 | −6.2751 | 0.9995 | 12046 | 0.010 | −11.2507 | −0.0232 | −4.4612 | 0.9779 | 266 | 0.031 |
| 30 | −30.1099 | −0.0763 | −7.6480 | 0.9996 | 13413 | 0.012 | −14.9553 | −0.0323 | −5.4933 | 0.9814 | 317 | 0.038 |
| 34 | −25.7792 | −0.0615 | −7.6401 | 0.9999 | 48330 | 0.005 | −13.1747 | −0.0244 | −6.0226 | 0.9618 | 151 | 0.048 |
| Log Phyper | HE | Polarizability | SA | Vol | µ | HF | HOMO | LUMO | ALOGPs | AC logP | ALOG P | MLOG P | XLOG P2 | XLOG P3 | SVM BBB | AdaBoost BBB |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cross-correlation with ΔG° estimated in methanol/water eluent system | ||||||||||||||||
| 0.005 | 0.000 | 0.921 | 0.037 | 0.670 | 0.043 | 0.001 | 0.406 | 0.011 | 0.143 | 0.361 | 0.282 | 0.385 | 0.302 | 0.339 | 0.397 | 0.119 |
| Cross-correlation with ΔG° estimated in acetonitrile/water eluent system | ||||||||||||||||
| 0.113 | 0.105 | 0.684 | 0.006 | 0.333 | 0.000 | 0.139 | 0.167 | 0.129 | 0.394 | 0.626 | 0.584 | 0.687 | 0.685 | 0.618 | 0.215 | 0.007 |
| Cross-correlation with log kw estimated in methanol/water eluent system | ||||||||||||||||
| 0.036 | 0.015 | 0.886 | 0.007 | 0.607 | 0.015 | 0.003 | 0.420 | 0.003 | 0.245 | 0.427 | 0.377 | 0.458 | 0.434 | 0.417 | 0.328 | 0.043 |
| Cross-correlation with log kw estimated in acetonitrile/water eluent system | ||||||||||||||||
| 0.110 | 0.094 | 0.689 | 0.016 | 0.335 | 0.002 | 0.068 | 0.202 | 0.065 | 0.406 | 0.642 | 0.598 | 0.681 | 0.687 | 0.630 | 0.234 | 0.009 |
| Ligand | Binding Energy, Docking (kJ/mol) | Binding Energy, DFT (kJ/mol) | Ligand | Binding Energy, Docking (kJ/mol) | Binding Energy, DFT (kJ/mol) |
|---|---|---|---|---|---|
| TRP-2 | −5.15 ± 0.53 | −10.22 | TRP-20 | −5.05 ± 0.29 | −11.47 |
| TRP-4 | −4.95 ± 0.33 | −10.88 | TRP-22 | −5.30 ± 0.49 | −13.57 |
| TRP-6 | −4.98 ± 0.11 | −12.62 | TRP-24 | −5.38 ± 0.29 | −12.10 |
| TRP-8 | −4.90 ± 0.41 | −13.47 | TRP-26 | −5.05 ± 0.42 | −13.96 |
| TPR-10 | −4.68 ± 0.23 | −14.93 | TPR-28 | −5.05 ± 0.38 | −12.56 |
| TRP-14 | −4.88 ± 0.31 | −12.98 | TRP-30 | −4.80 ± 0.37 | −16.46 |
| TRP-16 | −4.65 ± 0.21 | −14.59 | TRP-34 | −5.30 ± 0.44 | −15.96 |
| Prediction: Active/Nonactive | PR—Regularized Logistic Regression | RF—Random Forest | PLS-DA |
|---|---|---|---|
| Accuracy | 0.8 | 0.8 | 0.8 |
| 95% CI | (0.2836, 0.9949) | (0.2836, 0.9949) | (0.2836, 0.9949) |
| No Information Rate | 0.6 | 0.6 | 0.6 |
| p-Value (Acc > NIR) | 0.337 | 0.337 | 0.337 |
| Kappa | 0.5455 | 0.5455 | 0.5455 |
| McNemar’s Test p-Value | 1.000 | 1.000 | 1.000 |
| Sensitivity | 1.00 | 1.00 | 1.00 |
| Specificity | 0.50 | 0.50 | 0.50 |
| Pos Pred Value | 0.75 | 0.75 | 0.75 |
| Neg Pred Value | 1.00 | 1.00 | 1.00 |
| Prevalence | 0.60 | 0.60 | 0.60 |
| Detection Rate | 0.60 | 0.60 | 0.60 |
| Detection Prevalence | 0.80 | 0.80 | 0.80 |
| Balanced Accuracy | 0.75 | 0.75 | 0.75 |
| Positive class | active | active | active |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Flieger, J.; Orzeł, A.; Kowalska-Kępczyńska, A.; Pizoń, M.; Trębacz, H.; Majerek, D.; Plech, T.; Płaziński, W. Teicoplanin-Modified HPLC Column as a Source of Experimental Parameters for Prediction of the Anticonvulsant Activity of 1,2,4-Triazole-3-Thiones by the Regression Models. Materials 2020, 13, 2650. https://doi.org/10.3390/ma13112650
Flieger J, Orzeł A, Kowalska-Kępczyńska A, Pizoń M, Trębacz H, Majerek D, Plech T, Płaziński W. Teicoplanin-Modified HPLC Column as a Source of Experimental Parameters for Prediction of the Anticonvulsant Activity of 1,2,4-Triazole-3-Thiones by the Regression Models. Materials. 2020; 13(11):2650. https://doi.org/10.3390/ma13112650
Chicago/Turabian StyleFlieger, Jolanta, Anna Orzeł, Anna Kowalska-Kępczyńska, Magdalena Pizoń, Hanna Trębacz, Dariusz Majerek, Tomasz Plech, and Wojciech Płaziński. 2020. "Teicoplanin-Modified HPLC Column as a Source of Experimental Parameters for Prediction of the Anticonvulsant Activity of 1,2,4-Triazole-3-Thiones by the Regression Models" Materials 13, no. 11: 2650. https://doi.org/10.3390/ma13112650
APA StyleFlieger, J., Orzeł, A., Kowalska-Kępczyńska, A., Pizoń, M., Trębacz, H., Majerek, D., Plech, T., & Płaziński, W. (2020). Teicoplanin-Modified HPLC Column as a Source of Experimental Parameters for Prediction of the Anticonvulsant Activity of 1,2,4-Triazole-3-Thiones by the Regression Models. Materials, 13(11), 2650. https://doi.org/10.3390/ma13112650