Determination of Formation Energies and Phase Diagrams of Transition Metal Oxides with DFT+U


Abstract
:1. Introduction
- The molecular state is the natural reference phase for oxygen. The well-known over-binding of the O2 molecule in LDA and GGA [17] introduces an error in the formation energies of oxides. A correction scheme was proposed by Wang et al. [13], which builds on a comparison between the theoretical and experimental values of formation energies for a series of non-TM oxide compounds. In the Materials Project (MP) database [18], for example, this scheme is applied and denoted by the term anion corrections.
- The uncompensated electronic self-interaction imposed by approximate exchange-correlation functionals immanent in DFT methods, especially in the case of TMOs, where strongly correlated TM-d-electrons form the valence band, leads to incorrect total energies and underestimated band gaps. There are different approaches to cope with this inaccuracy, such as the use of hybrid functionals [19], a self-interaction correction (SIC) [20], or a Hubbard-U correction that acts on the d-electrons of the TM as an effective potential (DFT+U) [21].
- In case that the DFT+U method is used to obtain a corrected total energy of a TMO, the formation energy contains an error if the total energies of the elemental reference phases were calculated by LDA or GGA, as it is generally done for the elemental phases and for compounds not containing the TM elements. The error can be systematically corrected by applying a method worked out by Jain et al. [14], where the total energies from the DFT+U calculations are shifted by a constant amount per TM atom. The approach is used in the MP database [18], denoted by the term advanced corrections, to obtain formation energies of TM-containing compounds.
2. Methods
2.1. DFT(+U) Calculations of Total Energies
2.2. Correction of the O2 Over-Binding
2.3. Energy Correction Method for the Combined DFT and DFT+U Results
2.4. Derivation of Phase Diagrams
3. Results and Discussion
3.1. O2 Correction
3.2. Energy Correction for the Combined DFT and DFT+U Results
3.3. Determination of U
3.4. Band Gaps of the Iron Compounds
3.5. Phase Diagrams of LaFeO3, Li5FeO4, and NaFeO2
3.5.1. LaFeO3
3.5.2. Li5FeO4
3.5.3. NaFeO2
4. Summary and Conclusions
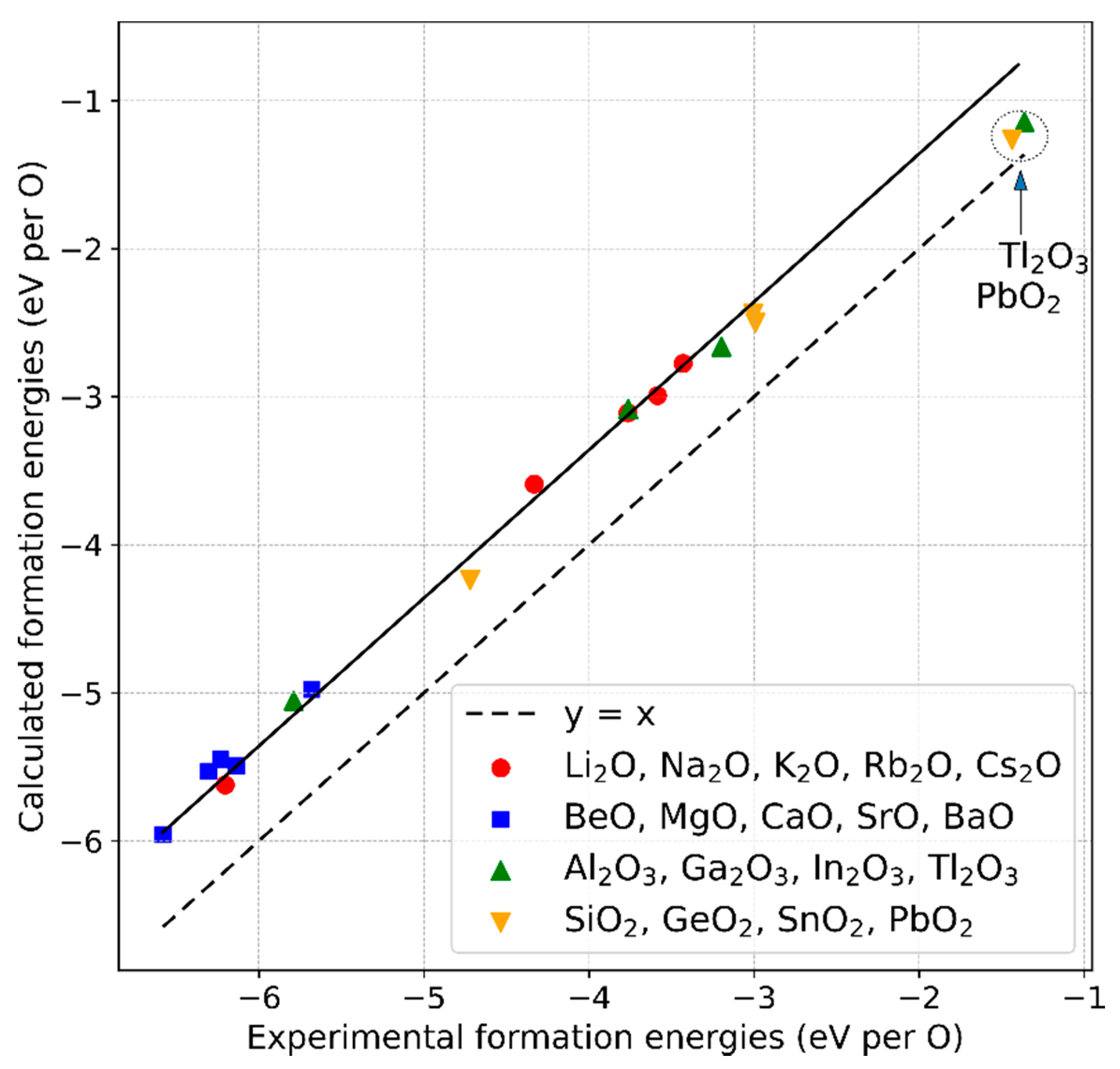
- The method to correct the formation energy error due to the over-binding of the O2 molecule remains valid if a larger set of non-transition metal oxide compounds from the groups I to IV of the periodic table is considered instead of the previously chosen smaller subset. The magnitude of the energy correction we derived (0.64 eV) agrees within 0.1 eV with the reported values, which reflects the uncertainty range we determined for the approach. Experimental enthalpies of the formations for the considered compounds can be reasonably reproduced within 0% to 5%, except for Tl2O3 and PbO2, for which the values deviate more. A possible explanation is given.
- For the binary iron oxide compounds, we confirmed that it is well-justified to correct the error arising in combining the results of DFT and DFT+U calculations by adding a value proportional to the Fe content to the formation energy. While it was originally derived considering only the binary oxides for a fixed U value for Fe, we strengthened and generalized the scheme by (a) taking into account ternary compounds, (b) not a priori constraining the correction value to be zero for hypothetical compounds without Fe, and (c) considering different values of U.
- Our U-dependent correction value offers a new possibility to determine an optimal U value for Fe for which the experimental formation energies are reproduced best. With this approach, we confirmed the frequently used value of U ≈ 4 eV, which additionally turned out to reproduce experimental band gaps of the considered compounds within 0.3 eV.
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A: List of Calculated and Experimental Formation Energies

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | SG | |||||
|---|---|---|---|---|---|---|
| Non-TM oxides | ||||||
| Li2O | −6.21 | −5.62 | −6.26 | - | 0.8 | |
| Na2O | −4.33 | −3.59 | −4.23 | - | 2.3 | |
| K2O | −3.76 | −3.11 | −3.75 | - | 0.3 | |
| Rb2O | −3.43 | −2.77 | −3.42 | - | 0.3 | |
| Cs2O | −3.59 | −2.99 | −3.63 | - | 1.1 | |
| BeO | −6.31 | −5.53 | −6.17 | - | 2.2 | |
| MgO | −6.23 | −5.45 | −6.09 | - | 2.2 | |
| CaO | −6.58 | −5.96 | −6.60 | - | 0.3 | |
| SrO | −6.14 | −5.49 | −6.14 | - | 0.0 | |
| BaO | −5.68 | −4.98 | −5.62 | - | 1.1 | |
| Al2O3 | −17.37 | −15.16 | −17.08 | - | 1.7 | |
| Ga2O3 | −11.29 | −9.25 | −11.17 | - | 1.1 | |
| In2O3 | −9.60 | −7.99 | −9.91 | - | 3.2 | |
| Tl2O3 | −4.09 | −3.43 | −5.35 | - | 30.8 | |
| SiO2 | −9.44 | −8.48 | −9.76 | - | 3.4 | |
| GeO2 | −6.01 | −4.88 | −6.16 | - | 2.5 | |
| SnO2 | −5.99 | −5.00 | −6.28 | - | 4.8 | |
| PbO2 | −2.88 | −2.52 | −3.80 | - | 31.9 | |
| TM oxides | ||||||
| FeO | −2.82 | −0.31 | −0.95 | −2.83 | 0.4 | |
| Fe2O3 | −8.56 | −3.04 | −4.96 | −8.69 | 1.5 | |
| Fe3O4 | −11.62 | −3.35 | −5.92 | −11.53 | 0.8 | |
| LaFeO3 | −14.24 | −10.59 | −12.51 | −14.27 | 0.2 | |
| Li5FeO4 | −20.21 | −16.34 | −18.91 | −20.45 | 1.2 | |
| NaFeO2 | −7.23 | −3.96 | −5.24 | −7.05 | 2.5 | |
Appendix B: Dependence of Oxidation Energies on U
Appendix C: Comparison of Methods for the Determination of U

Appendix D: Phase Diagram with Respect to the Precursor Compounds
References
- Rao, C.N.R. Transition Metal Oxides. Annu. Rev. Phys. Chem. 1989, 40, 291–326. [Google Scholar] [CrossRef]
- Fang, S.; Bresser, D.; Passerini, S. Transition Metal Oxide Anodes for Electrochemical Energy Storage in Lithium- and Sodium-Ion Batteries. Adv. Energy Mater. 2020, 10, 1902485. [Google Scholar] [CrossRef]
- Liu, Q.; Hu, Z.; Chen, M.; Zou, C.; Jin, H.; Wang, S.; Chou, S.-L.; Dou, S.-X. Recent Progress of Layered Transition Metal Oxide Cathodes for Sodium-Ion Batteries. Small 2019, 15, 1805381. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Jaffer, S.; Yu, H. Transition Metal Oxides for Sodium-Ion Batteries. Energy Storage Mater. 2016, 5, 116–131. [Google Scholar] [CrossRef]
- Zhang, J.; Yu, A. Nanostructured Transition Metal Oxides as Advanced Anodes for Lithium-Ion Batteries. Sci. Bull. 2015, 60, 823–838. [Google Scholar] [CrossRef] [Green Version]
- Jun, A.; Kim, J.; Shin, J.; Kim, G. Perovskite as a Cathode Material: A Review of Its Role in Solid-Oxide Fuel Cell Technology. ChemElectroChem 2016, 3, 511–530. [Google Scholar] [CrossRef]
- Goswami, C.; Hazarika, K.K.; Bharali, P. Transition Metal Oxide Nanocatalysts for Oxygen Reduction Reaction. Mater. Sci. Energy Technol. 2018, 1, 117–128. [Google Scholar] [CrossRef]
- Kısa, A.E.; Demircan, O. Transition Metal Doped Solid Oxide Fuel Cell Cathodes. J. Turk. Chem. Soc. 2018, 5, 1153–1168. [Google Scholar] [CrossRef]
- Chen, D.; Chen, C.; Baiyee, Z.M.; Shao, Z.; Ciucci, F. Nonstoichiometric Oxides as Low-Cost and Highly-Efficient Oxygen Reduction/Evolution Catalysts for Low-Temperature Electrochemical Devices. Chem. Rev. 2015, 115, 9869–9921. [Google Scholar] [CrossRef]
- Kuklja, M.M.; Kotomin, E.A.; Merkle, R.; Mastrikov, Y.A.; Maier, J. Combined Theoretical and Experimental Analysis of Processes Determining Cathode Performance in Solid Oxide Fuel Cells. Phys. Chem. Chem. Phys. 2013, 15, 5443–5471. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhou, W.; Yu, J.; Chen, Y.; Liu, M.; Shao, Z. Enhancing Electrocatalytic Activity of Perovskite Oxides by Tuning Cation Deficiency for Oxygen Reduction and Evolution Reactions. Chem. Mater. 2016, 28, 1691–1697. [Google Scholar] [CrossRef]
- Kirklin, S.; Saal, J.E.; Meredig, B.; Thompson, A.; Doak, J.W.; Aykol, M.; Rühl, S.; Wolverton, C. The Open Quantum Materials Database (OQMD): Assessing the Accuracy of DFT Formation Energies. npj Comp. Mater. 2015, 1, 15010. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Maxisch, T.; Ceder, G. Oxidation Energies of Transition Metal Oxides within the GGA+U Framework. Phys. Rev. B 2006, 73, 195107. [Google Scholar] [CrossRef] [Green Version]
- Jain, A.; Hautier, G.; Ong, S.P.; Moore, C.J.; Fischer, C.C.; Persson, K.A.; Ceder, G. Formation Enthalpies by Mixing GGA and GGA + U Calculations. Phys. Rev. B 2011, 84, 045115. [Google Scholar] [CrossRef] [Green Version]
- Hautier, G.; Ong, S.P.; Jain, A.; Moore, C.J.; Ceder, G. Accuracy of Density Functional Theory in Predicting Formation Energies of Ternary Oxides from Binary Oxides and Its Implication on Phase Stability. Phys. Rev. B 2012, 85, 155208. [Google Scholar] [CrossRef] [Green Version]
- Deml, A.M.; O’Hayre, R.; Wolverton, C.; Stevanović, V. Predicting Density Functional Theory Total Energies and Enthalpies of Formation of Metal-Nonmetal Compounds by Linear Regression. Phys. Rev. B 2016, 93, 085142. [Google Scholar] [CrossRef] [Green Version]
- Patton, D.C.; Porezag, D.V.; Pederson, M.R. Simplified Generalized-Gradient Approximation and Anharmonicity: Benchmark Calculations on Molecules. Phys. Rev. B 1997, 55, 7454–7459. [Google Scholar]
- Jain, A.; Ong, S.P.; Hautier, G.; Chen, W.; Richards, W.D.; Dacek, S.; Cholia, S.; Gunter, D.; Skinner, D.; Ceder, G.; et al. Commentary: The Materials Project: A Materials Genome Approach to Accelerating Materials Innovation. APL Mater. 2013, 1, 011002. [Google Scholar] [CrossRef] [Green Version]
- Garza, A.J.; Scuseria, G.E. Predicting Band Gaps with Hybrid Density Functionals. J. Phys. Chem. Lett. 2016, 7, 4165–4170. [Google Scholar]
- Perdew, J.P.; Zunger, A. Self-Interaction Correction to Density-Functional Approximations for Many-Electron Systems. Phys. Rev. B 1981, 23, 5048. [Google Scholar]
- Anisimov, V.I.; Zaanen, J.; Andersen, O.K. Band Theory and Mott Insulators: Hubbard U Instead of Stoner I. Phys. Rev. B 1991, 44, 943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rollmann, G.; Rohrbach, A.; Entel, P.; Hafner, J. First-Principles Calculation of the Structure and Magnetic Phases of Hematite. Phys. Rev. B 2004, 69, 165107. [Google Scholar] [CrossRef] [Green Version]
- Toroker, M.C.; Kanan, D.K.; Alidoust, N.; Isseroff, L.Y.; Liao, P.; Carter, E.A. First Principles Scheme to Evaluate Band Edge Positions in Potential Transition Metal Oxide Photocatalysts and Photoelectrodes. Phys. Chem. Chem. Phys. 2011, 13, 16644–16654. [Google Scholar] [CrossRef] [PubMed]
- Aykol, M.; Wolverton, C. Local Environment Dependent GGA+U Method for Accurate Thermochemistry of Transition Metal Compounds. Phys. Rev. B 2014, 90, 115105. [Google Scholar] [CrossRef] [Green Version]
- Noh, J.; Osman, O.I.; Aziz, S.G.; Winget, P.; Brédas, J.L. A Density Functional Theory Investigation of the Electronic Structure and Spin Moments of Magnetite. Sci. Technol. Adv. Mater. 2014, 15, 044202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eom, T.; Lim, H.-K.; Goddard, W.A., III; Kim, H. First-Principles Study of Iron Oxide Polytypes: Comparison of GGA+ U and Hybrid Functional Method. J. Phys. Chem. C 2015, 119, 556–562. [Google Scholar] [CrossRef] [Green Version]
- Yabuuchi, N.; Yoshida, H.; Komaba, S. Crystal Structures and Electrode Performance of Alpha-NaFeO2 for Rechargeable Sodium Batteries. Electrochemistry 2012, 80, 716–719. [Google Scholar] [CrossRef] [Green Version]
- Kataoka, R.; Kuratani, K.; Kitta, M.; Takeichi, N.; Kiyobayashi, T.; Tabuchi, M. Influence of the Preparation Methods on the Electrochemical Properties and Structural Changes of Alpha-Sodium Iron Oxide as a Positive Electrode Material for Rechargeable Sodium Batteries. Electrochim. Acta 2015, 182, 871–877. [Google Scholar] [CrossRef]
- Zhan, C.; Yao, Z.; Lu, J.; Ma, L.; Maroni, V.A.; Li, L.; Lee, E.; Alp, E.E.; Wu, T.; Wen, J.; et al. Enabling the High Capacity of Lithium-Rich Anti- Fluorite Lithium Iron Oxide by Simultaneous Anionic and Cationic Redox. Nat. Energy 2017, 2, 963–971. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Blöchl, P.E.; Jepsen, O.; Andersen, O.K. Improved Tetrahedron Method for Brillouin-Zone Integrations. Phys. Rev. B 1994, 49, 16223–16233. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Joubert, D. From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Perdew, J.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dudarev, S.L.; Botton, G.A.; Savrasov, S.Y.; Humphreys, C.J.; Sutton, A.P. Electron-Energy-Loss Spectra and the Structural Stability of Nickel Oxide: An LSDA+U Study. Phys. Rev. B 1998, 57, 1505. [Google Scholar] [CrossRef]
- Samanta, A.; Weinan, E.; Zhang, S.B. Method for Defect Stability Diagram from Ab Initio Calculations: A Case Study of SrTiO3. Phys. Rev. B 2012, 86, 195107. [Google Scholar] [CrossRef] [Green Version]
- Chase, M.W.J. NIST-JANAF Themochemical Tables, Fourth Edition, J. Phys. Chem. Ref. Data, Monograph 9; American Institute of Physics: College Park, MD, USA, 1998. [Google Scholar]
- Haynes, W.M. (Ed.) CRC Handbook of Chemistry and Physics, 92nd ed.; CRC Press: Boca Raton, FL, USA, 2011. [Google Scholar]
- Holleman, A.F.; Wiberg, E.; Wiberg, N. Lehrbuch Der Anorganischen Chemie, 101st ed.; Walter de Gruyter: Berlin, Germany, 1995. [Google Scholar]
- Wade, K.; Banister, A.J. The Chemistry of Aluminium, Gallium, Indium and Thallium, 1st ed.; Pergamon Press: Oxford, UK, 1973. [Google Scholar]
- Gautam, G.S.; Carter, E.A. Evaluating Transition Metal Oxides within DFT-SCAN and SCAN+U Frameworks for Solar Thermochemical Applications. Phys. Rev. Mater. 2018, 2, 095401. [Google Scholar] [CrossRef]
- Xu, Z.; Joshi, Y.V.; Raman, S.; Kitchin, J.R. Accurate Electronic and Chemical Properties of 3d Transition Metal Oxides Using a Calculated Linear Response U and a DFT + U (V) Method. J. Chem. Phys. 2015, 142, 144701. [Google Scholar] [CrossRef]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. QUANTUM ESPRESSO: A Modular and Open-Source Software Project for Quantum Simulations of Materials. J. Phys. Condens. Matter 2009, 21, 395502. [Google Scholar] [CrossRef]
- Cococcioni, M.; de Gironcoli, S. Linear Response Approach to the Calculation of the Effective Interaction Parameters in the LDA+U Method. Phys. Rev. B 2005, 71, 035105. [Google Scholar] [CrossRef] [Green Version]
- Garrity, K.F.; Bennett, J.W.; Rabe, K.M.; Vanderbilt, D. Pseudopotentials for High-Throughput DFT Calculations. Comp. Mater. Sci. 2014, 81, 446–452. [Google Scholar] [CrossRef] [Green Version]
- Glasscock, J.A.; Barnes, P.R.F.; Plumb, I.C.; Bendavid, A.; Martin, P.J. Structural, Optical and Electrical Properties of Undoped Polycrystalline Hematite Thin Films Produced Using Filtered Arc Deposition. Thin Solid Films 2008, 516, 1716–1724. [Google Scholar] [CrossRef]
- Gilbert, B.; Frandsen, C.; Maxey, E.R.; Sherman, D.M. Band-Gap Measurements of Bulk and Nanoscale Hematite by Soft x-Ray Spectroscopy. Phys. Rev. B Condens. Matter Mater. Phys. 2009, 79. [Google Scholar] [CrossRef] [Green Version]
- Schrettle, F.; Kant, C.; Lunkenheimer, P.; Mayr, F.; Deisenhofer, J.; Loidl, A. Wüstite: Electric, Thermodynamic and Optical Properties of FeO. Eur. Phys. J. B 2012, 85. [Google Scholar] [CrossRef] [Green Version]
- Arima, T.; Tokura, Y.; Torrance, J.B. Variation of Optical Gaps in Perovskite-Type 3d Transition-Metal Oxides. Phys. Rev. B 1993, 48, 17006–17009. [Google Scholar] [CrossRef]
- Scafetta, M.D.; Cordi, A.M.; Rondinelli, J.M.; May, S.J. Band Structure and Optical Transitions in LaFeO3: Theory and Experiment. J. Phys. Condens. Matter 2014, 26, 505502. [Google Scholar] [CrossRef]
- Ran, F.Y.; Tsunemaru, Y.; Hasegawa, T.; Takeichi, Y.; Harasawa, A.; Yaji, K.; Kim, S.; Kakizaki, A. Angle-Resolved Photoemission Study of Fe3O4 (001) Films across Verwey Transition. J. Phys. D. Appl. Phys. 2012, 45, 1–6. [Google Scholar] [CrossRef]
- Hoang, K.; Oh, M.; Choi, Y. Electronic Structure and Properties of Lithium-Rich Complex Oxides. ACS Appl. Electron. Mater. 2019, 1, 75–81. [Google Scholar] [CrossRef]
- Kuganathan, N.; Kelaidis, N.; Chroneos, A. Defect Chemistry, Sodium Diffusion and Doping Behaviour in NaFeO2 Polymorphs as Cathode Materials for Na-Ion Batteries: A Computational Study. Materials 2019, 12, 3243. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.; Navrotsky, A.; Zhou, X.-D.; Anderson, H.U. Enthalpies of Formation of LaMO3 Perovskites (M = Cr, Fe, Co, and Ni). J. Mater. Res. 2005, 20, 191–200. [Google Scholar] [CrossRef]
- Jacob, K.T.; Ranjani, R. Thermodynamic Properties of LaFeO3-d and LaFe12O19. Mater. Sci. Eng. B 2011, 176, 559–566. [Google Scholar] [CrossRef]
- Heifets, E.; Kotomin, E.A.; Bagaturyants, A.A.; Maier, J. Thermodynamic Stability of Stoichiometric LaFeO3 and BiFeO3: A Hybrid DFT Study. Phys. Chem. Chem. Phys. 2017, 19, 3738–3755. [Google Scholar] [CrossRef] [PubMed]
- Lany, S. Semiconducting Transition Metal Oxides. J. Phys. Condens. Matter 2015, 27, 283203. [Google Scholar] [CrossRef] [PubMed]
- Körbel, S.; Marton, P.; Elsässer, C. Formation of Vacancies and Copper Substitutionals in Potassium Sodium Niobate under Various Processing Conditions. Phys. Rev. B 2010, 81, 174115. [Google Scholar] [CrossRef]
- Erhart, P.; Albe, K. Modeling the Electrical Conductivity in BaTiO3 on the Basis of First-Principles Calculations. J. Appl. Phys. 2008, 104, 044315. [Google Scholar] [CrossRef] [Green Version]
- Pohl, J.; Albe, K. Intrinsic Point Defects in CuInSe2 and CuGaSe2 as Seen via Screened-Exchange Hybrid Density Functional Theory. Phys. Rev. B 2013, 87, 245203. [Google Scholar] [CrossRef]
- Takeshita, H.; Ohmichi, T.; Nasu, S.; Watanabe, H.; Sasayama, T.; Maeda, A.; Miyake, M.; Sano, T. Mass Spectrometric Study of the Evaporation of Li5FeO4 as a Corrosion Product in the Compatibility Experiment of Li2O Pellets with Fe-Ni-Cr Alloys. J. Nucl. Mater. 1978, 78, 281–288. [Google Scholar]
- Koehler, M.F.; Barany, R.; Kelley, K.K. Heats and Free Energies of Formation of Ferrites and Aluminates of Calcium, Magnesium, Sodium, and Lithium. Rep. Investig. RI 5711 1961, 5711, 14. [Google Scholar]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mutter, D.; Urban, D.F.; Elsässer, C. Determination of Formation Energies and Phase Diagrams of Transition Metal Oxides with DFT+U. Materials 2020, 13, 4303. https://doi.org/10.3390/ma13194303
Mutter D, Urban DF, Elsässer C. Determination of Formation Energies and Phase Diagrams of Transition Metal Oxides with DFT+U. Materials. 2020; 13(19):4303. https://doi.org/10.3390/ma13194303
Chicago/Turabian StyleMutter, Daniel, Daniel F. Urban, and Christian Elsässer. 2020. "Determination of Formation Energies and Phase Diagrams of Transition Metal Oxides with DFT+U" Materials 13, no. 19: 4303. https://doi.org/10.3390/ma13194303
APA StyleMutter, D., Urban, D. F., & Elsässer, C. (2020). Determination of Formation Energies and Phase Diagrams of Transition Metal Oxides with DFT+U. Materials, 13(19), 4303. https://doi.org/10.3390/ma13194303