Adsorptive-Oxidative Removal of Sulfides from Water by MnO2-Loaded Carboxylic Cation Exchangers


Abstract
:
1. Introduction
2. Materials and Methods
3. Results and Discussion
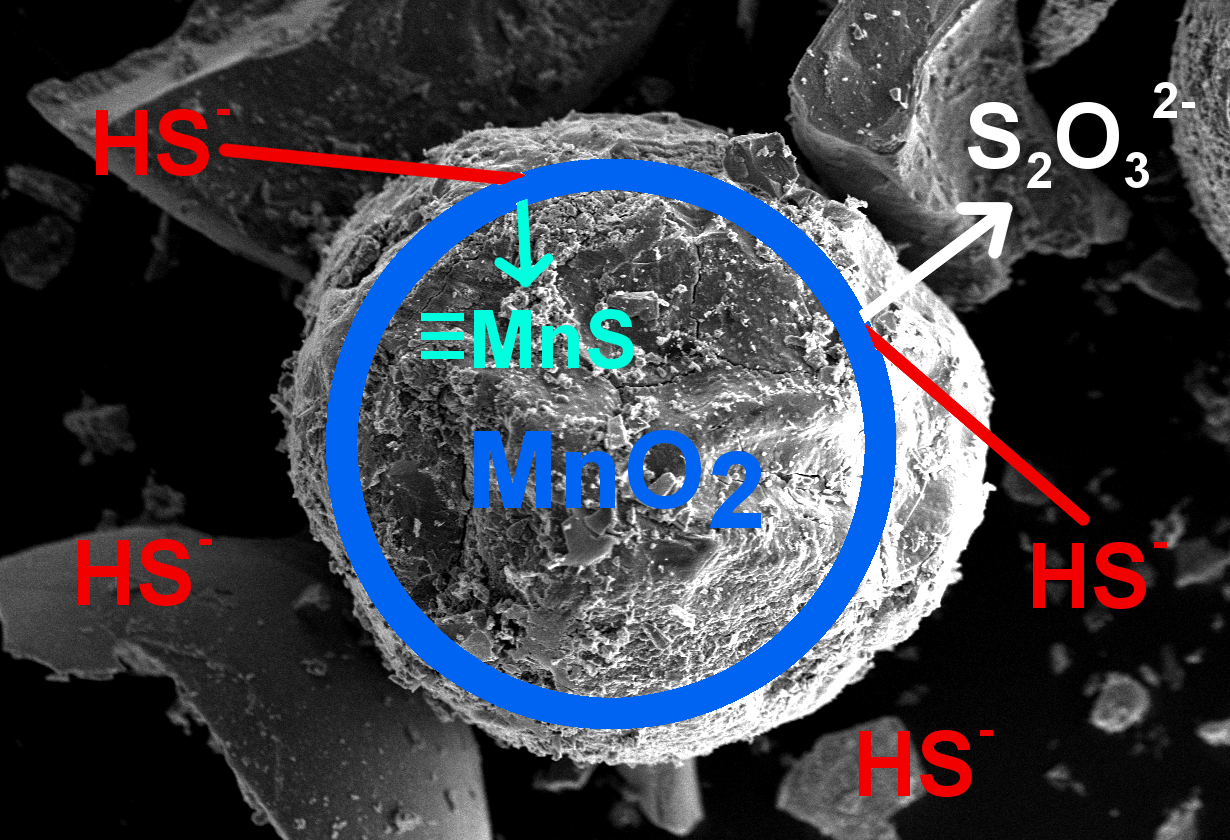
[P]-poly(acrylic-divinylbenzene) matrix, #—within the matrix
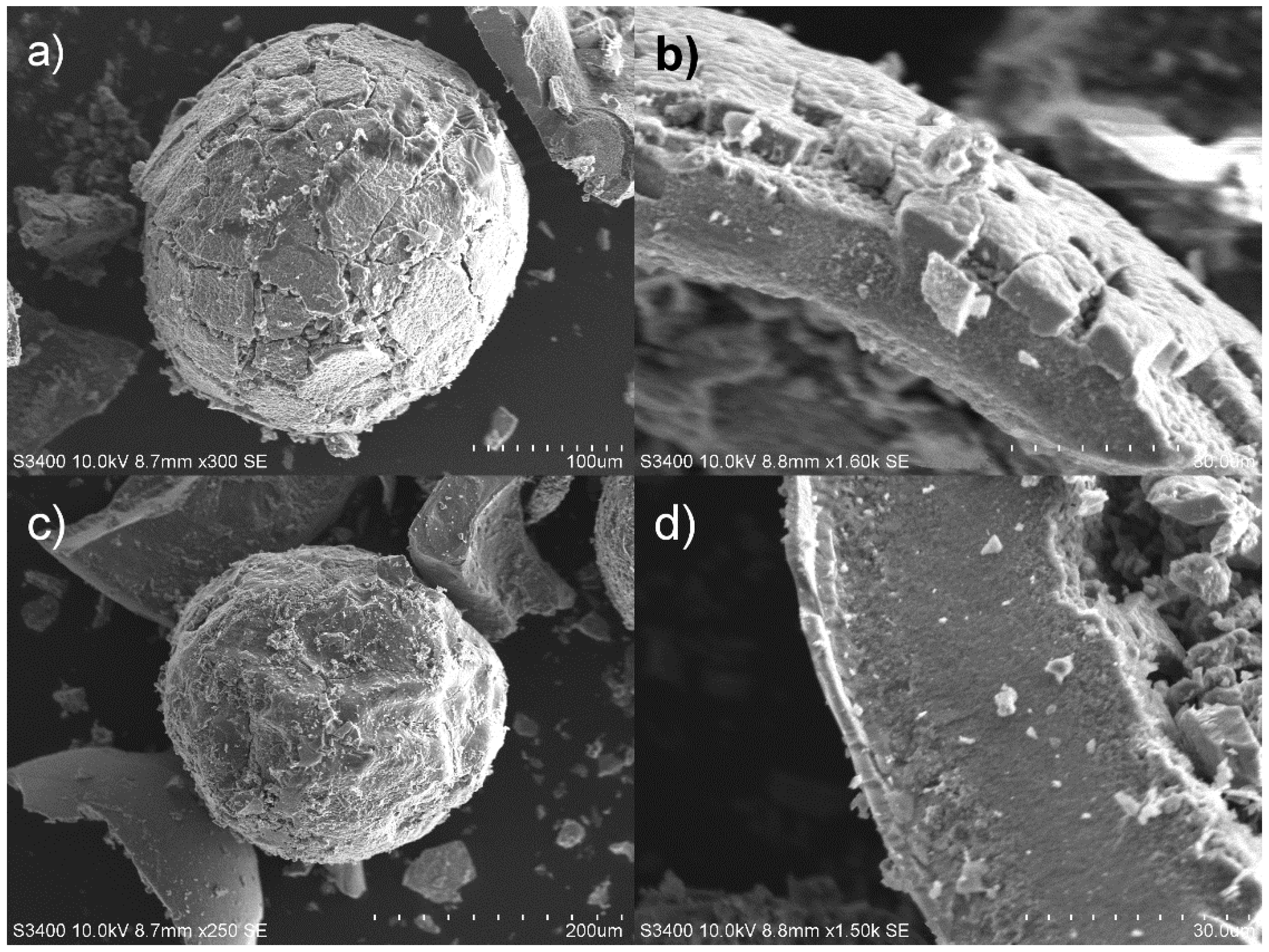
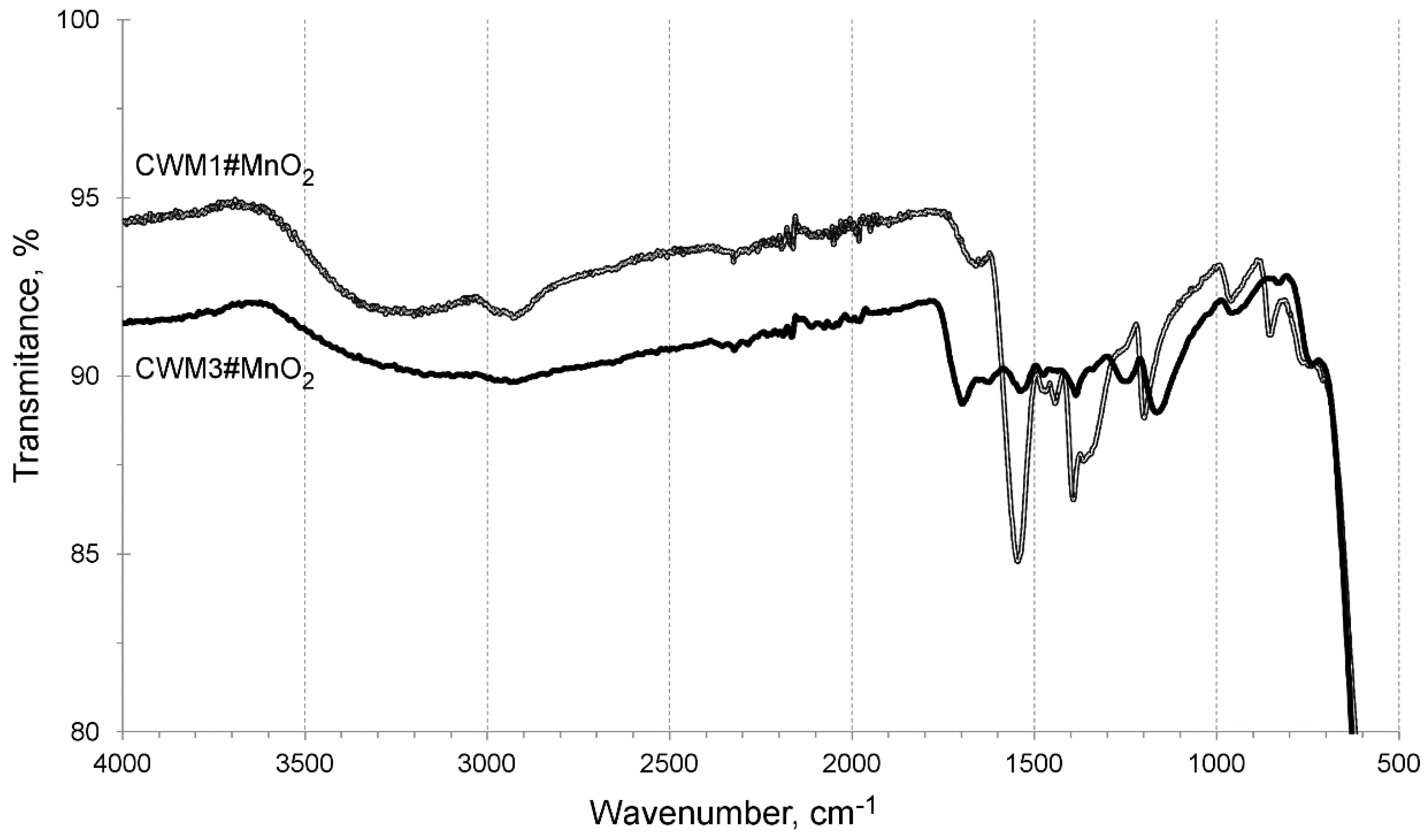
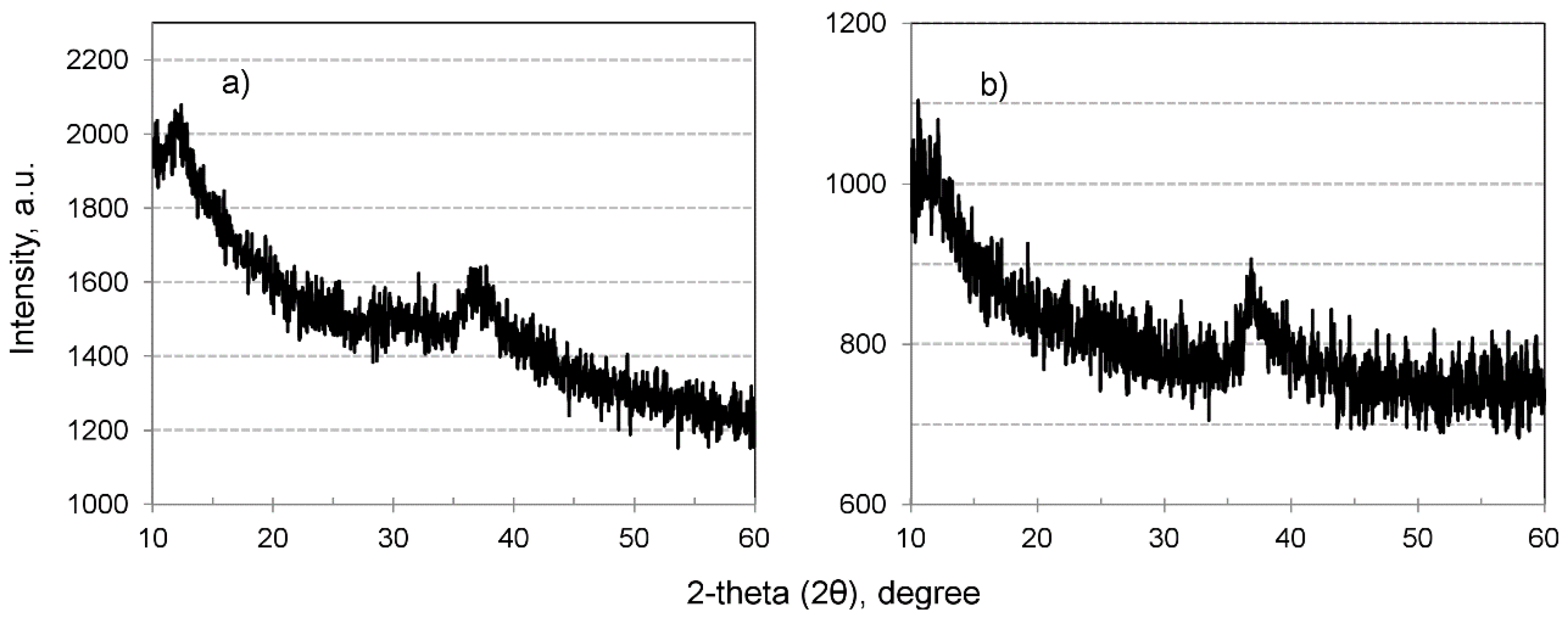
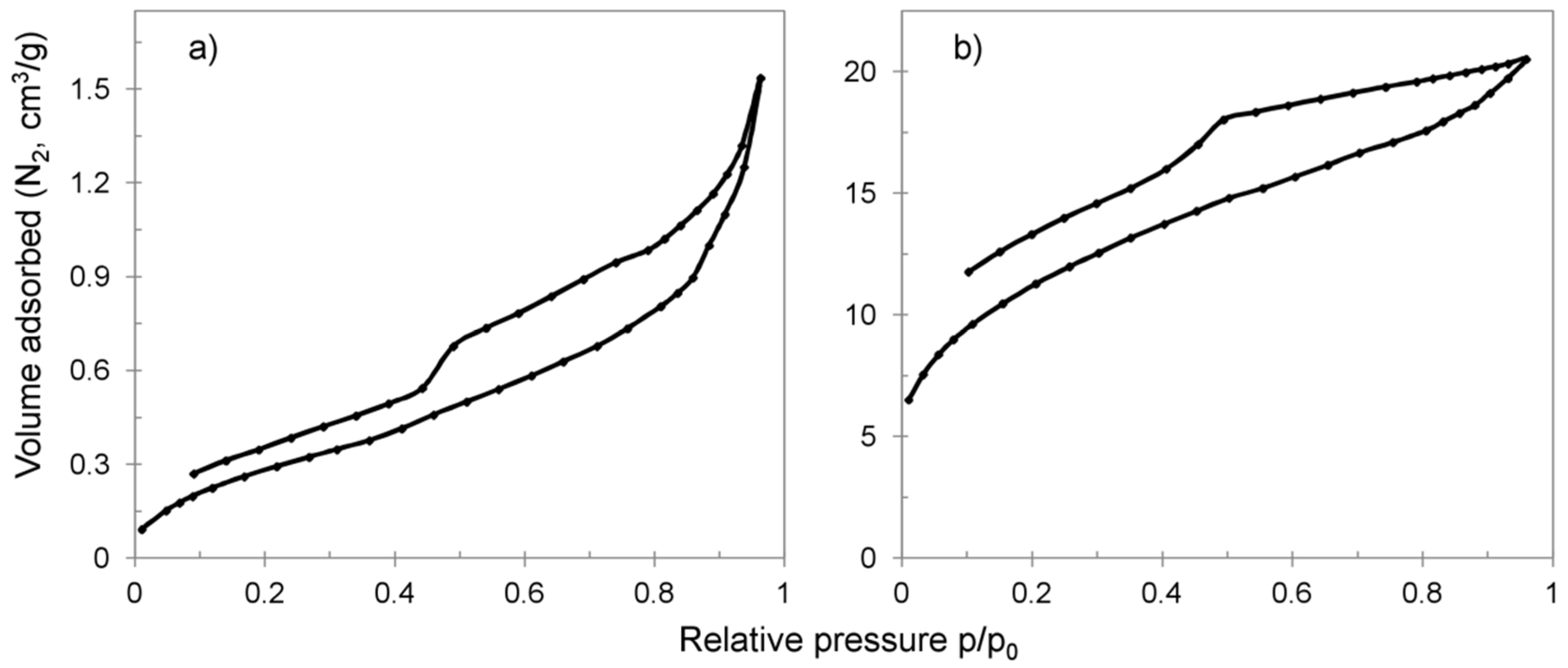
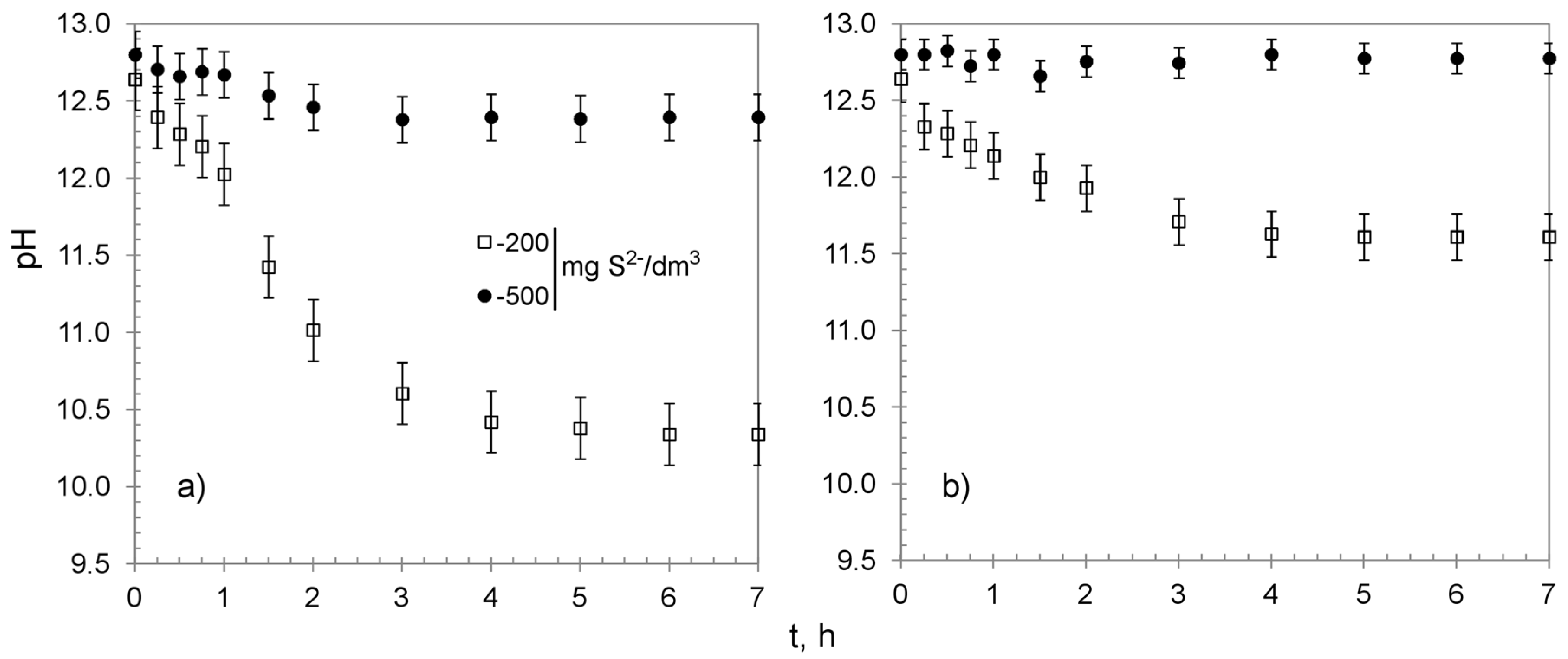
3.1. Characterization
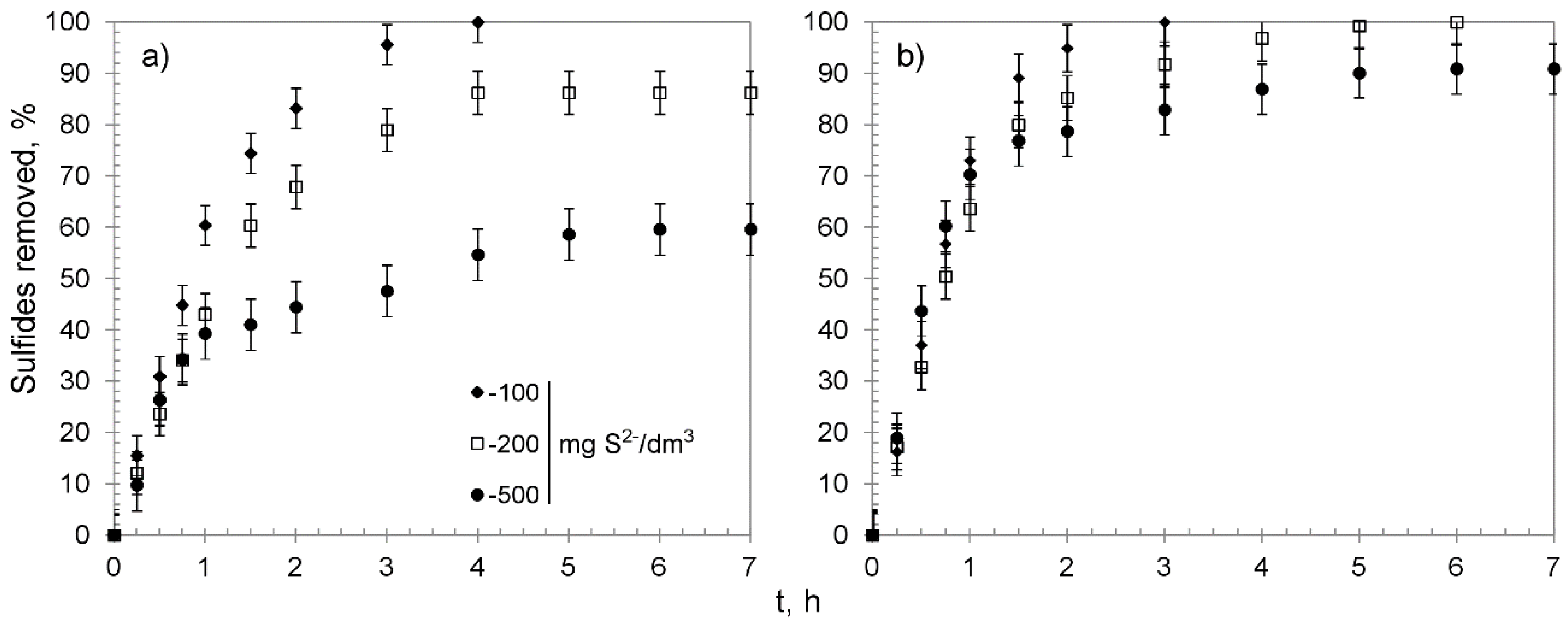
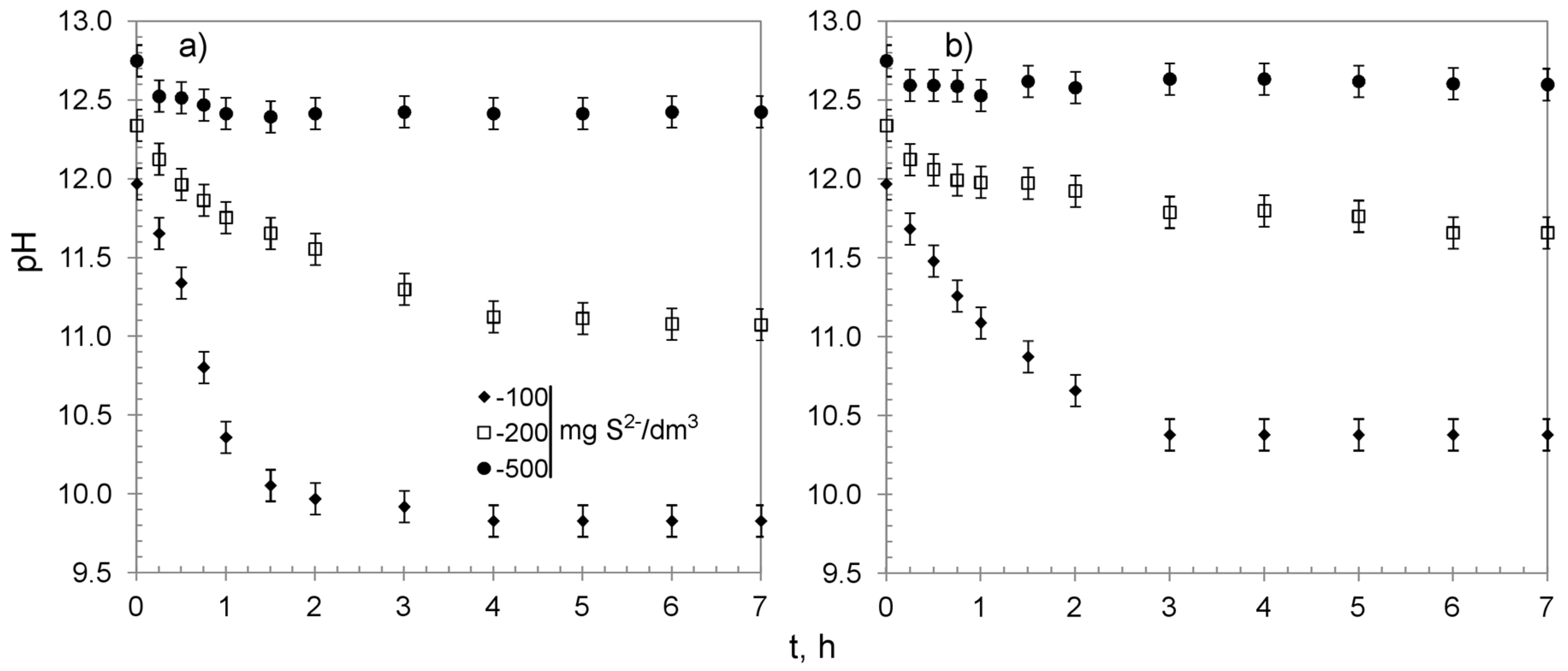
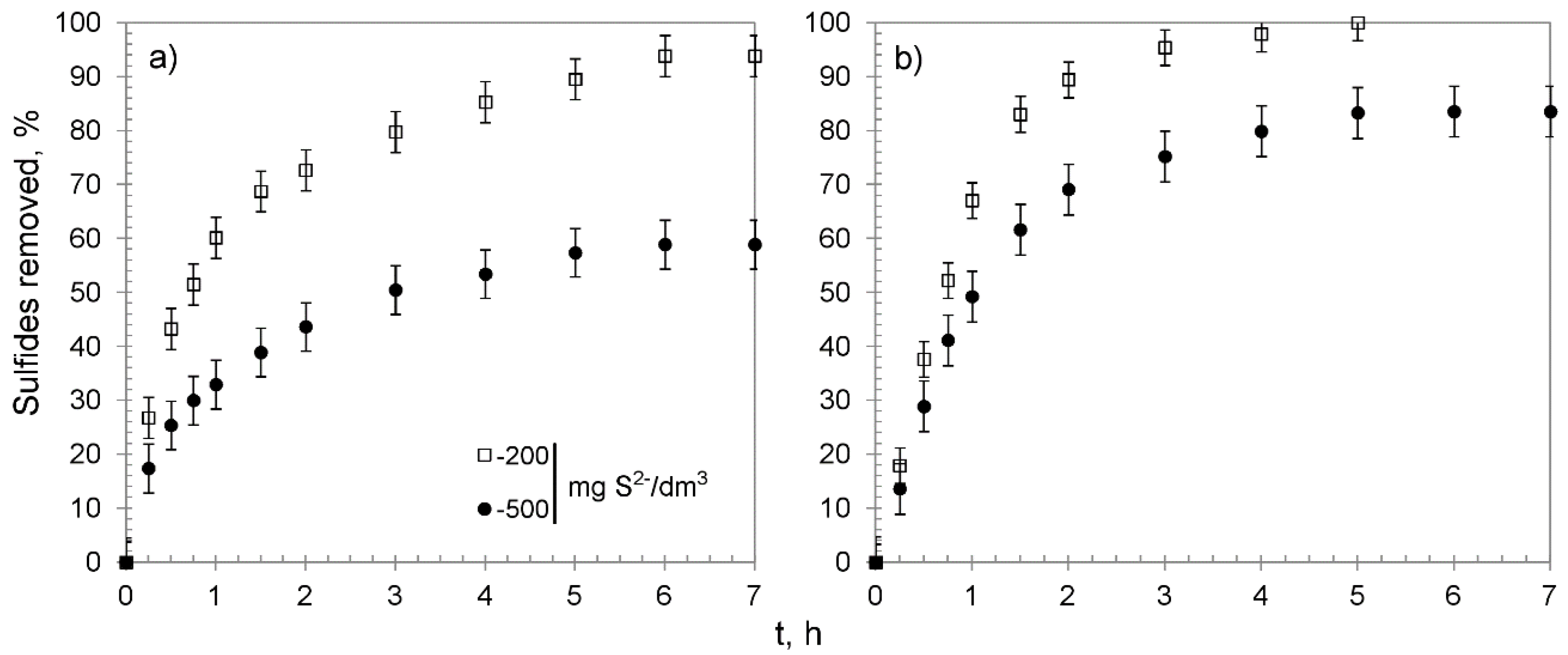
3.2. Chemisorption Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Zhen, G.; Lu, X.; Kato, H.; Zhao, Y.; Li, Y.-Y. Overview of pretreatment strategies for enhancing sewage sludge disintegration and subsequent anaerobic digestion: Current advances, full-scale application and future perspectives. Renew. Sustain. Energy Rev. 2017, 69, 559–577. [Google Scholar] [CrossRef]
- Blunden, J.; Aneja, V.P. Characterizing ammonia and hydrogen sulfide emissions from a swine waste treatment lagoon in North Carolina. Atmos. Environ. 2008, 42, 3277–3290. [Google Scholar] [CrossRef]
- Dai, X.-R.; Saha, C.K.; Ni, J.-Q.; Heber, A.J.; Blanes-Vidal, V.; Dunn, J.L. Characteristics of pollutant gas releases from swine, dairy, beef, and layer manure, and municipal wastewater. Water Res. 2015, 76, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Andriamanohiarisoamanana, F.J.; Sakamoto, Y.; Yamashiro, T.; Yasui, S.; Iwasaki, M.; Ihara, I.; Tsuji, O.; Umetsu, K. Effects of handling parameters on hydrogen sulfide emission from stored dairy manure. J. Environ. Manag. 2015, 154, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Appels, L.; Baeyens, J.; Degrève, J.; Dewil, R. Principles and potential of the anaerobic digestion of waste-activated sludge. Prog. Energy Combust. Sci. 2008, 34, 755–781. [Google Scholar] [CrossRef]
- O’Connell, M.; McNally, C.; Richardson, M.G. Biochemical attack on concrete in wastewater applications: A state of the art review. Cem. Concr. Compos. 2010, 32, 479–485. [Google Scholar] [CrossRef]
- Weil, E.D.; Sandler, S.R.; Gernon, M. Sulfur Compounds. In Kirk-Othmer Encyclopedia of Chemical Technology; Wiley: Hoboken, NJ, USA, 2006. [Google Scholar]
- Lewis, A.E. Review of metal sulphide precipitation. Hydrometallurgy 2010, 104, 222–234. [Google Scholar] [CrossRef]
- World Health Organization. Guidelines for Drinking-Water Quality: Fourth Edition Incorporating the First Addendum; World Health Organization: Geneva, Switzerland, 2014; ISBN 9789241549950. [Google Scholar]
- Marcus, P. Sulfur-assisted corrosion mechanisms and the role of alloyed elements. In Corrosion Mechanisms in Theory and Practice, 3rd ed.; Taylor & Francis Group: London, UK, 2011; ISBN 9781420094633. [Google Scholar]
- Massart, N.S.; Cooke, T.H. White’s Handbook of Chlorination and Alternative Disinfectants; Wiley: Hoboken, NJ, USA, 2010; ISBN 9780470180983. [Google Scholar]
- Qu, K.; Lee, S.W.; Bian, J.S.; Low, C.-M.; Wong, P.T.-H. Hydrogen sulfide: Neurochemistry and neurobiology. Neurochem. Int. 2008, 52, 155–165. [Google Scholar] [CrossRef]
- Grengg, C.; Mittermayr, F.; Baldermann, A.; Böttcher, M.E.; Leis, A.; Koraimann, G.; Grunert, P.; Dietzel, M. Microbiologically induced concrete corrosion: A case study from a combined sewer network. Cem. Concr. Res. 2015, 77, 16–25. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; De Schryver, P.; De Gusseme, B.; De Muynck, W.; Boon, N.; Verstraete, W. Chemical and biological technologies for hydrogen sulfide emission control in sewer systems: A review. Water Res. 2008, 42, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Jiang, G.; Sun, J.; Sharma, K.R.; Yuan, Z. Corrosion and odor management in sewer systems. Curr. Opin. Biotechnol. 2015, 33, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Rathnayake, D.; Kastl, G.; Sathasivan, A. Evaluation of a combined treatment to control gaseous phase H2S in sewer. Int. Biodeterior. Biodegrad. 2017, 124, 206–214. [Google Scholar] [CrossRef]
- Dos Santos Afonso, M.; Stumm, W. Reductive dissolution of iron(III) (hydr)oxides by hydrogen sulfide. Langmuir 1992, 8, 1671–1675. [Google Scholar] [CrossRef]
- Yao, W.; Millero, F.J. Oxidation of hydrogen sulfide by hydrous Fe(III) oxides in seawater. Mar. Chem. 1996, 52, 1–16. [Google Scholar] [CrossRef]
- Davydov, A.; Chuang, K.T.; Sanger, A.R. Mechanism of H2S Oxidation by Ferric Oxide and Hydroxide Surfaces. J. Phys. Chem. B 1998, 102, 4745–4752. [Google Scholar] [CrossRef]
- Poulton, S.W.; Krom, M.D.; Van Rijn, J.; Raiswell, R. The use of hydrous iron (III) oxides for the removal of hydrogen sulphide in aqueous systems. Water Res. 2002, 36, 825–834. [Google Scholar] [CrossRef]
- Poulton, S.W. Sulfide oxidation and iron dissolution kinetics during the reaction of dissolved sulfide with ferrihydrite. Chem. Geol. 2003, 202, 79–94. [Google Scholar] [CrossRef]
- Poulton, S.W.; Krom, M.D.; Raiswell, R. A revised scheme for the reactivity of iron (oxyhydr)oxide minerals towards dissolved sulfide. Geochim. Cosmochim. Acta 2004, 68, 3703–3715. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Zhou, J.; Shang, C.; Kikkert, G.A. Removal of aqueous hydrogen sulfide by granular ferric hydroxide—Kinetics, capacity and reuse. Chemosphere 2014, 117, 324–329. [Google Scholar] [CrossRef]
- Jacukowicz-Sobala, I.; Wilk, L.J.; Drabent, K.; Kociołek-Balawejder, E. Synthesis and characterization of hybrid materials containing iron oxide for removal of sulfides from water. J. Colloid Interface Sci. 2015, 460. [Google Scholar] [CrossRef]
- Luther, G.W. Inorganic Chemistry for Geochemistry and Environmental Sciences; Wiley: Hoboken, NJ, USA, 2016. [Google Scholar]
- Luther, G.W.; Thibault de Chanvalon, A.; Oldham, V.E.; Estes, E.R.; Tebo, B.M.; Madison, A.S. Reduction of Manganese Oxides: Thermodynamic, Kinetic and Mechanistic Considerations for One- Versus Two-Electron Transfer Steps. Aquat. Geochem. 2018, 24, 257–277. [Google Scholar] [CrossRef]
- Driehaus, W.; Seith, R.; Jekel, M. Oxidation of arsenate(III) with manganese oxides in water treatment. Water Res. 1995, 29, 297–305. [Google Scholar] [CrossRef]
- Ivanets, A.I.; Prozorovich, V.G.; Kouznetsova, T.F.; Radkevich, A.V.; Zarubo, A.M. Mesoporous manganese oxides prepared by sol-gel method: Synthesis, characterization and sorption properties towards strontium ions. Environ. Nanotechnol. Monit. Manag. 2016, 6, 261–269. [Google Scholar] [CrossRef]
- Qin, Q.; Wang, Q.; Fu, D.; Ma, J. An efficient approach for Pb(II) and Cd(II) removal using manganese dioxide formed in situ. Chem. Eng. J. 2011, 172, 68–74. [Google Scholar] [CrossRef]
- Su, Q.; Pan, B.; Wan, S.; Zhang, W.; Lv, L. Use of hydrous manganese dioxide as a potential sorbent for selective removal of lead, cadmium, and zinc ions from water. J. Colloid Interface Sci. 2010, 349, 607–612. [Google Scholar] [CrossRef]
- Zhang, Q.-H.; Li, S.-P.; Sun, S.-Y.; Yin, X.-S.; Yu, J.-G. Lithium selective adsorption on 1-D MnO2 nanostructure ion-sieve. Adv. Powder Technol. 2009, 20, 432–437. [Google Scholar] [CrossRef]
- Guo, Y.; Guo, H.; Wang, Y.; Liu, L.; Chen, W. Designed hierarchical MnO2 microspheres assembled from nanofilms for removal of heavy metal ions. RSC Adv. 2014, 4, 14048–14054. [Google Scholar] [CrossRef]
- Sun, H.; Xu, K.; Huang, M.; Shang, Y.; She, P.; Yin, S.; Liu, Z. One-pot synthesis of ultrathin manganese dioxide nanosheets and their efficient oxidative degradation of Rhodamine B. Appl. Surf. Sci. 2015, 357, 69–73. [Google Scholar] [CrossRef]
- Remucal, C.K.; Ginder-Vogel, M. A critical review of the reactivity of manganese oxides with organic contaminants. Environ. Sci. Process. Impacts 2014, 16, 1247–1266. [Google Scholar] [CrossRef]
- Maliyekkal, S.M.; Lisha, K.P.; Pradeep, T. A novel cellulose–manganese oxide hybrid material by in situ soft chemical synthesis and its application for the removal of Pb(II) from water. J. Hazard. Mater. 2010, 181, 986–995. [Google Scholar] [CrossRef]
- Wang, S.; Gao, B.; Li, Y.; Mosa, A.; Zimmerman, A.R.; Ma, L.Q.; Harris, W.G.; Migliaccio, K.W. Manganese oxide-modified biochars: Preparation, characterization, and sorption of arsenate and lead. Bioresour. Technol. 2015, 181, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Ouředníček, P.; Hudcová, B.; Trakal, L.; Pohořelý, M.; Komárek, M. Synthesis of modified amorphous manganese oxide using low-cost sugars and biochars: Material characterization and metal(loid) sorption properties. Sci. Total Environ. 2019, 670, 1159–1169. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Xu, F.; Xue, J.; Chen, S.; Wang, J.; Yang, Y. Enhanced removal of heavy metal ions from aqueous solution using manganese dioxide-loaded biochar: Behavior and mechanism. Sci. Rep. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dassanayake, R.S.; Rajakaruna, E.; Moussa, H.; Abidi, N. One-pot synthesis of MnO2–chitin hybrids for effective removal of methylene blue. Int. J. Biol. Macromol. 2016, 93, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Krivoshapkin, P.V.; Ivanets, A.I.; Torlopov, M.A.; Mikhaylov, V.I.; Srivastava, V.; Sillanpää, M.; Prozorovich, V.G.; Kouznetsova, T.F.; Koshevaya, E.D.; Krivoshapkina, E.F. Nanochitin/manganese oxide-biodegradable hybrid sorbent for heavy metal ions. Carbohydr. Polym. 2019, 210, 135–143. [Google Scholar] [CrossRef]
- Dinh, V.-P.; Le, N.-C.; Tuyen, L.A.; Hung, N.Q.; Nguyen, V.-D.; Nguyen, N.-T. Insight into adsorption mechanism of lead(II) from aqueous solution by chitosan loaded MnO2 nanoparticles. Mater. Chem. Phys. 2018, 207, 294–302. [Google Scholar] [CrossRef]
- Shim, J.; Kumar, M.; Mukherjee, S.; Goswami, R. Sustainable removal of pernicious arsenic and cadmium by a novel composite of MnO2 impregnated alginate beads: A cost-effective approach for wastewater treatment. J. Environ. Manag. 2019, 234, 8–20. [Google Scholar] [CrossRef]
- Kamran, U.; Heo, Y.-J.; Lee, J.W.; Park, S.-J. Chemically modified activated carbon decorated with MnO2 nanocomposites for improving lithium adsorption and recovery from aqueous media. J. Alloys Compd. 2019, 794, 425–434. [Google Scholar] [CrossRef]
- Pathan, S.; Pandita, N.; Kishore, N. Acid functionalized-nanoporous carbon/MnO2 composite for removal of arsenic from aqueous medium. Arab. J. Chem. 2019, 12, 5200–5211. [Google Scholar] [CrossRef]
- Hao, J.; Meng, X.; Fang, S.; Cao, H.; Lv, W.; Zheng, X.; Liu, C.; Chen, M.; Sun, Z. MnO2-Functionalized Amorphous Carbon Sorbents from Spent Lithium-Ion Batteries for Highly Efficient Removal of Cadmium from Aqueous Solutions. Ind. Eng. Chem. Res. 2020, 59, 10210–10220. [Google Scholar] [CrossRef]
- Zemskova, L.A.; Artemyanov, A.P.; Voit, A.V.; Shlyk, D.K. New composite materials based on activated carbon fibers with specific adsorption and catalytic properties. Mater. Today Proc. 2018, 5, 25997–26001. [Google Scholar] [CrossRef]
- Mohammadkhani, S.; Aghaie, M. Synthesis of a MnO2/multiwalled carbon nanotube nanocomposite and its application as a sorbent for removing Cu2+ ions from aqueous media. J. Chin. Chem. Soc. 2019, 66, 1436–1442. [Google Scholar] [CrossRef]
- Xu, H.; Qu, Z.; Zong, C.; Huang, W.; Quan, F.; Yan, N. MnOx/Graphene for the Catalytic Oxidation and Adsorption of Elemental Mercury. Environ. Sci. Technol. 2015, 49, 6823–6830. [Google Scholar] [CrossRef] [PubMed]
- Wan, S.; Ding, W.; Wang, Y.; Wu, J.; Gu, Y.; He, F. Manganese oxide nanoparticles impregnated graphene oxide aggregates for cadmium and copper remediation. Chem. Eng. J. 2018, 350, 1135–1143. [Google Scholar] [CrossRef]
- Yusuf, M.; Song, K.; Geng, S.; Fazhi, X. Adsorptive removal of anionic dyes by graphene impregnated with MnO2 from aqueous solution. Colloids Surfaces A Physicochem. Eng. Asp. 2020, 595, 124667. [Google Scholar] [CrossRef]
- Su, Q.; Pan, B.; Pan, B.; Zhang, Q.; Zhang, W.; Lv, L.; Wang, X.; Wu, J.; Zhang, Q. Fabrication of polymer-supported nanosized hydrous manganese dioxide (HMO) for enhanced lead removal from waters. Sci. Total Environ. 2009, 407, 5471–5477. [Google Scholar] [CrossRef]
- Lenoble, V.; Chabroullet, C.; Al Shukry, R.; Serpaud, B.; Deluchat, V.; Bollinger, J.-C. Dynamic arsenic removal on a MnO2-loaded resin. J. Colloid Interface Sci. 2004, 280, 62–67. [Google Scholar] [CrossRef]
- Mallakpour, S.; Motirasoul, F. Ultrasonication synthesis of PVA/PVP/α-MnO2-stearic acid blend nanocomposites for adsorbing CdII ion. Ultrason. Sonochem. 2018, 40, 410–418. [Google Scholar] [CrossRef]
- Wilk, Ł.J.; Ciechanowska, A.; Kociołek-Balawejder, E. Removal of sulfides from water using a hybrid ion exchanger containing manganese(IV) oxide. Sep. Purif. Technol. 2020, 231, 115882. [Google Scholar] [CrossRef]
- Wang, M.C.; Sheng, G.D.; Qiu, Y.P. A novel manganese-oxide/biochar composite for efficient removal of lead(II) from aqueous solutions. Int. J. Environ. Sci. Technol. 2015, 12, 1719–1726. [Google Scholar] [CrossRef]
- Lenoble, V.; Laclautre, C.; Serpaud, B.; Deluchat, V.; Bollinger, J.-C. As(V) retention and As(III) simultaneous oxidation and removal on a MnO2-loaded polystyrene resin. Sci. Total Environ. 2004, 326, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Rufino, E.S.; Monteiro, E.E.C. Infrared study on methyl methacrylate–methacrylic acid copolymers and their sodium salts. Polymer 2003, 44, 7189–7198. [Google Scholar] [CrossRef]
- Tsunashima, K.; Kutsumizu, S.; Hirasawa, E.; Yano, S. Far-infrared study on the zinc(II) complex salts of ethylene-methacrylic acid copolymer with 1,3-bis(aminomethyl)cyclohexane. Macromolecules 1991, 24, 5910–5913. [Google Scholar] [CrossRef]
- Zagorodni, A.A.; Kotova, D.L.; Selemenev, V.F. Infrared spectroscopy of ion exchange resins: Chemical deterioration of the resins. React. Funct. Polym. 2002, 53, 157–171. [Google Scholar] [CrossRef]
- Butyrskaya, E.V.; Shaposhnik, V.A. Interpretation of infrared spectra for ion-exchange systems. Opt. Spectrosc. 2002, 92, 370–374. [Google Scholar] [CrossRef]
- Ghosh, S.; Dhole, K.; Tripathy, M.K.; Kumar, R.; Sharma, R.S. FTIR spectroscopy in the characterization of the mixture of nuclear grade cation and anion exchange resins. J. Radioanal. Nucl. Chem. 2015, 304, 917–923. [Google Scholar] [CrossRef]
- Traboulsi, A.; Dupuy, N.; Rebufa, C.; Sergent, M.; Labed, V. Investigation of gamma radiation effect on the anion exchange resin Amberlite IRA-400 in hydroxide form by Fourier transformed infrared and 13C nuclear magnetic resonance spectroscopies. Anal. Chim. Acta 2012, 717, 110–121. [Google Scholar] [CrossRef]
- Balan, L.; Matei Ghimbeu, C.; Vidal, L.; Vix-Guterl, C. Photoassisted synthesis of manganese oxide nanostructures using visible light at room temperature. Green Chem. 2013, 15, 2191–2199. [Google Scholar] [CrossRef]
- Okay, O. Macroporous copolymer networks. Prog. Polym. Sci. 2000, 25, 711–779. [Google Scholar] [CrossRef]
- Sing, K. The use of nitrogen adsorption for the characterisation of porous materials. Colloids Surfaces A Physicochem. Eng. Asp. 2001, 187–188, 3–9. [Google Scholar] [CrossRef]
- Grosman, A.; Ortega, C. Capillary Condensation in Porous Materials. Hysteresis and Interaction Mechanism without Pore Blocking/Percolation Process. Langmuir 2008, 24, 3977–3986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horikawa, T.; Do, D.D.; Nicholson, D. Capillary condensation of adsorbates in porous materials. Adv. Colloid Interface Sci. 2011, 169, 40–58. [Google Scholar] [CrossRef] [PubMed]
- Cumbal, L.; SenGupta, A.K. Arsenic Removal Using Polymer-Supported Hydrated Iron(III) Oxide Nanoparticles: Role of Donnan Membrane Effect. Environ. Sci. Technol. 2005, 39, 6508–6515. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; SenGupta, A.K.; Prakash, P. The Donnan Membrane Principle: Opportunities for Sustainable Engineered Processes and Materials. Environ. Sci. Technol. 2010, 44, 1161–1166. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.W. The surface chemistry of hydrous manganese dioxide. J. Colloid Interface Sci. 1974, 46, 357–371. [Google Scholar] [CrossRef]
- Balistrieri, L.S.; Murray, J.W. The surface chemistry of δMnO2 in major ion sea water. Geochim. Cosmochim. Acta 1982, 46, 1041–1052. [Google Scholar] [CrossRef]
- Yao, W.; Millero, F.J. The rate of sulfide oxidation by δMnO2 in seawater. Geochim. Cosmochim. Acta 1993, 57, 3359–3365. [Google Scholar] [CrossRef]
- Burdige, D.J. The biogeochemistry of manganese and iron reduction in marine sediments. Earth-Science Rev. 1993, 35, 249–284. [Google Scholar] [CrossRef]
- Nico, P.S.; Zasoski, R.J. Mn(III) Center Availability as a Rate Controlling Factor in the Oxidation of Phenol and Sulfide on δ-MnO2. Environ. Sci. Technol. 2001, 35, 3338–3343. [Google Scholar] [CrossRef]
- Herszage, J.; dos Santos Afonso, M. Mechanism of Hydrogen Sulfide Oxidation by Manganese(IV) Oxide in Aqueous Solutions. Langmuir 2003, 19, 9684–9692. [Google Scholar] [CrossRef]
- Wang, C.; Pei, Y. The removal of hydrogen sulfide in solution by ferric and alum water treatment residuals. Chemosphere 2012, 88, 1178–1183. [Google Scholar] [CrossRef] [PubMed]
- Haimour, N.; El-Bishtawi, R.; Ail-Wahbi, A. Equilibrium adsorption of hydrogen sulfide onto CuO and ZnO. Desalination 2005, 181, 145–152. [Google Scholar] [CrossRef]
- Edathil, A.A.; Kannan, P.; Banat, F. Adsorptive oxidation of sulfides catalysed by δ-MnO2 decorated porous graphitic carbon composite. Environ. Pollut. 2020, 266, 115218. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Property | Cation Exchanger | |
|---|---|---|
| Amberlite IRC50 | Amberlite IRC86 | |
| Matrix | poly(acrylic-divinylbenzene) | poly(acrylic-divinylbenzene) |
| Matrix structure | macroporous | gel |
| Functional groups | carboxylic | carboxylic |
| Ionic form | H+ | H+ |
| Exchange capacity 1, meq/g | 10.8 | 10.7 |
| Physical form | beads | beads |
| Diameter, mm | 0.28–0.70 | 0.58–0.78 |
| Appearance | creamy, opaque, matt | browhish-red, translucent, low sheen |
| Point | Content, wt.% | |||
|---|---|---|---|---|
| Mn | O | Na | K | |
| CWM1#MnO2, Figure 2a | ||||
| 1 | - | 36.9 | 15.8 | 0.7 |
| 2 | - | 37.1 | 18.0 | 0.8 |
| 3 | 36.8 | 39.0 | 7.5 | 1.3 |
| 4 | 31.9 | 37.3 | 8.2 | 1.0 |
| CWM1#MnO2, Figure 2b | ||||
| 1 | 41.0 | 32.0 | 6.5 | 1.0 |
| 2 | 48.2 | 21.7 | 6.8 | 1.3 |
| 3 | 50.0 | 34.8 | 5.5 | 1.5 |
| CWM3#MnO2, Figure 2c | ||||
| 1 | 40.3 | 34.8 | - | 5.6 |
| 2 | 40.3 | 38.6 | - | 4.7 |
| 3 | 48.9 | 38.1 | - | 5.0 |
| 4 | 69.9 | 6.4 | - | 3.0 |
| Parameter | Sample | ||
|---|---|---|---|
| CWM | CWM1#MnO2 | CWM3#MnO2 | |
| Brunauer-Emmet-Teller (BET) surface area, m2/g | 1.67 | 1.15 | 40.03 |
| BET total pore volume, cm3/g | 0.0024 | 0.0024 | 0.0317 |
| BET average pore diameter, nm | 2.9 | 4.1 | 3.2 |
| Barret-Joyner-Halenda (BJH) surface area, m2/g | 1.61 | 1.79 | 29.51 |
| BJH total pore volume, cm3/g | 0.002 | 0.003 | 0.023 |
| BJH average pore diameter, nm | 1.68 | 3.74 | 3.81 |
| Parameter | Sample | ||
|---|---|---|---|
| CWM | CWM1#MnO2 | CWM3#MnO2 | |
| Total surface area, m2/g | 25.70 | 3.45 | 18.29 |
| Total porosity, % | 7.73 | 8.17 | 14.29 |
| Apparent density, g/cm3 | 1.157 | 1.739 | 1.781 |
| Skeletal density, g/cm3 | 1.352 | 1.896 | 2.079 |
| Total intrusion volume, cm3 Hg/g | 0.124 | 0.047 | 0.080 |
| Hybrid Material | Sulfide Initial Concentration, mg S2−/dm3 | Sulfides Removed: % | Manganese Released from the Beads to the Solution 1: % | pH of the Solution: | Overall Average Removal Capacity, mg S2−/g | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Overall | Bound to the Beads Structure | Present in the Solution in the S2O32− Form | Unbal-anced | Overall | Dissolved in the Solution | Bound in the Sediment | Initial | Final | |||
| CWM1#MnO2/K+ | 100 | 100 | 4.0 | 89.7 | 6.3 | 0.32 | 0.17 | 0.15 | 12.0 | 9.8 | 34.1 2 |
| 200 | 86.2 | 3.4 | 76.7 | 6.1 | 2.24 | 0.52 | 1.72 | 12.3 | 11.1 | 54.8 | |
| 500 | 59.6 | 1.6 | 49.9 | 8.1 | 10.71 | 0.41 | 10.30 | 12.7 | 12.4 | 94.7 | |
| CWM3#MnO2/K+ | 100 | 100 | 20.8 | 75.1 | 4.1 | 1.98 | 0.32 | 1.66 | 12.0 | 10.4 | 34.1 2 |
| 200 | 100 | 28.4 | 61.5 | 9.9 | 10.82 | 1.85 | 8.97 | 12.3 | 11.6 | 63.3 2 | |
| 500 | 90.9 | 15.8 | 60.9 | 14.2 | 25.84 | 0.21 | 25.64 | 12.7 | 12.6 | 144.3 | |
| CWM1#MnO2/H+ | 200 | 93.8 | 3.2 | 79.3 | 11.3 | 2.24 | 0.52 | 1.72 | 12.6 | 10.3 | 65.2 |
| 500 | 58.9 | 3.6 | 40.3 | 15.0 | 5.37 | 0.63 | 4.74 | 12.8 | 12.4 | 99.0 | |
| CWM3#MnO2/H+ | 200 | 100 | 25.7 | 70.1 | 4.2 | 2.40 | 1.60 | 0.80 | 12.6 | 11.6 | 63.3 2 |
| 500 | 83.5 | 23.2 | 50.4 | 9.9 | 5.44 | 1.92 | 3.52 | 12.8 | 12.8 | 140.4 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wilk, Ł.J.; Ciechanowska, A.; Kociołek-Balawejder, E. Adsorptive-Oxidative Removal of Sulfides from Water by MnO2-Loaded Carboxylic Cation Exchangers. Materials 2020, 13, 5124. https://doi.org/10.3390/ma13225124
Wilk ŁJ, Ciechanowska A, Kociołek-Balawejder E. Adsorptive-Oxidative Removal of Sulfides from Water by MnO2-Loaded Carboxylic Cation Exchangers. Materials. 2020; 13(22):5124. https://doi.org/10.3390/ma13225124
Chicago/Turabian StyleWilk, Łukasz J., Agnieszka Ciechanowska, and Elżbieta Kociołek-Balawejder. 2020. "Adsorptive-Oxidative Removal of Sulfides from Water by MnO2-Loaded Carboxylic Cation Exchangers" Materials 13, no. 22: 5124. https://doi.org/10.3390/ma13225124
APA StyleWilk, Ł. J., Ciechanowska, A., & Kociołek-Balawejder, E. (2020). Adsorptive-Oxidative Removal of Sulfides from Water by MnO2-Loaded Carboxylic Cation Exchangers. Materials, 13(22), 5124. https://doi.org/10.3390/ma13225124