Thermal Modification Effect on Supported Cu-Based Activated Carbon Catalyst in Hydrogenolysis of Glycerol



Abstract
:
1. Introduction
2. Materials and Methods
2.1. Catalyst Preparation
2.2. Characterization of Supports and Catalysts
2.3. Catalytic Tests
3. Results
3.1. Catalysts and Support Characterization
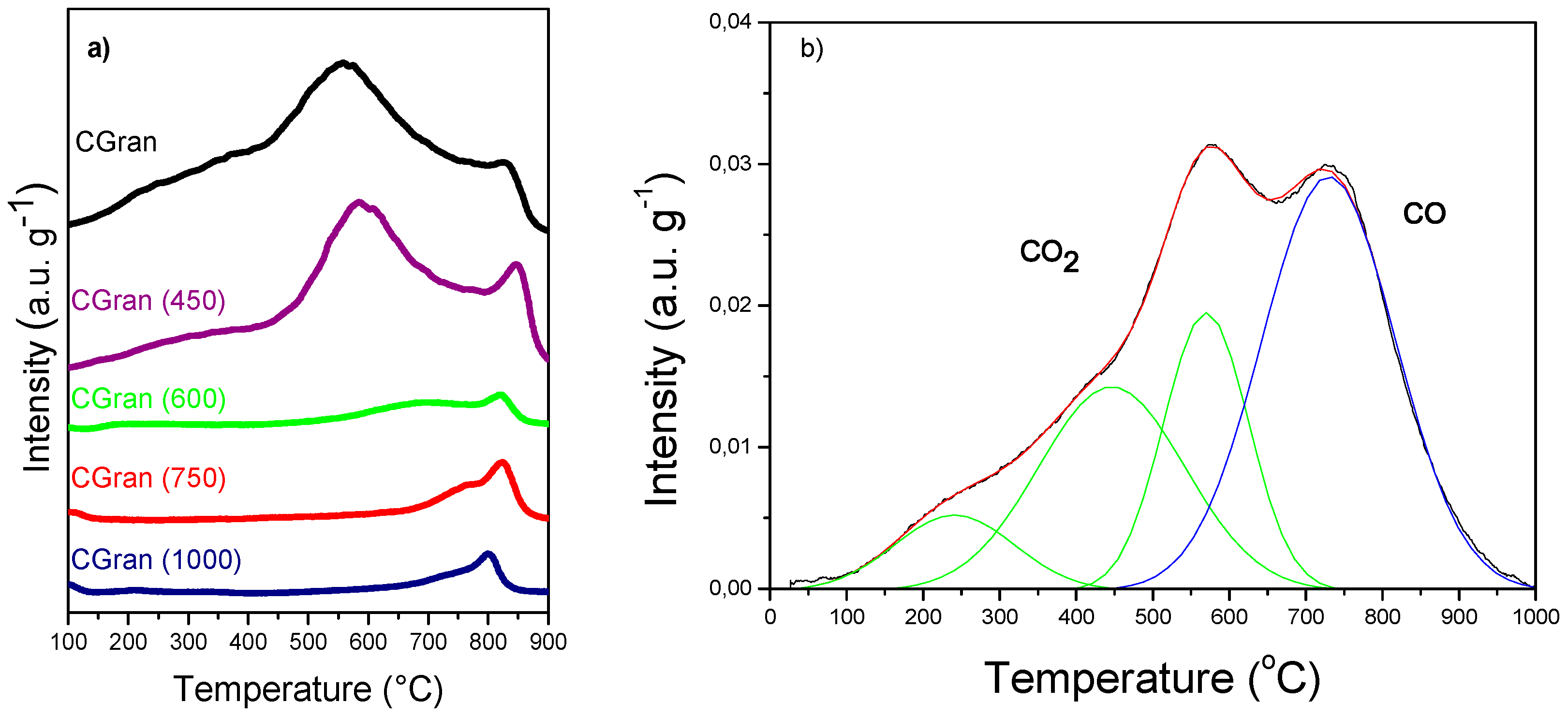
3.1.1. Temperature Programmed Decomposition–Mass Spectrometry (TPD–MS)
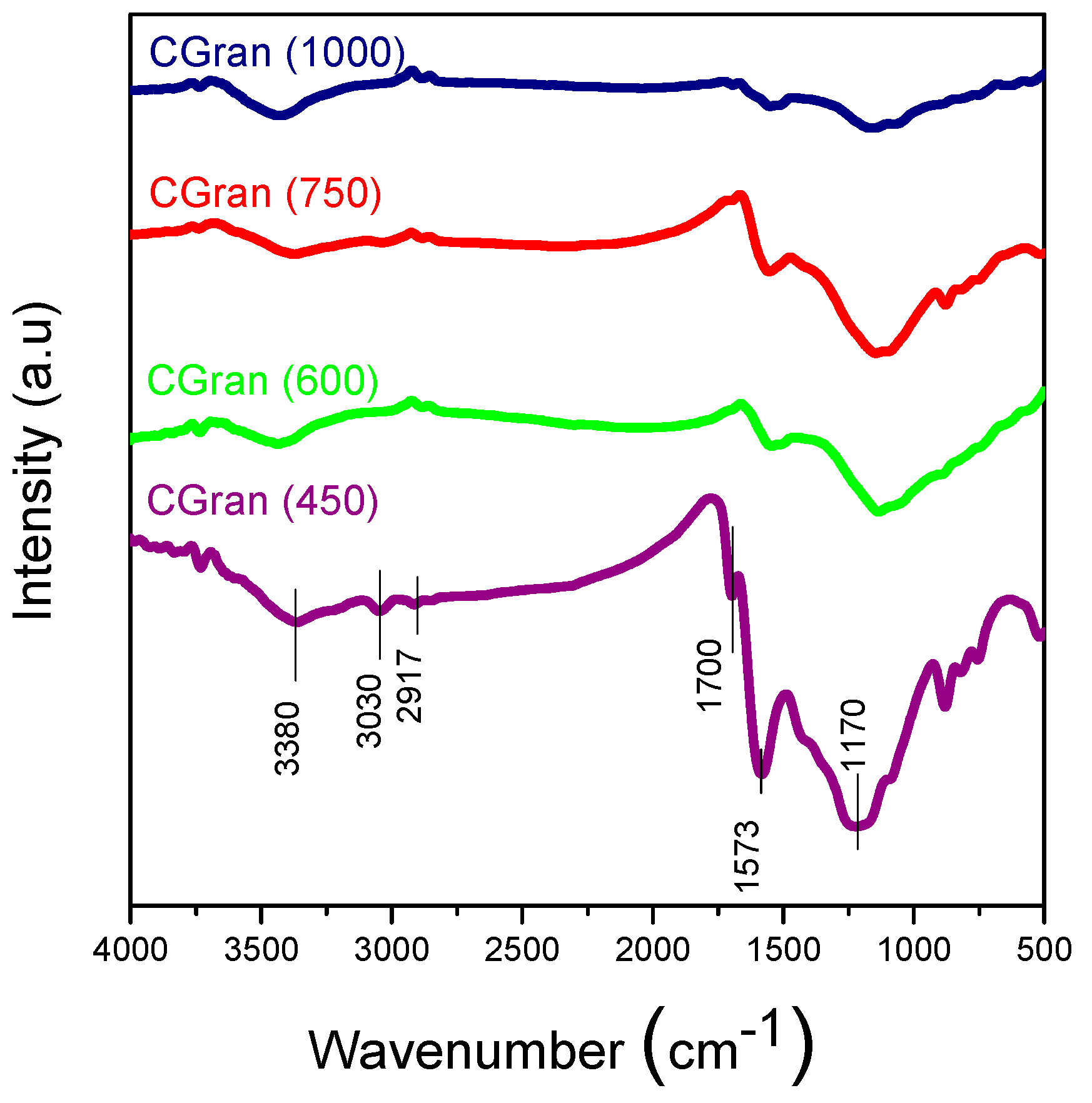
3.1.2. Fourier Transformed Infrared Spectroscopy (FT-IR)
3.1.3. Total Potentiometric Acidity
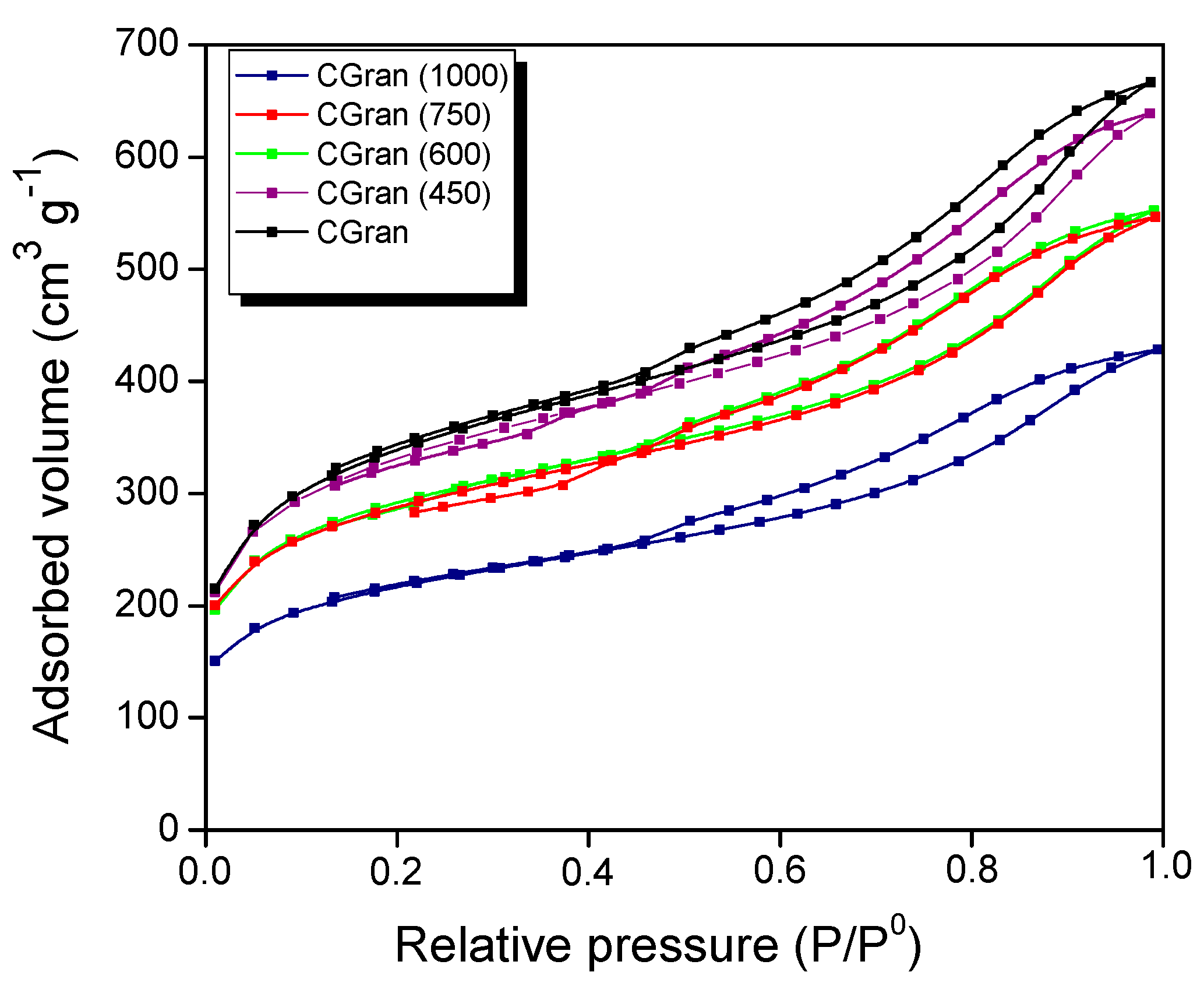
3.1.4. Nitrogen Sorption at 77 K
3.1.5. Temperature Programmed Reduction (TPR)
3.1.6. N2O Titration (N2O-TPR)
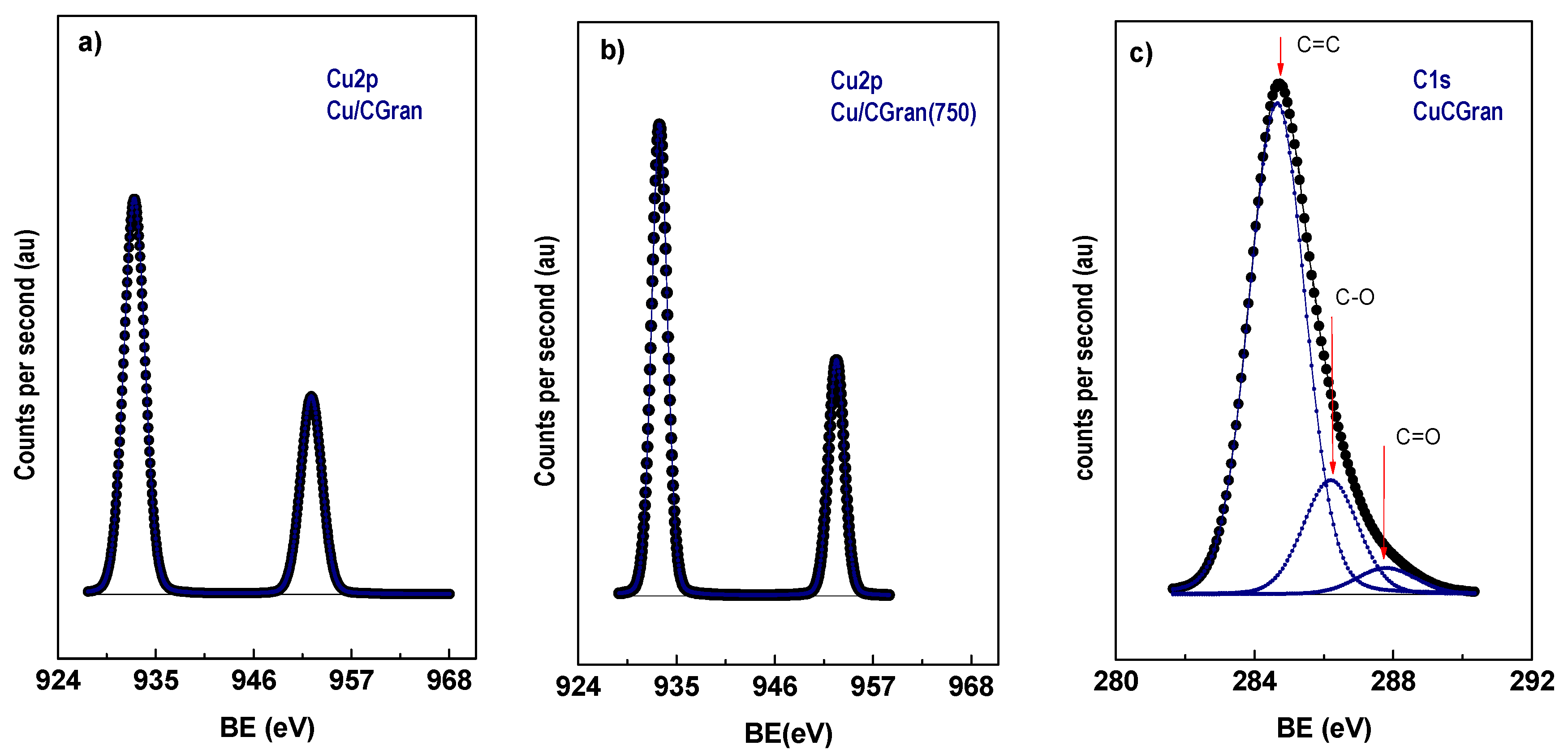
3.1.7. X-ray Photoelectron Spectroscopy (XPS)
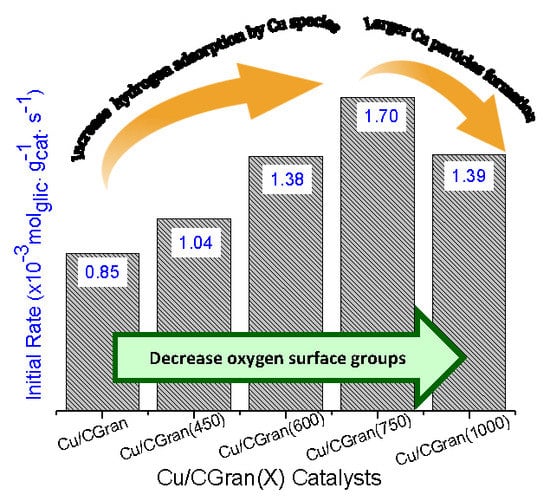
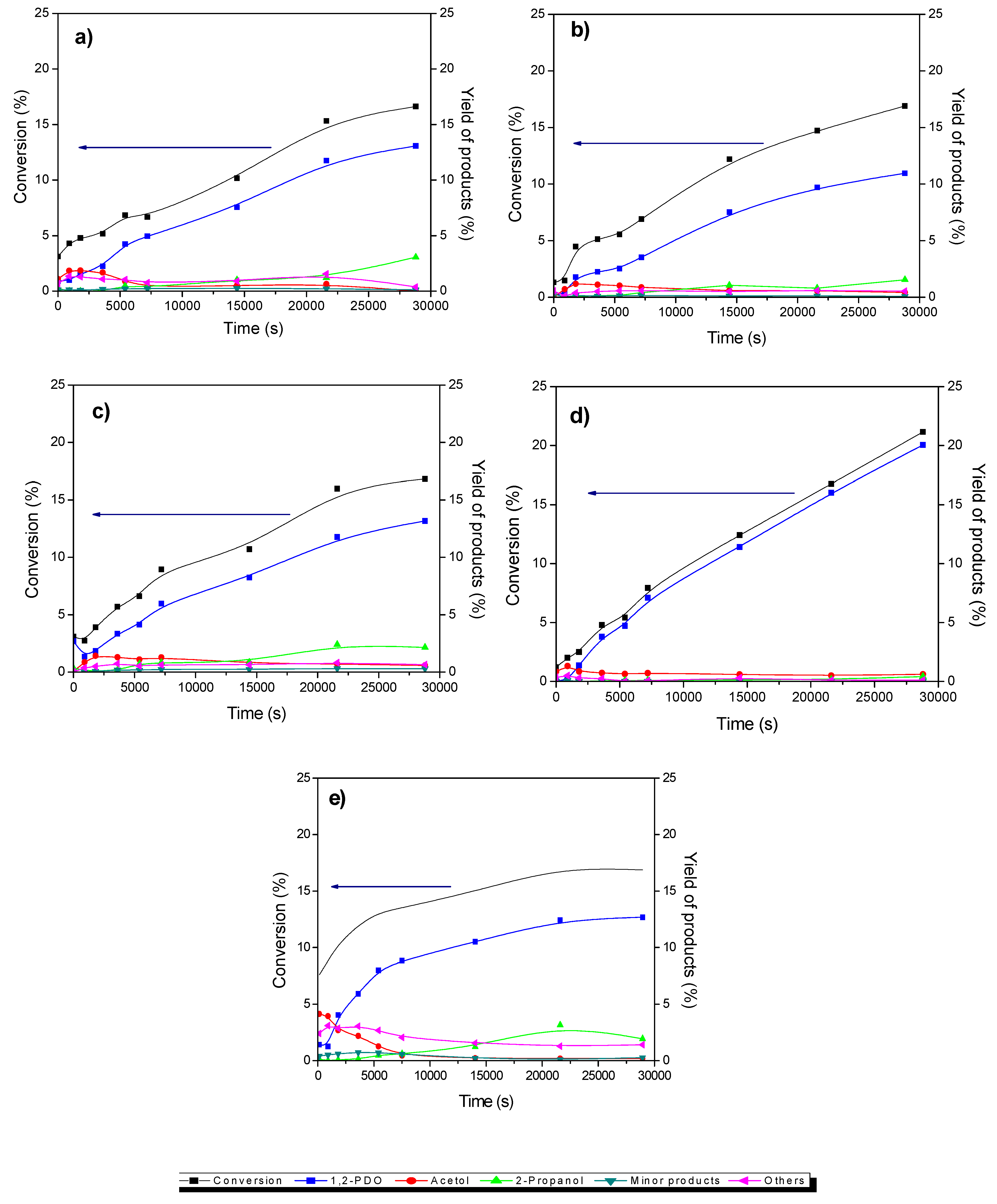
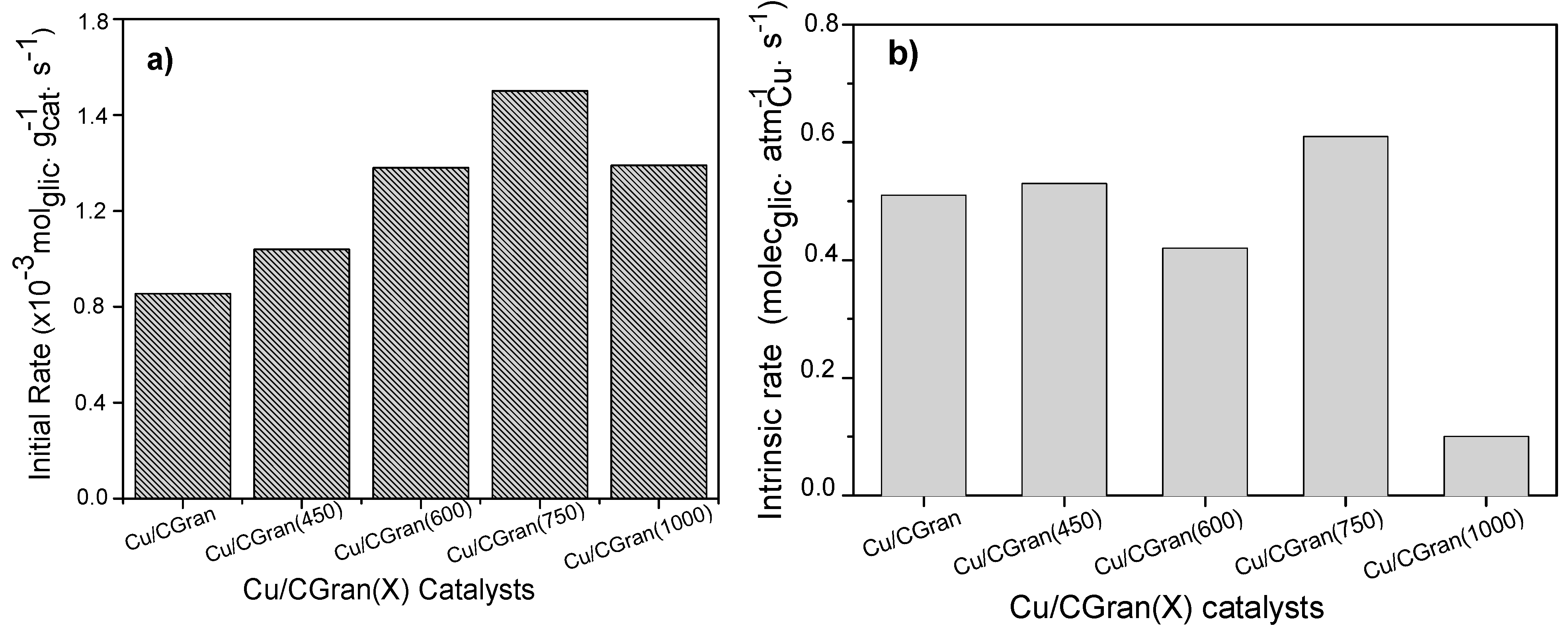
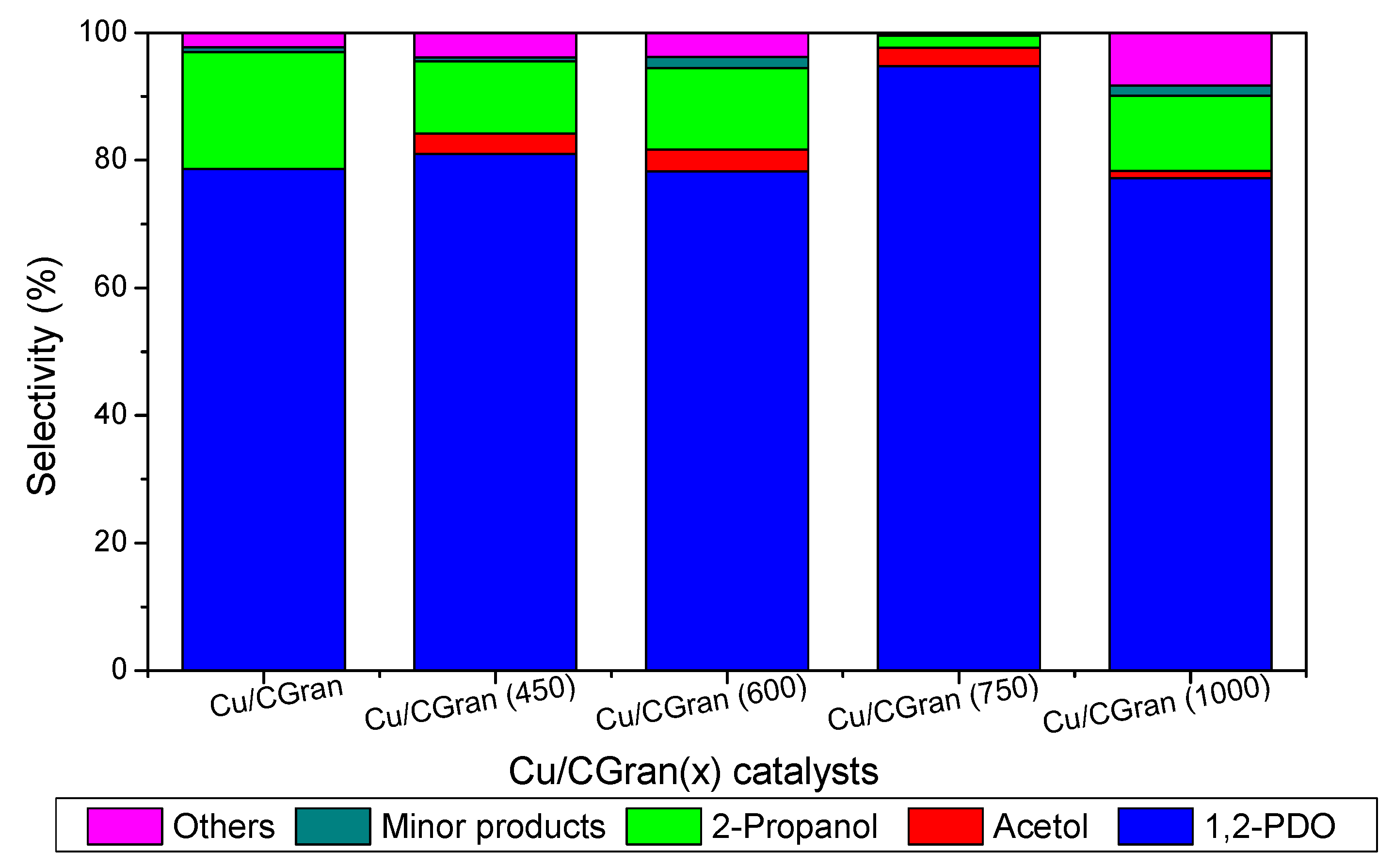
3.2. Catalytic Activity of Cu/CGran and CCGran(x) Catalysts
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Zhou, C.-H.; Beltramini, J.N.; Fan, Y.-X.; Lu, G.Q. Chemoselective catalytic conversion of glycerol as a biorenewable source to valuable commodity chemicals. Chem. Soc. Rev. 2008, 37, 527–549. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.; Prasad, R. Triglycerides-based diesel fuels. Renew. Sustain. Energy Rev. 2000, 4, 111–133. [Google Scholar] [CrossRef]
- Werpy, T.; Petersen, G. Top Value Added Chemicals from Biomass: Volume I—Results of Screening for Potential Candidates from Sugars and Synthesis Gas; DOE/GO-102004-1992, TRN: US200427%%671; National Renewable Energy Lab., Golden, CO (US): Golden, CO, USA, 2004.
- Flaxbart, D. Kirk−Othmer Encyclopedia of Chemical Technology, Fourth Edition, 27-Volume Set Wiley Interscience: New York, NY, USA, 1992−1998. ISBN 0-471-52704-1. J. Am. Chem. Soc. 1999, 121, 2339. [Google Scholar]
- Alhanash, A.; Kozhevnikova, E.F.; Kozhevnikov, I.V. Hydrogenolysis of Glycerol to Propanediol Over Ru: Polyoxometalate Bifunctional Catalyst. Catal. Lett. 2008, 120, 307–311. [Google Scholar] [CrossRef]
- Rose, J. Industrial Organic Chemistry; 3rd Edition, K.W., H-J.A. Eds; Wiley-VCH: United States, ISBN 3-527-28838-4. Appl. Organomet. Chem. 1999, 13, 857–858. [Google Scholar]
- Lahr, D.G.; Shanks, B.H. Kinetic Analysis of the Hydrogenolysis of Lower Polyhydric Alcohols: Glycerol to Glycols. Ind. Eng. Chem. Res. 2003, 42, 5467–5472. [Google Scholar] [CrossRef] [Green Version]
- Montassier, C.; Giraud, D.; Barbier, J. Polyol Conversion by Liquid Phase Heterogeneous Catalysis Over Metals; Elsevier: Amsterdam, The Netherlands, 1988; Volume 41, pp. 165–170. [Google Scholar]
- Dasari, M.A.; Kiatsimkul, P.-P.; Sutterlin, W.R.; Suppes, G.J. Low-pressure hydrogenolysis of glycerol to propylene glycol. Appl. Catal. A Gen. 2005, 281, 225–231. [Google Scholar] [CrossRef]
- Zhao, H.; Zheng, L.; Li, X.; Chen, P.; Hou, Z. Hydrogenolysis of glycerol to 1,2-propanediol over Cu-based catalysts: A short review. Catal. Today 2019, in press. [Google Scholar] [CrossRef]
- Gabrysch, T.; Muhler, M.; Peng, B. The kinetics of glycerol hydrodeoxygenation to 1,2-propanediol over Cu/ZrO2 in the aqueous phase. Appl. Catal. A Gen. 2019, 576, 47–53. [Google Scholar] [CrossRef]
- Freitas, I.C.; Manfro, R.L.; Souza, M.M. Hydrogenolysis of glycerol to propylene glycol in continuous system without hydrogen addition over Cu-Ni catalysts. Appl. Catal. B Environ. 2018, 220, 31–41. [Google Scholar] [CrossRef]
- Zheng, L.; Li, X.; Du, W.; Shi, D.; Ning, W.; Lu, X.; Hou, Z. Metal-organic framework derived Cu/ZnO catalysts for continuous hydrogenolysis of glycerol. Appl. Catal. B Environ. 2017, 203, 146–153. [Google Scholar] [CrossRef]
- Sepúlveda, C.; Delgado, L.; García, R.; Melendrez, M.; Fierro, J.; Ghampson, I.; Escalona, N. Effect of phosphorus on the activity of Cu/SiO2 catalysts in the hydrogenolysis of glycerol. Catal. Today 2017, 279, 217–223. [Google Scholar] [CrossRef]
- Tsukuda, E.; Sato, S.; Takahashi, R.; Sodesawa, T. Production of acrolein from glycerol over silica-supported heteropoly acids. Catal. Commun. 2007, 8, 1349–1353. [Google Scholar] [CrossRef]
- Chai, S.-H.; Wang, H.-P.; Liang, Y.; Xu, B.-Q. Sustainable production of acrolein: investigation of solid acid–base catalysts for gas-phase dehydration of glycerol. Green Chem. 2007, 9, 1130. [Google Scholar] [CrossRef]
- Jia, C.-J.; Liu, Y.; Schmidt, W.; Lu, A.-H.; Schuth, F. Small-sized HZSM-5 zeolite as highly active catalyst for gas phase dehydration of glycerol to acrolein. J. Catal. 2010, 269, 71–79. [Google Scholar] [CrossRef]
- Kim, Y.T.; Jung, K.-D.; Park, E.D. Gas-phase dehydration of glycerol over ZSM-5 catalysts. Microporous Mesoporous Mater. 2010, 131, 28–36. [Google Scholar] [CrossRef]
- Chaminand, J.; Djakovitch, L.; Gallezot, P.; Marion, P.; Pinel, C. Glycerol hydrogenolysis on heterogeneous catalysts. Green Chem. 2004, 6, 359–361. [Google Scholar] [CrossRef]
- Maris, E.P.; Ketchie, W.C.; Oleshko, V.; Davis, R.J. Metal Particle Growth during Glucose Hydrogenation over Ru/SiO2Evaluated by X-ray Absorption Spectroscopy and Electron Microscopy. J. Phys. Chem. B 2006, 110, 7869–7876. [Google Scholar] [CrossRef]
- Kusunoki, Y.; Miyazawa, T.; Kunimori, K.; Tomishige, K. Highly active metal–acid bifunctional catalyst system for hydrogenolysis of glycerol under mild reaction conditions. Catal. Commun. 2005, 6, 645–649. [Google Scholar] [CrossRef]
- Miyazawa, T.; Kusunoki, Y.; Kunimori, K.; Tomishige, K. Glycerol conversion in the aqueous solution under hydrogen over Ru/C + an ion-exchange resin and its reaction mechanism. J. Catal. 2006, 240, 213–221. [Google Scholar] [CrossRef]
- Montassier, C.; Ménézo, J.; Hoang, L.; Renaud, C.; Barbier, J. Aqueous polyol conversions on ruthenium and on sulfur-modified ruthenium. J. Mol. Catal. 1991, 70, 99–110. [Google Scholar] [CrossRef]
- Cerro-Alarcón, M.; Maroto-Valiente, A.; Rodríguez-Ramos, I.; Guerrero-Ruiz, A. Further insights into the Ru nanoparticles–carbon interactions and their role in the catalytic properties. Carbon 2005, 43, 2711–2722. [Google Scholar] [CrossRef]
- Strelko, V.; Malik, D.J. Characterization and Metal Sorptive Properties of Oxidized Active Carbon. J. Colloid Interface Sci. 2002, 250, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Reinoso, F.; Rodríguez-Ramos, I.; Moreno-Castilla, C.; Guerrero-Ruiz, A.; López-González, J.D. Platinum catalysts supported on activated carbons: I. Preparation and characterization. J. Catal. 1986, 99, 171–183. [Google Scholar] [CrossRef]
- Gurevich, S.V.; Simonov, P.A.; Lisitsyn, A.S.; Likholobov, V.A.; Moroz, E.M.; Chuvilin, A.L.; Kolomiichuk, V.N. Influence of carbon support pretreatment on properties of Pd/C catalysts. React. Kinet. Catal. Lett. 1990, 41, 211–216. [Google Scholar] [CrossRef]
- Ehrburger, P.; Mahajan, O.; Walkerjr, P.W., Jr. Carbon as a support for catalystsI. Effect of surface heterogeneity of carbon on dispersion of platinum. J. Catal. 1976, 43, 61–67. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, T.; Liu, X.; Ding, Y. Cu-promoted Pt/activated carbon catalyst for glycerol oxidation to lactic acid. J. Mol. Catal. A Chem. 2016, 424, 91–97. [Google Scholar] [CrossRef]
- Cid, R.; Pecchi, G. Potentiometric method for determining the number and relative strength of acid sites in colored catalysts. Appl. Catal. 1985, 14, 15–21. [Google Scholar] [CrossRef]
- Figueiredo, J.; Pereira, M.; Freitas, M.; Órfão, J. Modification of the surface chemistry of activated carbons. Carbon 1999, 37, 1379–1389. [Google Scholar] [CrossRef]
- Zielke, U.; Hüttinger, K.; Hoffman, W. Surface-oxidized carbon fibers: I. Surface structure and chemistry. Carbon 1996, 34, 983–998. [Google Scholar] [CrossRef]
- Marchon, B.; Carrazza, J.; Heinemann, H.; Somorjai, G.A. Carbon. Interface Anal. 1990, 15, 627–634. [Google Scholar]
- Sepúlveda, C.; Garcia, R.; Reyes, P.; Ghampson, I.; Fierro, J.; Laurenti, D.; Vrinat, M.; Escalona, N. Hydrodeoxygenation of guaiacol over ReS2/activated carbon catalysts. Support and Re loading effect. Appl. Catal. A Gen. 2014, 475, 427–437. [Google Scholar] [CrossRef]
- Fanning, P.E.; Vannice, M. A DRIFTS study of the formation of surface groups on carbon by oxidation. Carbon 1993, 31, 721–730. [Google Scholar] [CrossRef]
- Oliveira, L.C.; Silva, C.N.; Yoshida, M.I.; Lago, R.M. The effect of H2 treatment on the activity of activated carbon for the oxidation of organic contaminants in water and the H2O2 decomposition. Carbon 2004, 42, 2279–2284. [Google Scholar] [CrossRef]
- Pradoburguete, C. The effect of oxygen surface groups of the support on platinum dispersion in Pt/carbon catalysts. J. Catal. 1989, 115, 98–106. [Google Scholar] [CrossRef]
- Lagos, G.; Garcia, R.; Agudo, A.L.; Yates, M.; Fierro, J.; Gil-Llambias, F.; Escalona, N. Characterisation and reactivity of Re/carbon catalysts in hydrodesulphurisation of dibenzothiophene: Effect of textural and chemical properties of support. Appl. Catal. A Gen. 2009, 358, 26–31. [Google Scholar] [CrossRef]
- Chary, K.V.R.; Seela, K.K.; Sagar, G.V.; Sreedhar, B. Characterization and Reactivity of Niobia Supported Copper Oxide Catalysts. J. Phys. Chem. B 2004, 108, 658–663. [Google Scholar] [CrossRef]
- Marchi, A.; Fierro, J.; Santamaria, J.; Monzon, A. Dehydrogenation of isopropylic alcohol on a Cu/SiO2 catalyst: a study of the activity evolution and reactivation of the catalyst. Appl. Catal. A Gen. 1996, 142, 375–386. [Google Scholar] [CrossRef]
- Lueking, A.; Yang, R.T. Hydrogen storage in carbon nanotubes: Residual metal content and pretreatment temperature. AIChE J. 2003, 49, 1556–1568. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Yang, R.T. Hydrogen Storage Properties of N-Doped Microporous Carbon. J. Phys. Chem. C 2009, 113, 21883–21888. [Google Scholar] [CrossRef]
- Wang, Z.; Yang, F.H.; Yang, R.T. Enhanced Hydrogen Spillover on Carbon Surfaces Modified by Oxygen Plasma. J. Phys. Chem. C 2010, 114, 1601–1609. [Google Scholar] [CrossRef]
- Lachawiec, A.J.; Yang, R.T. Reverse Spillover of Hydrogen on Carbon-Based Nanomaterials: Evidence of Recombination Using Isotopic Exchange. J. Phys. Chem. C 2009, 113, 13933–13939. [Google Scholar] [CrossRef]
- Psofogiannakis, G.M.; Froudakis, G.E. Fundamental studies and perceptions on the spillover mechanism for hydrogen storage. Chem. Commun. 2011, 47, 7933. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Yin, Y.; Liu, G.; Chen, S.; Hong, X.; Tsang, S.C.E. Hydrogen spillover enabled active Cu sites for methanol synthesis from CO2 hydrogenation over Pd doped CuZn catalysts. J. Catal. 2018, 359, 17–26. [Google Scholar] [CrossRef]
- López-Suárez, F.E.; López, A.B.; Illán-Gómez, M. Cu/Al2O3 catalysts for soot oxidation: Copper loading effect. Appl. Catal. B Environ. 2008, 84, 651–658. [Google Scholar]
- Agrell, J. Production of hydrogen from methanol over Cu/ZnO catalysts promoted by ZrO2 and Al2O3. J. Catal. 2003, 219, 389–403. [Google Scholar] [CrossRef]
- Jernigan, G.; Somorjai, G. Carbon Monoxide Oxidation over Three Different Oxidation States of Copper: Metallic Copper, Copper (I) Oxide, and Copper (II) Oxide—A Surface Science and Kinetic Study. J. Catal. 1994, 147, 567–577. [Google Scholar] [CrossRef]
- Sun, D.; Yamada, Y.; Sato, S.; Ueda, W. Glycerol hydrogenolysis into useful C3 chemicals. Appl. Catal. B Environ. 2016, 193, 75–92. [Google Scholar] [CrossRef] [Green Version]
- Vissers, J.; Bouwens, S.; De Beer, V.; Prins, R. Carbon black-supported molybdenum sulfide catalysts. Carbon 1987, 25, 485–493. [Google Scholar] [CrossRef] [Green Version]
- Biniak, S.; Szymański, G.; Siedlewski, J.; Świątkowski, A. The characterization of activated carbons with oxygen and nitrogen surface groups. Carbon 1997, 35, 1799–1810. [Google Scholar] [CrossRef]
- Gardner, S.D.; Singamsetty, C.S.; Booth, G.L.; He, G.-R.; Pittman, C.U. Surface characterization of carbon fibers using angle-resolved XPS and ISS. Carbon 1995, 33, 587–595. [Google Scholar] [CrossRef]
- Sun, Y.; Tian, P.; Ding, D.; Yang, Z.; Wang, W.; Xin, H.; Xu, J.; Han, Y.-F. Revealing the active species of Cu-based catalysts for heterogeneous Fenton reaction. Appl. Catal. B Environ. 2019, 258, 117985. [Google Scholar] [CrossRef]
- Bienholz, A.; Hofmann, H.; Claus, P. Selective hydrogenolysis of glycerol over copper catalysts both in liquid and vapour phase: Correlation between the copper surface area and the catalyst’s activity. Appl. Catal. A Gen. 2011, 391, 153–157. [Google Scholar] [CrossRef]
- Huang, Z.; Cui, F.; Xue, J.; Zuo, J.; Chen, J.; Xia, C. Cu/SiO2 catalysts prepared by hom- and heterogeneous deposition–precipitation methods: Texture, structure, and catalytic performance in the hydrogenolysis of glycerol to 1,2-propanediol. Catal. Today 2012, 183, 42–51. [Google Scholar] [CrossRef]
- Sharma, R.V.; Kumar, P.; Dalai, A.K. Selective hydrogenolysis of glycerol to propylene glycol by using Cu:Zn:Cr:Zr mixed metal oxides catalyst. Appl. Catal. A Gen. 2014, 477, 147–156. [Google Scholar] [CrossRef]
- Vila, F.; Granados, M.L.; Ojeda, M.; Fierro, J.; Mariscal, R. Glycerol hydrogenolysis to 1,2-propanediol with Cu/γ-Al2O3: Effect of the activation process. Catal. Today 2012, 187, 122–128. [Google Scholar] [CrossRef]
- Vasiliadou, E.; Eggenhuisen, T.; Munnik, P.; De Jongh, P.; De Jong, K.; Lemonidou, A. Synthesis and performance of highly dispersed Cu/SiO2 catalysts for the hydrogenolysis of glycerol. Appl. Catal. B Environ. 2014, 145, 108–119. [Google Scholar] [CrossRef]
- Gallegos-Suarez, E.; Perez-Cadenas, M.; Guerrero-Ruiz, A.; Rodríguez-Ramos, I.; Arcoya, A. Effect of the functional groups of carbon on the surface and catalytic properties of Ru/C catalysts for hydrogenolysis of glycerol. Appl. Surf. Sci. 2013, 287, 108–116. [Google Scholar] [CrossRef]
- Montassier, C.; Dumas, J.; Granger, P.; Barbier, J. Deactivation of supported copper based catalysts during polyol conversion in aqueous phase. Appl. Catal. A Gen. 1995, 121, 231–244. [Google Scholar] [CrossRef]
- Gandarias, I.; Arias, P.L.; Requies, J.; Güemez, M.; Fierro, J. Hydrogenolysis of glycerol to propanediols over a Pt/ASA catalyst: The role of acid and metal sites on product selectivity and the reaction mechanism. Appl. Catal. B Environ. 2010, 97, 248–256. [Google Scholar] [CrossRef]
- Foo, G.S.; Wei, D.; Sholl, D.S.; Sievers, C. Role of Lewis and Brønsted Acid Sites in the Dehydration of Glycerol over Niobia. ACS Catal. 2014, 4, 3180–3192. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SUPPORTS | |||||||
|---|---|---|---|---|---|---|---|
| Cu Loading (wt%) | SBET (m2 g-1) | Vp (cm3 g-1) | Vm (cm3 g-1) | Vo (cm3 g-1) | E0 (mV) | Reducibility * (%) | |
| CGran | - | 1477 | 1.11 | 0.72 | 0.39 | 171.5 | - |
| CGran (450) | 1022 | 0.95 | 0.58 | 0.37 | |||
| CGran (600) | 893 | 0.84 | 0,51 | 0.33 | |||
| CGran (750) | 881 | 0.82 | 0.50 | 0.32 | |||
| CGran (1000) | 667 | 0.64 | 0.38 | 0.26 | |||
| CATALYSTS | |||||||
| Cu/CGran | 7.4 | 559 | 0.52 | 0.32 | 0.20 | −134.9 | 86 |
| Cu/CGran (450) | 5.8 | 687 | 0.66 | 0.39 | 0.27 | −140.8 | 94 |
| Cu/CGran (600) | 5.8 | 652 | 0.62 | 0.37 | 0.25 | −144.5 | 95 |
| Cu/CGran (750) | 5.7 | 579 | 0.55 | 0.33 | 0.22 | −149.4 | 97 |
| Cu/CGran (1000) | 6.3 | 423 | 0.44 | 0.24 | 0.20 | −158.3 | 81 |
| Sample | Consumed H2 (μmol/gcat) | Cu Active Sites × 1021 (N2O TPR) | Cu Bulk × 1020 (AAS) |
|---|---|---|---|
| Cu/CGran | 829 | 1.00 | 7.02 |
| Cu/CGran (450) | 983 | 1.18 | 5.50 |
| Cu/CGran (600) | 1635 | 1.97 | 5.50 |
| Cu/CGran (750) | 1376 | 1.66 | 5.41 |
| Cu/CGran (1000) | 723 | 8.71 | 6.00 |
| Catalysts | C1s (eV) | Cu2p3/2 (eV) | Cu/C (exp.) | Cu/C (nominal) | αCu (eV) |
|---|---|---|---|---|---|
| Cu/CGran | 284.8 (76) 286.2 (21) 287.8 (3) | 933.0 | 0.022 | 0.015 | 1851.1 |
| Cu/CGran (450) | 284.8 (77) 286.2 (20) 287.8 (3) | 933.0 | 0.016 | 0.012 | 1851.0 |
| Cu/CGran (600) | 284.8 (78) 286.2 (20) 287.8 (2) | 933.0 | 0.018 | 0.012 | 1851.2 |
| Cu/CGran (750) | 284.8 (77) 286.2 (20) 287.8 (3) | 933.0 | 0.017 | 0.012 | 1851.0 |
| Cu/CGran (1000) | 284.8 (76) 286.2 (20) 287.8 (4) | 933.0 | 0.021 | 0.013 | 1850.9 |
| Catalysts | Conversion (%) | Initial Rate (×10−3 molglyc gCat-1 s-1) | Intrinsic Rate (molecglyc ASCu-1 s−1) | Selectivity (%) (10% of glyc Conversion) | ||||
|---|---|---|---|---|---|---|---|---|
| 1,2-PDO | acetol | 2-Pro | Other | Minor Products | ||||
| Cu/CGran | 16.6 | 0.85 | 0.51 | 78.6 | 0.00 | 18.4 | 2.26 | 0.75 |
| Cu/CGran (450) | 13.6 | 1.04 | 0.53 | 81.0 | 3.13 | 11.4 | 3.91 | 0.60 |
| Cu/CGran (600) | 16.8 | 1.38 | 0.42 | 78.3 | 3.42 | 12.8 | 3.82 | 1.73 |
| Cu/CGran (750) | 21.8 | 1.70 | 0.61 | 94.7 | 2.88 | 1.91 | 0.44 | 0.00 |
| Cu/CGran (1000) | 16.9 | 1.39 | 0.10 | 77.2 | 1.12 | 11.8 | 8.57 | 1.61 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seguel, J.; García, R.; Chimentão, R.J.; García-Fierro, J.L.; Ghampson, I.T.; Escalona, N.; Sepúlveda, C. Thermal Modification Effect on Supported Cu-Based Activated Carbon Catalyst in Hydrogenolysis of Glycerol. Materials 2020, 13, 603. https://doi.org/10.3390/ma13030603
Seguel J, García R, Chimentão RJ, García-Fierro JL, Ghampson IT, Escalona N, Sepúlveda C. Thermal Modification Effect on Supported Cu-Based Activated Carbon Catalyst in Hydrogenolysis of Glycerol. Materials. 2020; 13(3):603. https://doi.org/10.3390/ma13030603
Chicago/Turabian StyleSeguel, Juan, Rafael García, Ricardo José Chimentão, José Luis García-Fierro, I. Tyrone Ghampson, Néstor Escalona, and Catherine Sepúlveda. 2020. "Thermal Modification Effect on Supported Cu-Based Activated Carbon Catalyst in Hydrogenolysis of Glycerol" Materials 13, no. 3: 603. https://doi.org/10.3390/ma13030603
APA StyleSeguel, J., García, R., Chimentão, R. J., García-Fierro, J. L., Ghampson, I. T., Escalona, N., & Sepúlveda, C. (2020). Thermal Modification Effect on Supported Cu-Based Activated Carbon Catalyst in Hydrogenolysis of Glycerol. Materials, 13(3), 603. https://doi.org/10.3390/ma13030603