Extended Polysaccharide Analysis within the Liposomal Encapsulation of Polysaccharides System

 ,
, 

Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials and Reagents
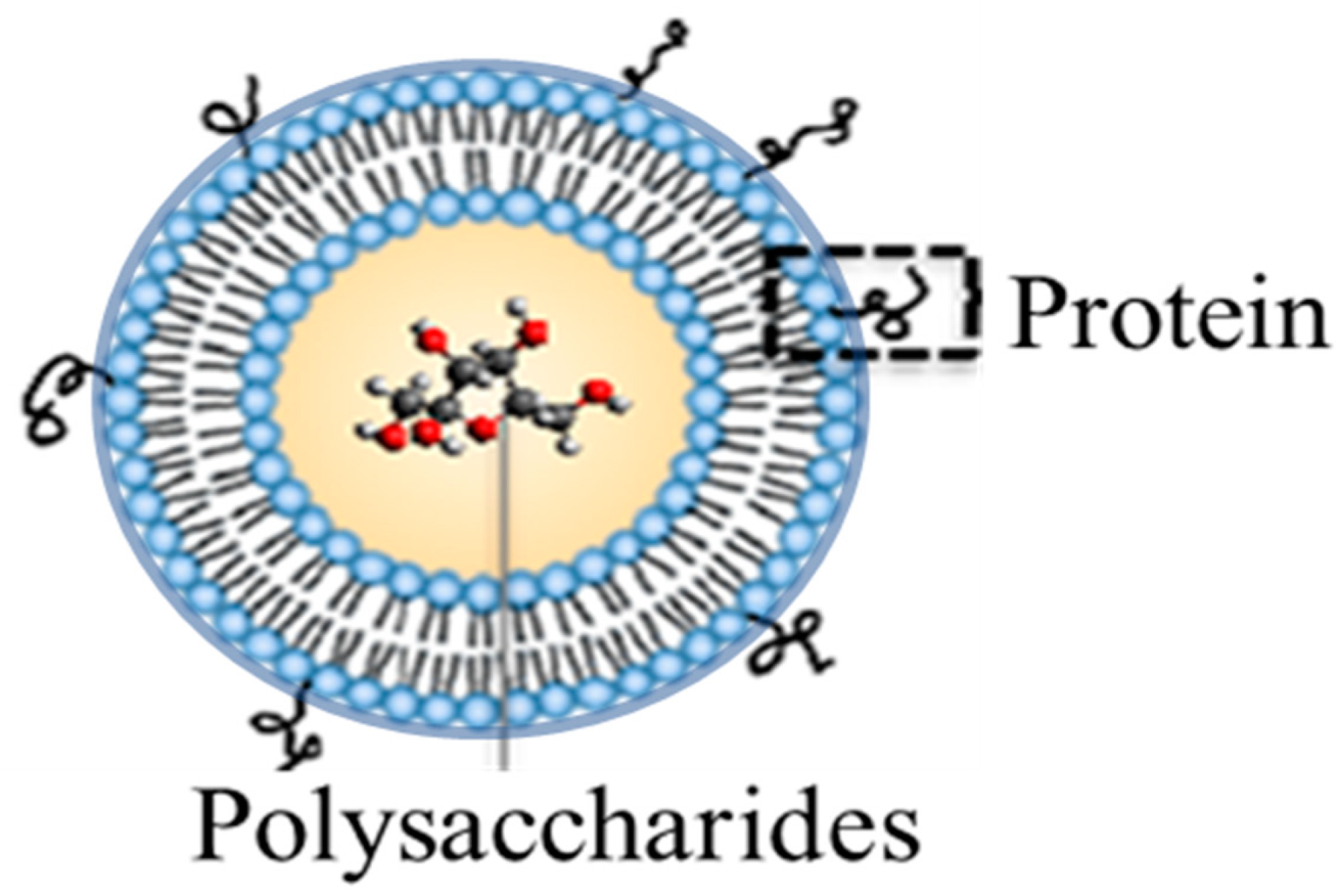
2.2. Liposomal Preparation
2.3. Polysaccharide Encapsulation Analysis
2.4. Size and Zeta Potential Analysis
2.5. Liposomal Protein Surface Assessment
2.6. Experimental Repetition
3. Results
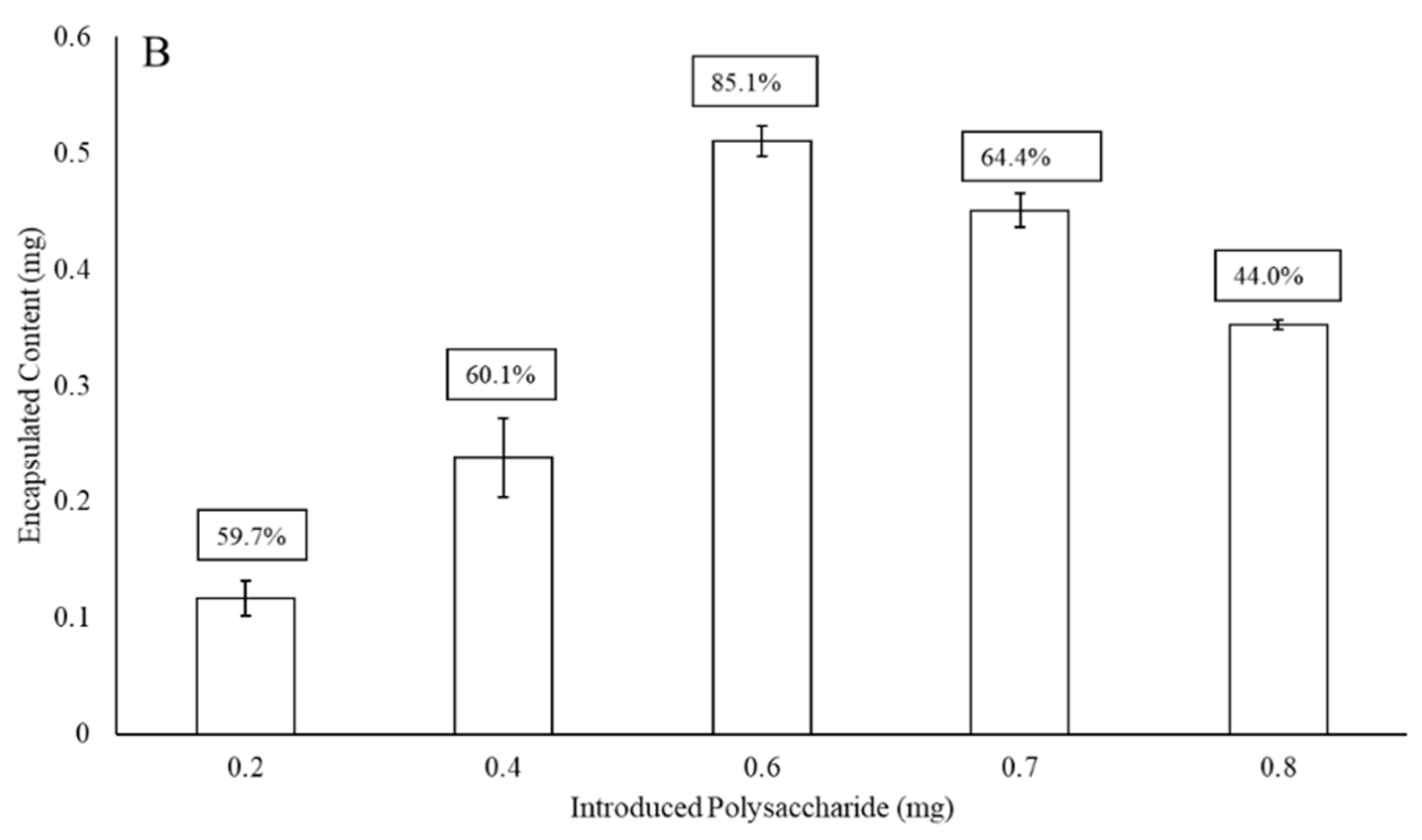
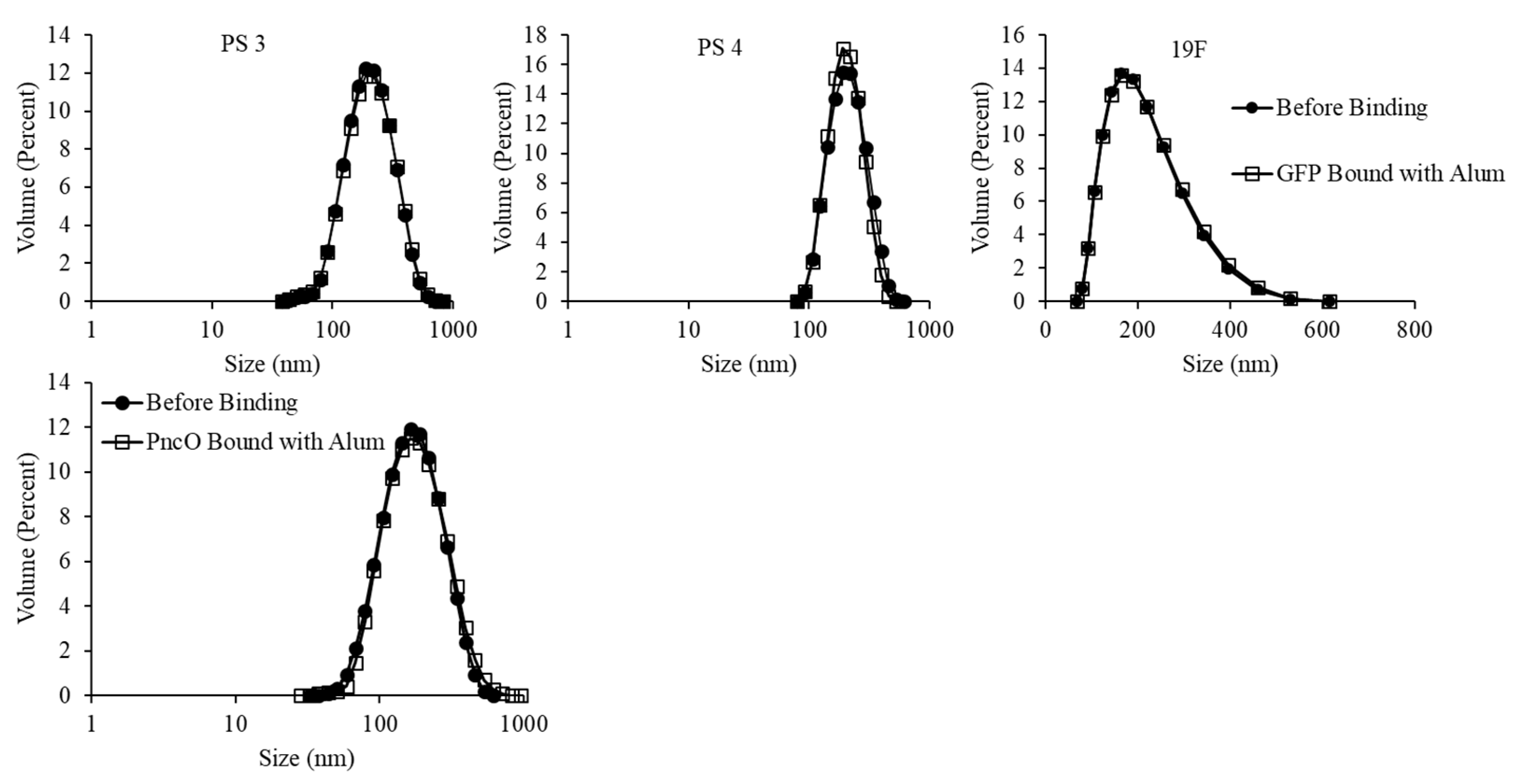
3.1. Liposomal Encapsulation of Streptococcus pneumoniae Serotype Capsular Polysaccharides 3 and 4
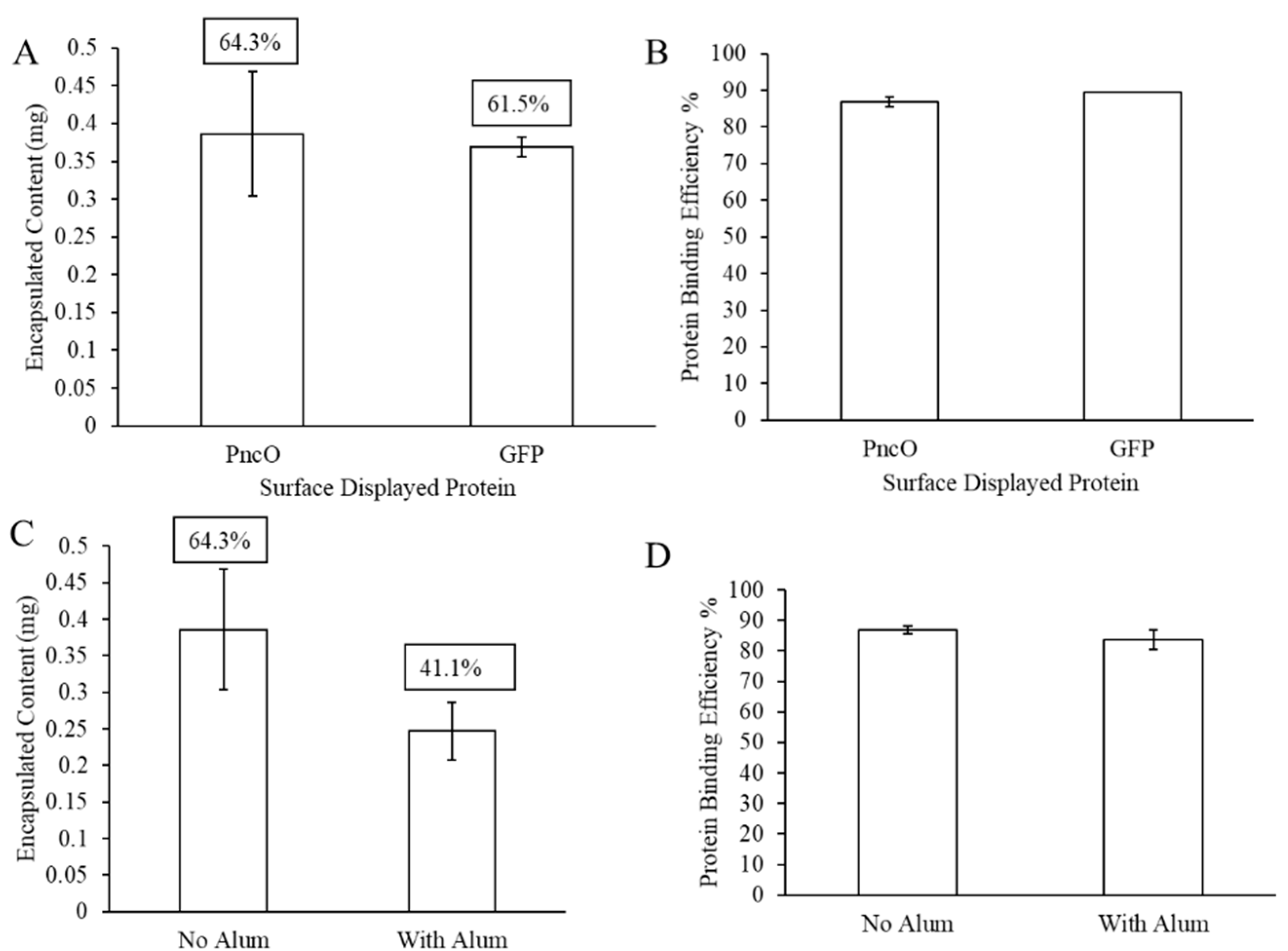
3.2. LEPS Surface Protein Binding Impact upon Polysaccharide Encapsulation Efficiency
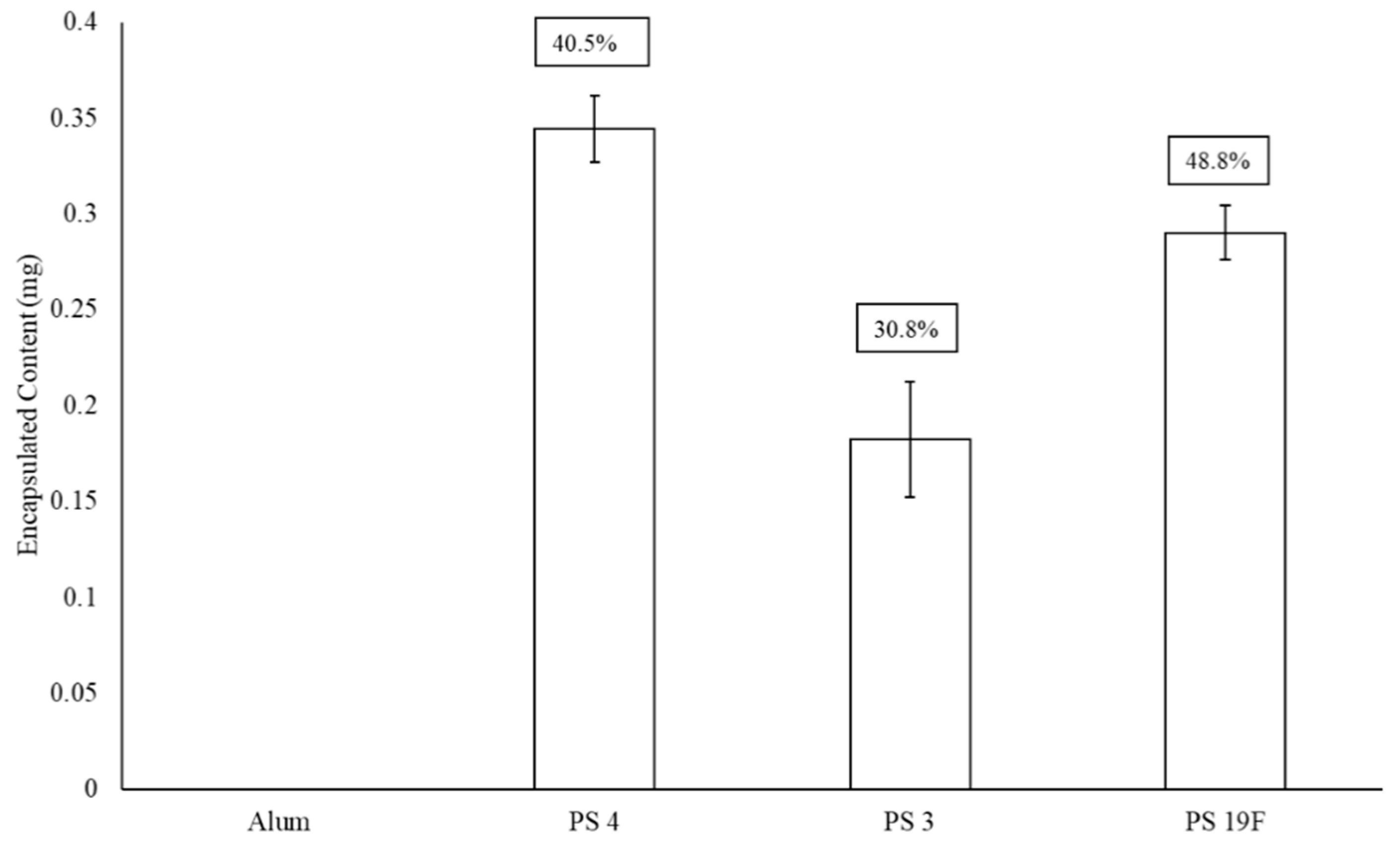
3.3. The Addition of Adjuvant upon LEPS Formulation Parameters
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Klugman, K.P.; Black, S.; Dagan, R.; Malley, R.; Whitney, C. Pneumococcal Conjugate Vaccine and Pneumococcal Common Protein Vaccines, 6th ed.; Elsevier: Beijing, China, 2013. [Google Scholar]
- Bogaert, D.; de Groot, R.; Hermans, P.W.M. Streptococcus pneumoniae colonisation: The key to pneumococcal disease. Lancet Infect Dis. 2004, 4, 144–154. [Google Scholar] [CrossRef]
- Vergison, A.; Dagan, R.; Arguedas, A.; Bonhoeffer, J.; Cohen, R.; DHooge, I.; Haberman, A.; Liese, J.; Marchisio, P.; Palmu, A.A.; et al. Otitis media and its consequences: Beyond the earache. Lancet Infect Dis. 2010, 10, 195–203. [Google Scholar] [CrossRef]
- O’Brien, K.L.; Wolfson, L.J.; Watt, J.P.; Henkle, E.; Deloria-Knoll, M.; McCall, N.; Lee, E.; Mulholland, K.; Levine, O.S.; Cherian, T.; et al. Burden of disease caused by Streptococcus pneumoniae in children younger than 5 years: Global estimates. Lancet 2009, 374, 893–902. [Google Scholar] [CrossRef]
- Henriques-Normark, B.; Normark, S. Bacterial vaccines and antibiotic resistance. Ups. J. Med. Sci. 2014, 119, 205–208. [Google Scholar] [CrossRef] [PubMed]
- Bonten, M.J.; Huijts, S.M.; Bolkenbaas, M.; Webber, C.; Patterson, S.; Gault, S.; van Werkhoven, C.H.; van Deursen, A.M.; Sanders, E.A.; Verheij, T.J.; et al. Polysaccharide conjugate vaccine against pneumococcal pneumonia in adults. N. Engl. J. Med. 2015, 372, 1114–1125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feldman, C.; Anderson, R. Review: Current and new generation pneumococcal vaccines. J. Infect. 2014, 69, 309–325. [Google Scholar] [CrossRef] [Green Version]
- Hill, A.B.; Beitelshees, M.; Nayerhoda, R.; Pfeifer, B.A.; Jones, C.H. Engineering a next-generation glycoconjugate-like Streptococcus pneumoniae vaccine. ACS Infect Dis. 2018, 4, 1553–1563. [Google Scholar] [CrossRef]
- Eskola, J.; Black, S.; Shinefield, H. Pneumococcal Conjugate Vaccines; W. B. Saunders: Philadelphia, PA, USA, 2004. [Google Scholar]
- Hausdorff, W.P.; Hoet, B.; Adegbola, R.A. Predicting the impact of new pneumococcal conjugate vaccines: Serotype composition is not enough. Expert Rev. Vaccines 2015, 14, 413–428. [Google Scholar] [CrossRef]
- Tin Tin Htar, M.; Christopoulou, D.; Schmitt, H.J. Pneumococcal serotype evolution in Western Europe. BMC Infect Dis. 2015, 15, 419. [Google Scholar] [CrossRef] [Green Version]
- Jones, C.H.; Zhang, G.; Nayerhoda, R.; Beitelshees, M.; Hill, A.; Rostami, P.; Li, Y.; Davidson, B.A.; Knight, P., 3rd; Pfeifer, B.A. Comprehensive vaccine design for commensal disease progression. Sci. Adv. 2017, 3, e1701797. [Google Scholar] [CrossRef] [Green Version]
- Gruber, W.C.; Scott, D.A.; Emini, E.A. Development and clinical evaluation of Prevnar 13, a 13-valent pneumocococcal CRM197 conjugate vaccine. Ann. N. Y. Acad. Sci. 2012, 1263, 15–26. [Google Scholar] [CrossRef] [PubMed]
- McFetridge, R.; Meulen, A.S.; Folkerth, S.D.; Hoekstra, J.A.; Dallas, M.; Hoover, P.A.; Marchese, R.D.; Zacholski, D.M.; Watson, W.J.; Stek, J.E.; et al. Safety, tolerability, and immunogenicity of 15-valent pneumococcal conjugate vaccine in healthy adults. Vaccine 2015, 33, 2793–2799. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Hill, A.; Beitelshees, M.; Shao, S.; Lovell, J.F.; Davidson, B.A.; Knight, P.R., 3rd; Hakansson, A.P.; Pfeifer, B.A.; Jones, C.H. Directed vaccination against pneumococcal disease. Proc. Natl. Acad. Sci. USA 2016, 113, 6898–6903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beitelshees, M.; Li, Y.; Pfeifer, B.A. Enhancing vaccine effectiveness with delivery technology. Curr. Opin. Biotechnol. 2016, 42, 24–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacNair, J.E.; Desai, T.; Teyral, J.; Abeygunawardana, C.; Hennessey, J.P., Jr. Alignment of absolute and relative molecular size specifications for a polyvalent pneumococcal polysaccharide vaccine (PNEUMOVAX 23). Biologicals 2005, 33, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Laskowich, E.R.; Arumugham, R.G.; Kaiser, R.E.; MacMichael, G.J. Determination of saccharide content in pneumococcal polysaccharides and conjugate vaccines by GC-MSD. Anal. Biochem. 2005, 347, 262–274. [Google Scholar] [CrossRef]
- Hill, A.; Beitelshees, M.; Pfeifer, B.A.; Jones, C.H. Standardizing pneumococcal biofilm release to PncO expression, a predictive measurement of virulence. Infect Immun. 2018, 86, e00494-18. [Google Scholar] [CrossRef] [Green Version]
- Nayerhoda, R.; Hill, A.; Beitelshees, M.; Jones, C.; Pfeifer, B. Design variation of a dual-antigen liposomal vaccine carrier system. Materials (Basel) 2019, 12, 2809. [Google Scholar] [CrossRef] [Green Version]
- Apostolico Jde, S.; Lunardelli, V.A.; Coirada, F.C.; Boscardin, S.B.; Rosa, D.S. Adjuvants: Classification, Modus Operandi, and Licensing. J. Immunol. Res. 2016, 2016, 1459394. [Google Scholar] [CrossRef] [Green Version]
- Reed, S.G.; Orr, M.T.; Fox, C.B. Key roles of adjuvants in modern vaccines. Nat. Med. 2013, 19, 1597–1608. [Google Scholar] [CrossRef]
- Di Pasquale, A.; Preiss, S.; Tavares Da Silva, F.; Garcon, N. Vaccine Adjuvants: From 1920 to 2015 and beyond. Vaccines (Basel) 2015, 3, 320–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marks, L.R.; Davidson, B.A.; Knight, P.R.; Hakansson, A.P. Interkingdom signaling induces Streptococcus pneumoniae biofilm dispersion and transition from asymptomatic colonization to disease. mBio 2013, 4, e00438-13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettigrew, M.M.; Marks, L.R.; Kong, Y.; Gent, J.F.; Roche-Hakansson, H.; Hakansson, A.P. Streptococcus pneumoniae and influenza: Dynamic changes in the pneumococcal transcriptome during transition from biofilm formation to invasive disease. Infect Immun. 2014, 82, 4607–4619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beitelshees, M.; Hill, A.; Jones, C.H.; Pfeifer, B.A. Phenotypic variation during biofilm formation: Implications for anti-biofilm therapeutic design. Mater. (Basel) 2018, 11, 1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, C.H.; Hill, A.; Chen, M.; Pfeifer, B.A. Contemporary approaches for nonviral gene therapy. Discov. Med. 2015, 19, 447–454. [Google Scholar]
- Gomez, P.L.; Robinson, J.M.; Rogalewicz, J.A. Vaccine Manufacturing; Elsevier: Beijing, China, 2013. [Google Scholar]
- Jones, C.H.; Chen, M.; Gollakota, A.; Ravikrishnan, A.; Zhang, G.; Lin, S.; Tan, M.; Cheng, C.; Lin, H.; Pfeifer, B.A. Structure-Function Assessment of Mannosylated Poly (beta-amino esters) upon Targeted Antigen Presenting Cell Gene Delivery. Biomacromolecules 2015, 16, 1534–1541. [Google Scholar] [CrossRef]
- Jones, C.H.; Gollakota, A.; Chen, M.; Chung, T.C.; Ravikrishnan, A.; Zhang, G.; Pfeifer, B.A. Influence of molecular weight upon mannosylated bio-synthetic hybrids for targeted antigen presenting cell gene delivery. Biomaterials 2015, 58, 103–111. [Google Scholar] [CrossRef] [Green Version]
- Coffman, R.L.; Sher, A.; Seder, R.A. Vaccine Adjuvants: Putting Innate Immunity to Work. Immunity 2010, 33, 492–503. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| LEPS Particle | Zeta Potential (mV) |
|---|---|
| Encapsulating PS 19F (Pre-protein Binding) | −12.6 |
| Encapsulating PS 3 (Pre-protein Binding) | −28.7 |
| Encapsulating PS 4 (Pre-protein Binding) | −30.7 |
| Encapsulating PS 19F (Post-GFP Binding with Alum) | −17.3 |
| Encapsulating PS 3 (Post-GFP Binding with Alum) | −26.7 |
| Encapsulating PS 3 (Post-PncO Binding with Alum) | −8.5 |
| Encapsulating PS 4 (Post-GFP Binding with Alum) | −12.6 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nayerhoda, R.; Park, D.; Jones, C.; Bou Ghanem, E.N.; Pfeifer, B.A. Extended Polysaccharide Analysis within the Liposomal Encapsulation of Polysaccharides System. Materials 2020, 13, 3320. https://doi.org/10.3390/ma13153320
Nayerhoda R, Park D, Jones C, Bou Ghanem EN, Pfeifer BA. Extended Polysaccharide Analysis within the Liposomal Encapsulation of Polysaccharides System. Materials. 2020; 13(15):3320. https://doi.org/10.3390/ma13153320
Chicago/Turabian StyleNayerhoda, Roozbeh, Dongwon Park, Charles Jones, Elsa N. Bou Ghanem, and Blaine A. Pfeifer. 2020. "Extended Polysaccharide Analysis within the Liposomal Encapsulation of Polysaccharides System" Materials 13, no. 15: 3320. https://doi.org/10.3390/ma13153320
APA StyleNayerhoda, R., Park, D., Jones, C., Bou Ghanem, E. N., & Pfeifer, B. A. (2020). Extended Polysaccharide Analysis within the Liposomal Encapsulation of Polysaccharides System. Materials, 13(15), 3320. https://doi.org/10.3390/ma13153320