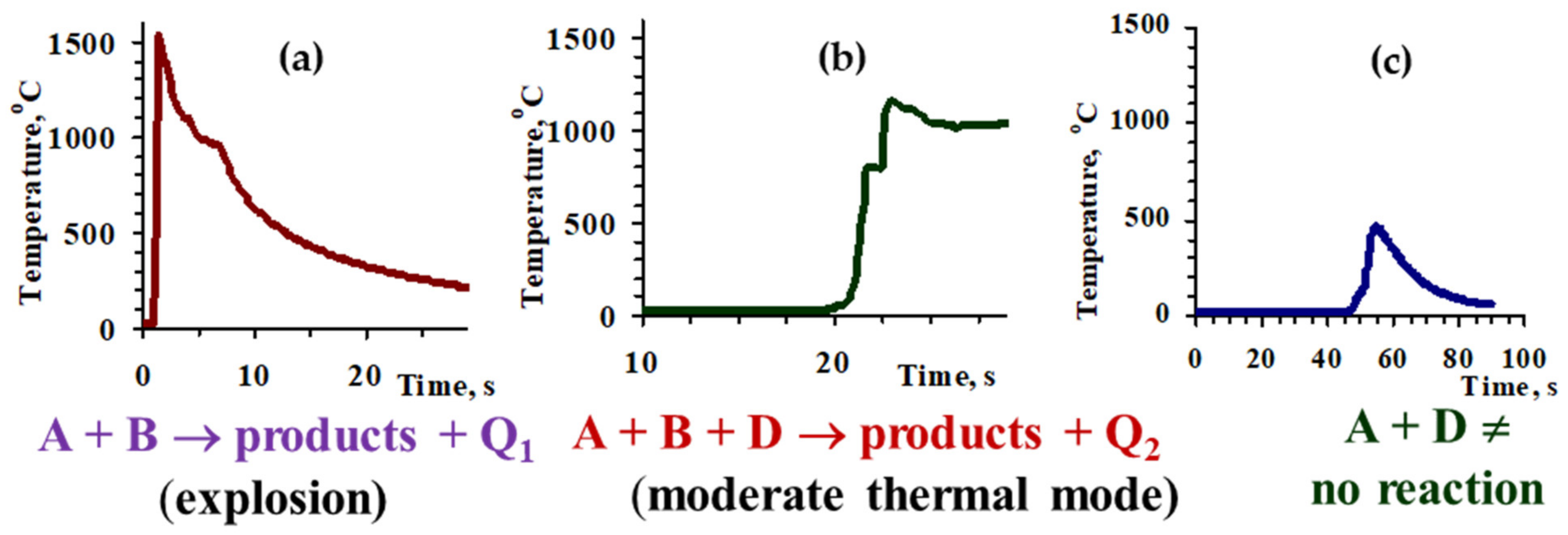
The role of coupling on the nature of combustion thermograms is demonstrated on the example of the MoO
3-CuO-Mg-C system. The combustion process in the (MoO
3-CuO)(A) + Mg(B) mixture proceeds in an explosion mode (
Figure 2a), and the pure carbothermic reaction (MoO
3-CuO)(A) + C(D) does not have enough exothermicity to self-propagate (
Figure 2c), whereas the introduction of carbon to the MoO
3-CuO-Mg mixture reduces combustion parameters and allows to perform the combustion process at a moderate combustion regime (A + B + D) [
24]. Therefore, the addition of carbon has a disproportionate influence on the combustion velocity and temperature, and thereby on the microstructure and phase composition of the products. In particular, the addition of a small amount of carbon (up to 0.7 wt%) to the MoO
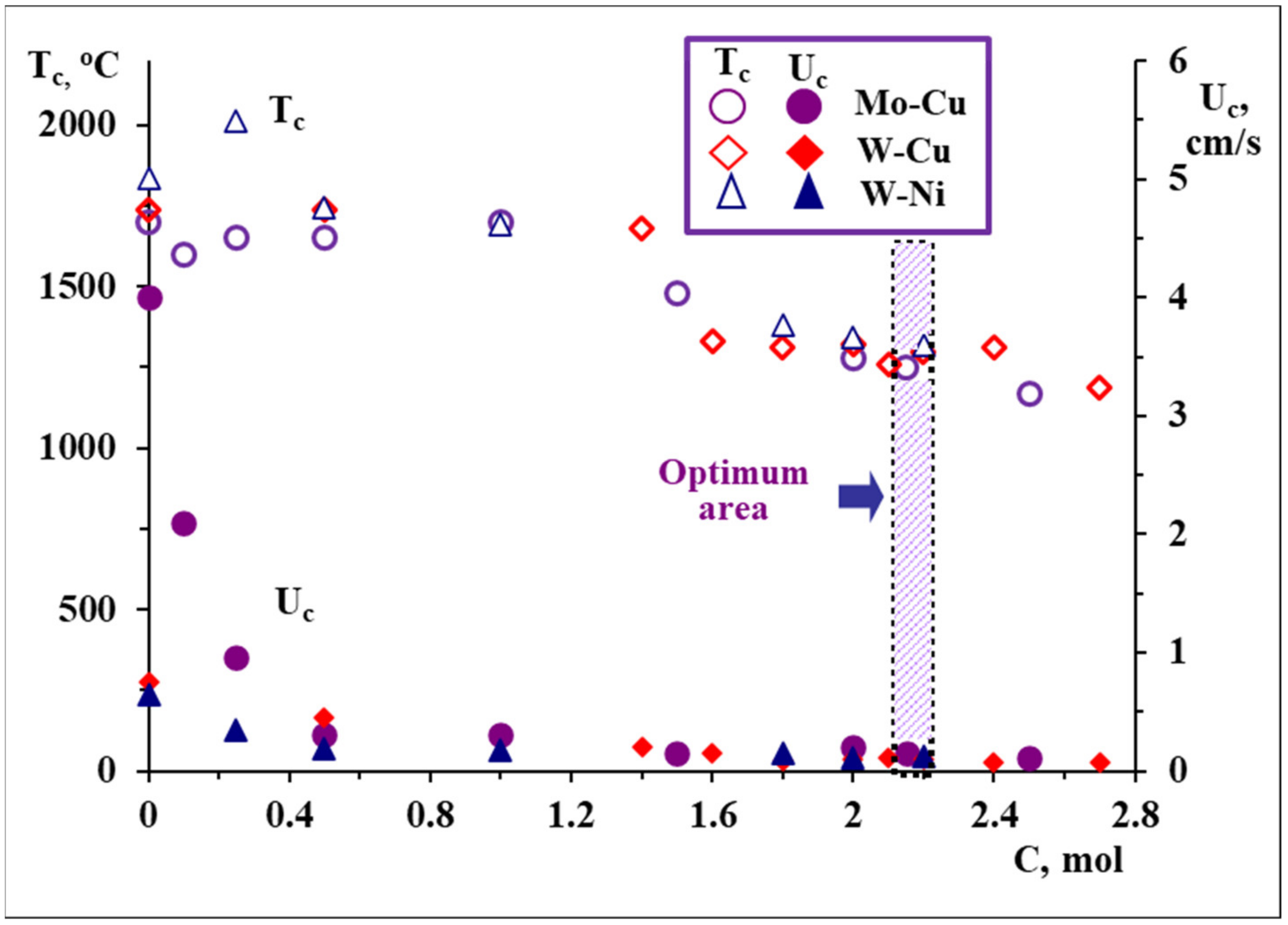
3 + CuO + Mg green mixture causes a decrease in combustion velocity of more than 10-fold (
Figure 2a and
Figure 3). At that velocity, the combustion temperature decreases by only 300 °C (
Figure 3). Note that, in parallel, a sharp decline of the heating rate of the reagents also occur (by one order in the CuO-MoO
3-Mg-C system) and a double-stage nature of the combustion wave appears on the combustion thermogram (
Figure 2b and
Figure 3).
2.2.1. Copper-Tungsten Bimetallic System
Copper (II) oxide (High grade, STANCHEM, Poland, <40 µm), tungsten (VI) (High grade, Krasniy khimik, Ukraine, <40 µm) oxide, carbon black (P-803, Russia, <1 µm) and magnesium (MPF-3, Russia, 150–300 µm) were used to prepare Cu-W powders in combustion experiments. Cylindrical samples were fabricated from the initial mixture of the reactants via uniaxial pressing (P = 1 kN) and placed in a constant pressure reactor (CPR-3l reaction chamber). The reactor was filled with nitrogen (purity 99.97%) to a pressure of up to 3.0 MPa. The combustion reaction was initiated by a short-term annealing of a tungsten coil. Temperature-time histories of the combustion process were recorded by W-Re thermocouples (5Re/20Re). The preparation of samples for the typical combustion experiment and registration of combustion parameters is thoroughly described elsewhere [
1,
3,
10]. A preliminary thermodynamic consideration of the WO
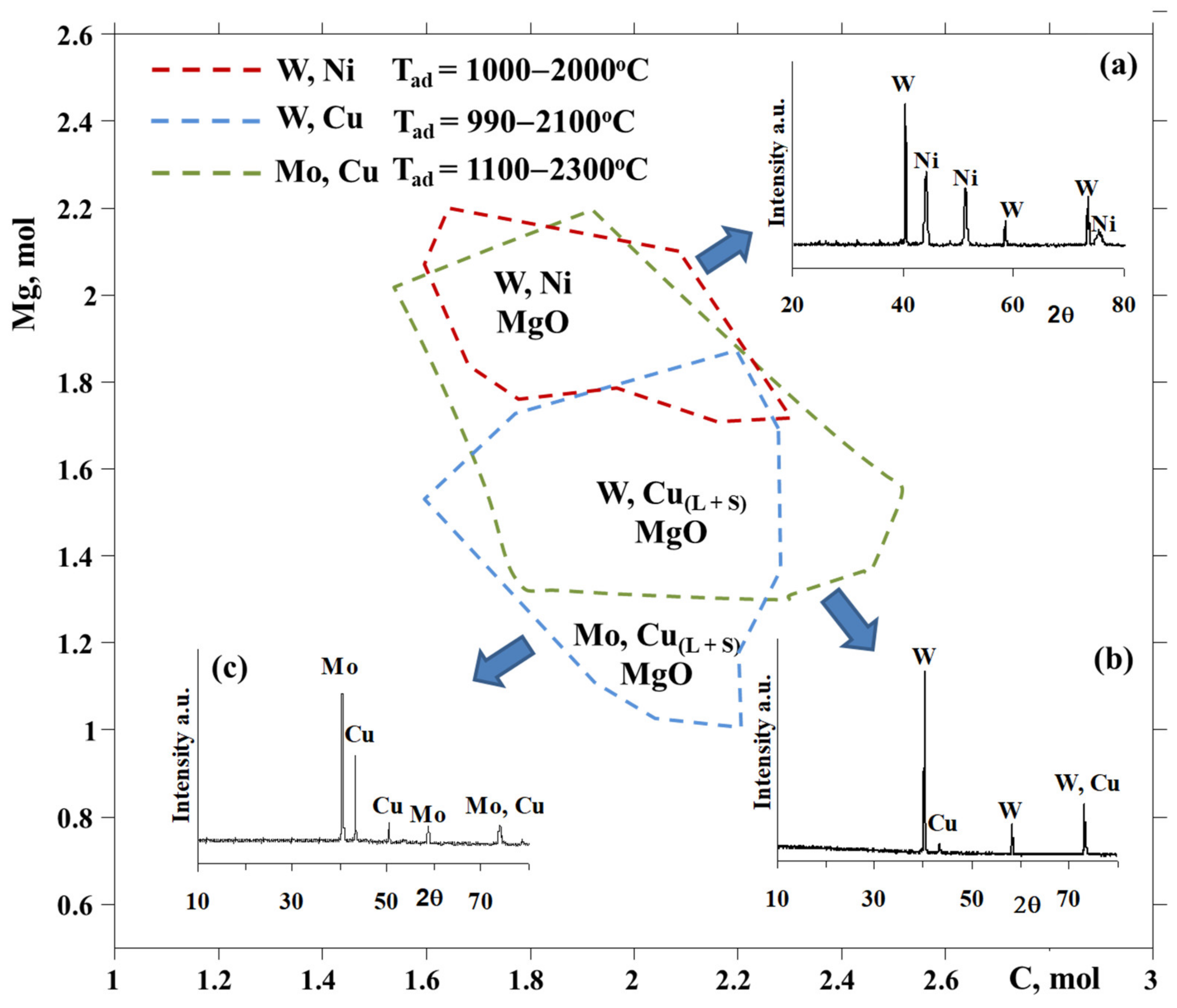
3-CuO-yMg-xC system showed that Cu:W of 1:1 composition can be synthesized due to the variation of the amount of Mg between 1.3 and 2.2 mol at a change in carbon amount in the range of 1.5 to 2.5 mol (
Figure 1).
The combustion reaction was implemented at a gas pressure of P = 0.3 MPa in order to avoid the evaporation of various WO
x oxides, magnesium and copper. According to thermodynamic modelling, the combined magnesio-carbothermic reduction of copper and tungsten oxides was performed in the WO
3-CuO-yMg-xC system at a fixed amount of Mg (y = 1.3 mol) and various amounts of the carbon reducer. Thermodynamic data were rather supported by experiments and indicated the reasonable choice of reducers’ amounts in order to provide a complete and joint reduction of metals at moderate temperatures (
Figure 3) [
10].
A number of SHS experiments were performed, with the aim to explore the overall (qualitative and quantitative) impact of carbon on the peculiarities of self-sustained reaction, morphology evolution and phase formation patterns. It was revealed that the decrease in combustion temperature and velocity versus the increase in carbon amount was observed as a result of the increased fraction of low-exothermic carbothermic process (
Figure 3).
XRD examinations of the solid combustion products for the WO
3 + CuO + 1.3Mg + xC mixtures indicated that a carbon amount of 2.1–2.2 mol makes it possible to produce target compounds of combustion (
Figure 1b), i.e., W-Cu and its by-product, MgO, which was leached by 10 wt% hydrochloric acid. The preparation of the W-Cu = 1:1 alloy becomes feasible at T
c = 1150–1300 °C, which is close to the optimum and low-temperature area of thermodynamic prediction.
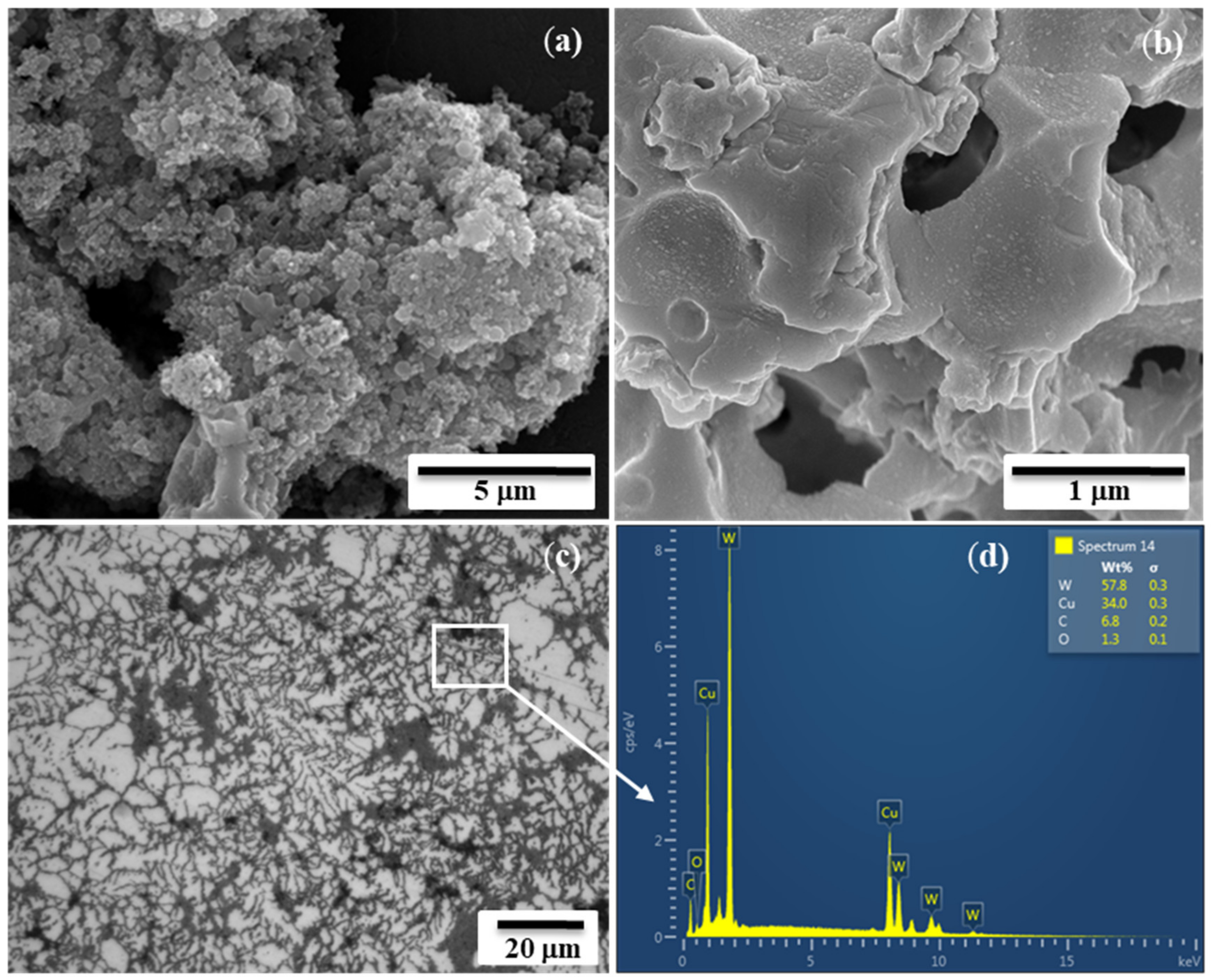
Microstructural examinations combined with XRD analysis demonstrated the presence of fine and snowflake-like particles of the W-Cu alloy in the submicron range and with ~1.4 m
2·g
−1 SSA (specific surface area) after the acid leaching of the combustion products (
Figure 4a). The partial merging of the particulates (up to tenths of microns) in the combustion wave was observed due to a higher combustion temperature than the melting point of copper (
Figure 4b). The presence of such agglomerates gives a predisposition for subsequent consolidation via promoting the emergence of intimate bindings among grains and boundaries, thereby reducing diffusion distances and significantly facilitating the sintering of SHS powders.
The fabrication of W-Cu green parts of a high relative density from the combustion synthesized powders was performed via the hot explosive consolidation (HEC) method. For the HEC procedure, the SHS-derived tungsten–copper composite powder was poured into a cylindrical tube container made of steel and pre-densified with a static pressure of 1.5 t load. The compacts of powdered samples were processed by the dynamic pre-densification of coupons at a room temperature and a hot explosive consolidation from 700 to 1050 °C (approximately 10 to 20 K∙s-1 within 0.3 to 1 min).
Cylindrical steel tubes of various dimensions were utilized to optimize the consolidation of W-Cu alloys at certain conditions of the detonation pressure, temperature and initial density. Optimized parameters were identified according to pre-densification at room temperature, firstly applying static pressure via compression at different intensities (1.5 t), and then explosive densification at 950, 1000 and 1050 °C, with a loading intensity of 10 GPa [
10,
11].
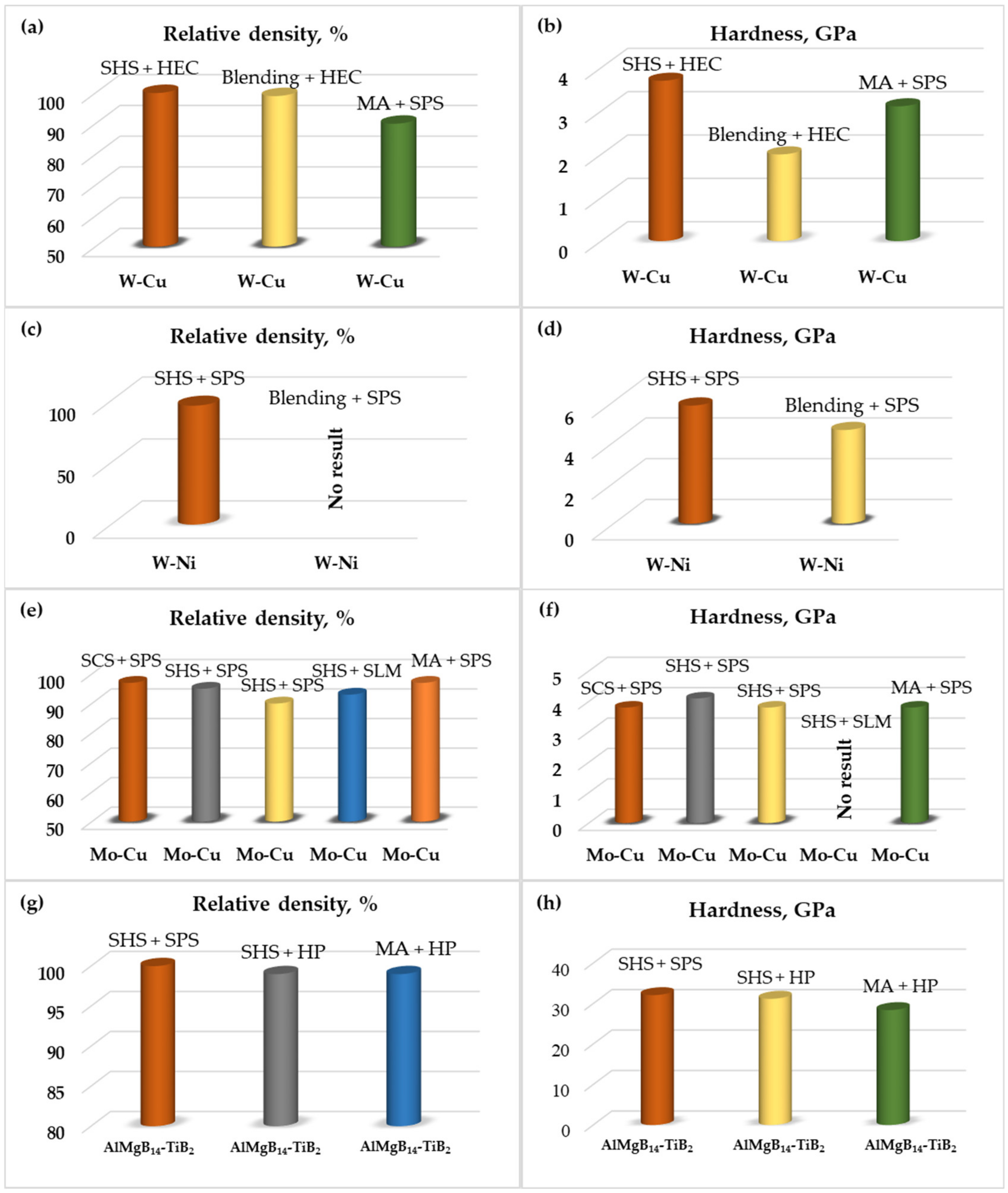
HEC-prepared billets from fine SHS-derived powders exhibited a severe diamagnetic susceptibility response. In addition, the microhardness and density measurements manifested that W-Cu alloys obtained by the SHS method via the coupling approach and subsequently compacted by explosive technology comprise an approximately twofold increased microhardness, as compared to those obtained by mechanical alloying (370 kg·mm−2 vs 200 kg·mm−2), and have a relative density near to the theoretical value (according to both the geometrical and Archimedes values).
The SEM analysis results indicated that 1000 °C is considered an optimum temperature for the complete annealing of microscopic defects and internal stresses from the W-Cu alloys. The optimum annealing temperature was also found to be 1000 °C, according to both the internal friction and Young modulus (E) values. However, they depend not only on the annealing conditions, but also on the history of the sample; in particular, the conditions of powder synthesis, post-treatment, distribution of dendrites, etc.
Microstructural and spectral analyses indicated a homogeneous structure of the obtained W-Cu composites (dark gray corresponds to copper, and light gray corresponds to tungsten) (
Figure 4c,d). According to those examinations, the surface of the compacted specimens was almost free of cracks and visible defects. Dendrite formation in W-Cu composite materials is conditioned by the distinct differences in the metals’ melting points, crystal structure and insolubility in each other. The supercooling of the material after HEC and the anisotropy in the surface energy of the tungsten–copper interface led the solid nucleus of tungsten-rich particles to grow in the under-sintered pool of copper in preferred growth directions of the tungsten. Such a highly textured dendrite microstructure of micron-size-W rich dendrites may provide a greater freedom of expansion and contraction (plasticity) when exposed to high thermal loads, suggesting them as the potential candidates for high temperature coatings. On the contrary, the utilization of mechanically alloyed commercial powders for shock wave consolidation has not allowed for the achievement of dense samples (<95%) [
25]. High-velocity wave propagation extrudes copper from the adjacent areas of W particles; they instantaneously impact each other and aggregate, creating both pores and the inhomogeneous distribution of constituents.
2.2.2. Nickel-Tungsten Bimetallic System
Nickel (II) oxide (Alfa Aesar, >99%, <44 μm), tungsten (VI) oxide (High grade, Krasniy khimik, Ukraine, <40 µm), carbon black (P-803, Russia, <1 µm) and magnesium (MPF-3, Russia, 150–300 µm) were used to prepare Ni-W powders in combustion experiments. Thermodynamic calculations in the NiO-WO
3-yMg-xC system demonstrated that the formation of a target product (Ni:W = 1:1 molar ratio) is achievable in a certain amount of carbon (from 1.6 to 2.3 mol) and magnesium (from 1.7 to 2.2 mol) at a 1000–2000 °C adiabatic temperature interval, allowing to design the optimum synthesis circumstances for the simultaneous and entire reduction of tungsten and nickel oxides in a wide range of compositions and thermal conditions (
Figure 1) [
26]. The probable formation of tungsten carbides (WC and W
2C) was ruled out during the thermodynamic calculations. According to the results of TC, the amount of magnesium selected was around 1.7–1.8 mol, in order to be sufficient to implement the self-sustaining reaction and provide the complete reduction of oxides at comparatively lower temperatures at a certain amount of carbon. By the next step, the influence of the carbon quantity on the combustion parameters and products’ characteristics was revealed, with the aim to find out the optimum composition of the NiO + WO
3 + yMg + xC mixture in order to produce the W-Ni alloy (
Figure 3). By the same token, the magnesio-carbothermic combined reduction of WO
3 and NiO was performed via changing the amount of carbon within a prescribed range, in accordance with the TC predicted area. It was revealed that the increase in the carbon amount is responsible for the reduction in the combustion parameters (temperature and velocity), conditioned by a consistent increase in the contribution of the low-caloric carbothermic reaction in the system in one hand, and the change in the interaction mechanism in another [
26]. Specifically, at a low amount of carbon, molten NiO participates in the process, and at a high carbon content, solid NiO participates in the process. The investigation of the interaction mechanism in the current mixture revealed that, in contrast to the CuO-WO
3-Mg-C system, the interaction is initiated with the magnesiothermic reduction of NiO, which is decisive for the heat release and self-propagation of the combustion wave. The reduction process is then continued with the magnesio-carbothermic reduction of WO
3, and accompanied by the Ni-rich phase (Ni
17W
3 or Ni
4W) formation. An unconsumed amount of oxides reduce up to the metals due to the carbothermic process. In addition, the combustion temperature in the system under consideration is comparatively lower (even lower than the melting temperature of nickel (fusible metal)), and at that temperature, the reduced metallic components are in a solid state and are of more homogeneity as compared to bimetallic systems with molten ingredients. Here, nickel and tungsten form nickel and/or tungsten-rich solid solutions or nickel-rich intermetallic with a particular ratio of elements (Ni
17W
3 or Ni
4W), along with the tungsten-rich phase corresponding to the W-Ni composition (1:1 molar ratio) (
Figure 1a).
Based on the XRD patterns of the products, the optimum conditions, according to the initial mixture composition and inert gas pressure, were determined for the preparation of the W-Ni bimetallic system. It was revealed that the conversion degree tends to increase when increasing the amount of carbon in the reactive mixture. The XRD showed that the reduction degree increases with an increase in carbon amount;), the optimum area of the target alloy preparation according to the carbon amount (x = 2.2–3 mol) was also determined. The absence of peaks of metallic nickel and nickel oxide from XRD patterns, as well as the absence of magnetic properties, confirm the formation of the W-Ni bimetallic system. Note that two different combustion modes were observed, according to the carbon amount in the NiO-WO
3-yMg-xC system: a steady state (with a carbon amount of around 2.2 mol) and an unsteady mode at a high-carbon content (5 mol). The latter corresponds to the spin combustion mode, demonstrating a change in both the microstructure and phase composition of the products [
26].
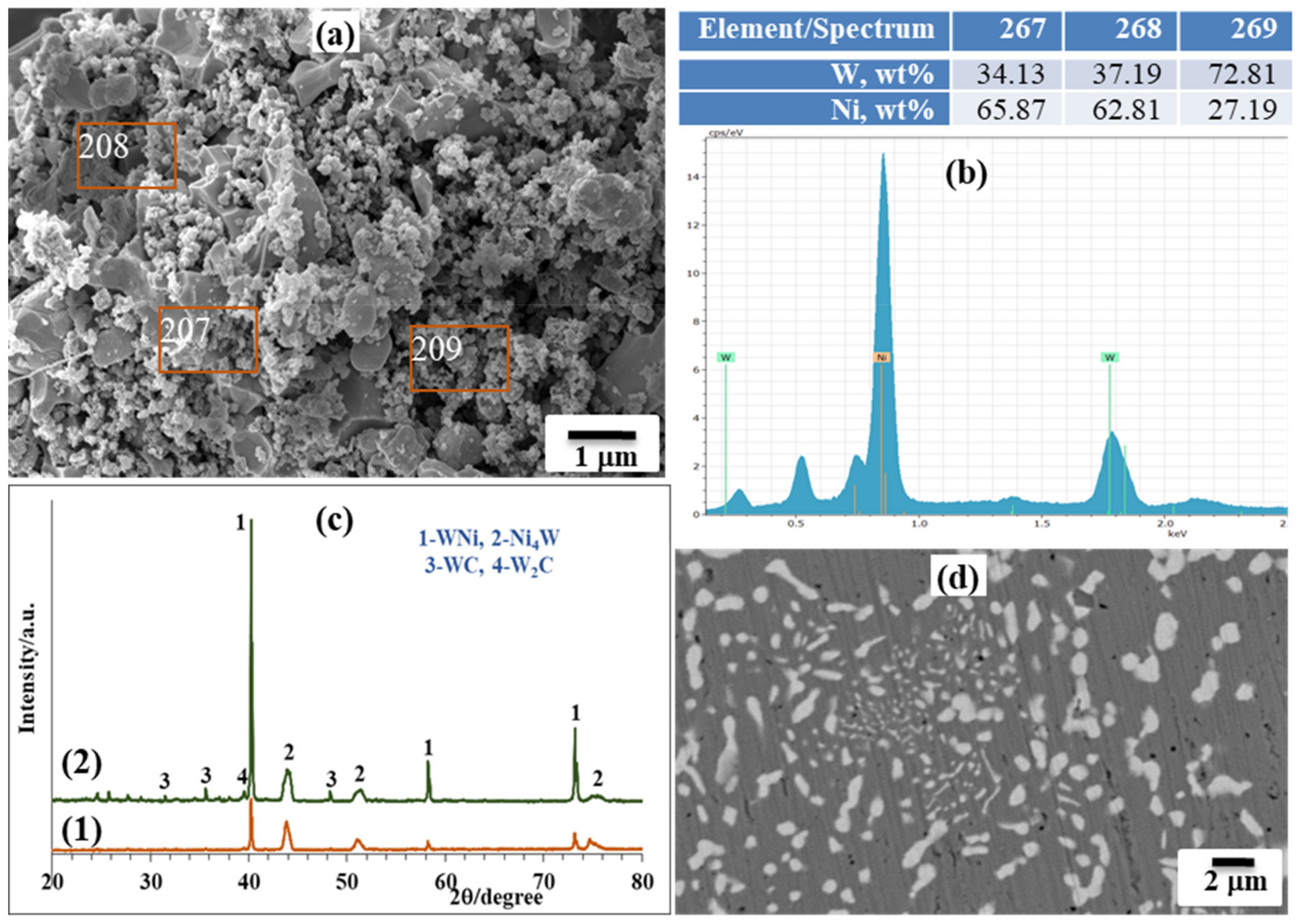
The target SHS product was crushed into a powder after cooling and hydrochloric acid leaching was used to eliminate byproduct magnesia. XRD and SEM/EDS examinations showed that the product after acid treatment contained mainly Ni and W elements, with an average particle size in the submicron range (
Figure 5a,b), illustrating the homogeneous dispersion of both metals throughout the sample. It is obvious that the characteristic peaks of tungsten are broadened and correspond to the W-Ni solid solution, and that the nickel peaks are right-shifted and broadened (Ni-W solid solution).
Emphasizing the influence of reactions’ coupling on the microstructure features, one may observe the fine microstructure formation during the addition of carbon, accompanied with a simultaneous decline in combustion temperature and velocity, and, as a result, the fabrication of the W-Ni bimetallic system at moderate and tailored conditions.
SHS-derived powders were ball milled for 30–60 min at a fixed rotation speed of 200 rpm at a ball-to-powder weight ratio of 4:1, and were subsequently compacted by a spark plasma sintering (SPS) apparatus (KCE®-FCT HP D 10-GB, FCT Systeme GmbH, Rauenstein, Germany) in a vacuum (5·10–2 mbar). Sintering conditions were as follows: sintering temperature of 950–1350 °C, 50–100 MPa pressure and dwell time of 3–10 min. The heating rate employed was 100 °C·min−1 for the ramp-up and the cooling rate was set up to ~200 °C·min−1. SPS-produced samples from the Ni-W bimetallic system sintered at 1200 °C temperature and at a 50 MPa pressure within a 5 min dwell time exhibited a 96% relative density.
XRD analysis demonstrated no dramatic change in phase composition after SPS processing (
Figure 5c). Trace amounts of tungsten carbides observed are conditioned by the carbon uptake during sintering due to the tungsten’s affinity to carbon at the sintering temperature; as powders were consolidated in graphite dies, graphite punches and protective graphite foil were also utilized. Vickers microhardness was measured to be 5.8 ± 0.6 GPa (HV1), which is comparable (or even higher) with results reported elsewhere [
27,
28,
29] for the Ni-W alloys (4.5–5.5 GPa). The increased microhardness value is solely conditioned by an increased W amount in the Ni lattice structure. The microstructure of the alloy after sintering illustrates the homogeneous distribution of the tungsten-rich phase (light grey) in the nickel-rich phase matrix (dark grey) (
Figure 5d). In addition, the influence of the sintering duration showed that 3 min of sintering is not enough to complete the process, as there are under-sintered areas; and 10 min resulted in fusion of the powder (
Figure 6).
EDS mapping of the fractured surface revealed the relatively homogeneous distribution of Ni and W elements (
Figure 7). An increase in dwelling time of up to 10 min allowed to reduce the porosity to 1%. A further increase in sintering duration is not desirable, as the Ni
2W
4C phase may be formed from the surface to throughout the thickness of the sintered compact [
30]. However, due to partial agglomeration and the pre-sintered state of the SHS-derived Ni-W powder, at a moderate sintering duration (up to 10 min), the diffusion of carbon occurs only on the surface of powder particles, in contrast to mechanically alloyed powders. In particular, interfaces between grains of initial powders remain structurally different from the volume, offer tracks for quick carbon diffusion and favor the carbides’ formation. In [
31], mechanical alloying was used to both in situ fabricate the Ni-W solid solution alloy matrix from a Ni-30 wt% W powder mixture and sinter coupons at 1000 °C during 3 min of a 96–97% relative density. Microstructural examinations showed the homogeneous distribution of phases in a micrometer range; however, the materials were characterized by some residual porosity. The authors suggested to utilize annealing prior to SPS, which may allow eliminating contaminations and increasing the relative density. Another approach to enhance the microstructural homogeneity and relative density of the Ni-W composite is using NiWO
4 as a precursor for Ni-W alloy synthesis, or avoiding graphite dies during the sintering, which changes the reciprocal diffusion in the sintered material paired with h-BN or aluminum, as suggested in [
32].
2.2.3. Copper-Molybdenum Bimetallic System
As Mo-Cu materials with a homogeneous microstructure are difficult to achieve with conventional methods, various attempts have been carried out to produce fine and homogeneously dispersed Mo-Cu powders, utilizing a similar pathway for W-Ni and W-Cu composite materials. The increased scientific intrigue in Mo-Cu materials is conditioned by the fact that Mo and Cu form a pseudo-alloy or composite material, where constituents are either the matrix or dispersing component depending on the ratio of Mo and Cu. In order to achieve the purpose, oxygen-containing compounds of molybdenum and copper were jointly reduced utilizing the reactions’ coupling approach and, in particular, the Mg + C mixture was used as a combined reducer [
24]. The real possibility of the joint and the complete reduction of Mo and Cu metals were manifested at a certain amount of reducing mixture (certain ratio of Mg and C) and at moderate propagation velocity of the combustion wave. The obtained powders were consolidated using SPS in a vacuum at a sintering temperature of 950–1050 °C, with the simultaneous exploitation of 100 MPa pressure for a fixed dwell time (3, 5 or 10 min). The mould of 10 mm in diameter was charged with SHS-derived powders and heated up to a defined sintering temperature with a heating rate of 100–200 °C·min
−1.
Three pathways of powder synthesis were utilized.
One of the pathways is the preparation of Mo-Cu nanocomposites by the combination of energy-efficient solution combustion and the self-propagation of high-temperature synthesis. In the first step, a MoO
2 + Cu nanopowder mixture with an average particle size of 50 nm was prepared by SCS using ammonium heptamolybdate, copper nitrate as precursors and citric acid as a chelating reagent. During the second step via the SHS reduction of a fine MoO
2 + Cu powder mixture by a Mg/C combined reducer, a Mo-Cu nanocomposite (with 50–70 nm size) was produced (T
c = 1250 °C) and subjected to SPS at 950 °C for 6 min and 100 MPa pressure. SEM images demonstrate the particle size and shape preservation during the sintering (
Figure 8a–c) [
33]. The relative density of the samples was 97% and the Vickers hardness was around 3.8 GPa.
Using the second approach, Mo-Cu powders of 1–3 µm particle size were produced by the SHS of the CuO-MoO
3-yMg-xC mixture (y = 1.2; x = 2.1–2.2 mol; T
c = 1300 °C) from copper (II) oxide (High grade, STANCHEM, Poland, <40 µm), molybdenum (VI) (High grade, Pobedit Company, Russia, <15 µm) oxide, carbon black (P-803, Russia, <1 µm) and magnesium (MPF-3, Russia, 150–300 µm) (
Figure 3). Powders consolidated at identical conditions are presented in
Figure 8d–f. Sintered counter bodies comprise a 4.1 GPa Vickers hardness and a 95% relative density. In contrast to the sample derived by the first pathway, the Mo-Cu compact here has more porosity, but a higher hardness, most likely conditioned by a coarser grain size.
A similar approach was utilized for the preparation of the Mo-Cu bimetallic system from copper molybdates derived by calcination (I) and coprecipitation (II) methods, with a subsequent consolidation [
34].
According to the thermodynamic modelling and primary combustion experiments of the copper molybdate reduction using a Mg + C combined reducer, the amount of magnesium was selected as 1.2–1.5 mol in order to achieve complete reduction. The combustion velocity and the combustion temperature tend to slightly decrease with an increase in the carbon content in the mixture with copper molybdate derived by coprecipitation, whereas using calcined CuMoO
4 caused a substantial drop in the combustion velocity, which was registered by the addition of an even insignificant amount of carbon. The observed pattern was explained by the composition of CuMoO
4(II) when it was calcined for 1 h, and copper hydroxymolybdate decomposed to copper molybdate during the calcination process and combustion parameters of the calcined CuMoO
4(II) powder resembled the behavior of copper molybdate(I). With the addition in carbon, the reduction was accomplished by the formation of both metals. Accordingly, the corresponding mixtures with both salts, CuMoO
4(I) + 1.2Mg + 2.2C and CuMoO
4(II) + 1.5Mg + 1.6C, were selected for Mo-Cu alloy preparation. Despite the different amounts of reducers in the reactive mixture, both salts followed a similar reduction pathway, firstly converting into salts of Cu
2MoO
5 and Cu
6Mo
5O
18, then Cu and MoO
2 by carbon, and, at the end, into molybdenum by the magnesiothermic reduction of MoO
2 [
34].
XRD examinations of the final products indicate the characteristic diffraction lines of molybdenum and copper after the acid leaching procedure, and the complete elimination of the MgO byproduct. SEM micrographs of the target Mo-Cu products derived from both salts display the existence of spherical nanoparticles (50–100 nm) of a narrow distribution (
Figure 8g).
For the consolidation of produced SHS nanopowders Mo-Cu (1:1), the spark plasma sintering (SPS) was utilized. The sintering temperature has a detrimental effect on the density of the produced specimens. Particularly, when the sintering temperature increases by 50 °C (from 950 to 1000 °C), the increase in relative density by 10% occurs. In parallel, a shorter dwelling duration causes grains refinement and, consequently, an increase in hardness. According to multiparametric studies, the optimum sintering temperature was considered to be 1000 °C applied during 3 min for Mo-CuSHS-derived powders. Geometrical and Archimedes densities of Mo-Cu coupons fabricated at optimized conditions were measured as 8.8 g cm
−3. Accordingly, the relative density was >90%. Microhardness measurements using the Vickers indentation method showed that the HV5 of the Mo-Cu composite makes 3.2 GPa, which exceeds the hardness of pure copper by around ten times and the hardness of molybdenum by more than two times. High-energy ball milling (HEBM) for 1 h certified the increase in hardness of up to 3.8 GPa due to grain refinement; however, the substantial influence of the pre-treatment on the relative density of the compacts was not established. The hardness values for these nanostructured alloys exceeds twice the hardness of the materials prepared by mechanical alloying. The difference is obviously ascribed to the preserved nanostructure at a combination of SHS and SPS techniques (
Figure 8h,i).
According to the literature data, Mo-Cu billets sintered without any mechanical pre-treatment comprised spheroidal particles of molybdenum, and the room between them was captured by copper; the maximum density achieved was 88%. A commercial powder mixture of Cu and Mo being subjected to intense HEBM for 1 h led to the formation of a fine structured composite, according to the XRD pattern [
35], and Cu and Mo constituents were intermixed at the submicron level; however, SPS at similar sintering conditions did not allow to achieve full density compacts. The comparative analysis of SEM images and the characterization of compacts obtained from SHS-derived powders and HEBM processes demonstrate the privilege of SHS powders by path 1 and path 3 in terms of the microstructural homogeneity of the sintered counterpart and the microhardness. However, for the fabrication of high-density Mo-Cu compacts (98–99%), the laborious and expensive method of the liquid phase sintering of pre-alloyed Mo-Cu nanopowder (100–200 nm) is still preferable [
36].
Further, it was proposed to utilize Mo-Cu composite powders with a molar ratio of constituent metals of Mo:Cu = 1:1, obtained from oxide precursors in a combustion mode (
Figure 1c and
Figure 3), for further densification by the selective laser melting (SLM) technique in order to obtain dense cubic and lattice structured Mo-Cu shapes with the help of the SLM apparatus ReaLizer 50 GmbH (Frankfurt, Germany). Sintering conditions, such as the laser current (mA), scan (mm·s
−1), point distance (μm) and exposure time (μs), were adjusted for each system under consideration. The SLM machine employs a high-powered continuous-wave laser, which is modulated to function like a pulsed laser system. The Yb:YAG fiber laser, with a maximum power of 120 W and wavelength of 1.07 μm, was used to solidify the structures. The process of SLM was performed in an argon atmosphere of a high purity (99.999 vol.%). Mo-Cu lattice structures showed poor sinterability, and the splashing of the powder and deterioration of the designed structure occurred due to the fine-grained powder. Bulk samples sintered at a 900 mA laser current comprised an 85% relative density. With an increase in the laser current of up to 2500 mA (exposure time 125 µs, point distance 10 µm), a relative density of 93% was achieved, which is comparable with the W-Cu composite obtained by the SLM method (91.6%) [
37]. The laser current was revealed to be the most determinative parameter to achieve high relative density compacts of Mo-Cu and W-Cu. Considering the higher laser absorptivity of tungsten and molybdenum compared to copper, it is assumed that refractory metals absorb more laser energy than copper, resulting in the formation of a Cu molten pool and the solid-state sintering of a refractory skeleton almost contemporaneously. Hence, the SLM method has a perspective for the preparation of pseudoalloys comprising two or more distinctly different metals.
2.2.4. Thermochemically Coupled Synthesis of AlMgB14-TiB2 System with Subsequent Sintering
The thermochemical coupling approach was used to deliver AlMgB
14-TiB
2 composite materials by self-propagating high-temperature synthesis for subsequent SPS compaction at 1470 °C for a duration of 5 min [
38]. Ti (OJSC Polema), (purity 99.2%, average particle size 140 μm), Al
12Mg
17 (Original powder) (purity 99.2%, average particle size 20 μm) and boron (OJSC Aviabor), (purity 98.8%, average particle size 0.6 μm) were used as precursors in combustion experiments to prepare AlMgB
14-TiB
2 composite materials. The powder mixture of Ti and B elements was taken as a donor mixture, and the acceptor mixture encompassed the intermetallic powder of Al
12Mg
17 with the amorphous boron powder. For the combustion experiments, (Ti + 2B) and Al
12Mg
17:B powder mixtures were mixed in a mass ratio of 70 wt% (Al
12Mg
17:B) + 30 wt% (Ti + 2B) in ethanol. Samples with a diameter of 23 mm were cold-pressed from the dried powder mixture. Then, the samples were placed in an SHS reactor. The reactor was evacuated and filled with argon to a pressure of 2.5 MPa. After the reaction initiation in the Ti-B-Al
12Mg
17 system, the heating zone was formed enough to melt Al
12Mg
17 particles. The melt dissolved the Ti and B elements and the temperature increased up to 1580 °C as a result of the exothermic reaction of the (Ti + 2B) donor mixture. The heat released contributed to the synthesis reaction in the acceptor mixture, with a parallel decrease in the temperature down to 1400 °C. As the donor reaction is exothermic, a further increase in the temperature up to 1550 °C was registered before cooling. Under the influence of the high temperature and with enough time for cooling, TiB
2 crystal grains grew. The SHS-derived powder material contained particles with a ~1 μm average size (
Figure 9). TiB
2 particles were in the form of agglomerates of up to 30 μm in size due to recrystallization under the influence of elevated temperatures.
The simultaneous consolidation and sintering of the mixture were conducted at a temperature (T
s) of 1450 and 1470 °C, with 70 MPa of pressure. Heating rates (V
h) of 50 and 250 °C/min were used during the sintering. After the sintering without holding, the estimated average particle size of both phases, AlMgB
14 and TiB
2, was around 3–5 μm. The ceramic structure was not uniform, but was fully dense and exhibited an enhanced hardness of 32.1 GPa. The composite material demonstrated comparable characteristics to the material obtained from the hot pressed (1400 °C, 2 h) mixture of SHS-derived AlMgB
14 and commercial TiB
2 [
39] in terms of initial powder (particle size and distribution) and final compact (density and hardness).
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}