
Single-Crystal X-ray and Solid-State NMR Characterisation of AND-1184 and Its Hydrochloride Form


Abstract
:1. Introduction
2. Materials and Methods
2.1. Synthesis and Crystallisation of MBS
2.2. Crystal Structure Determination
2.3. NMR Spectroscopy
2.4. QM Calculations
3. Results
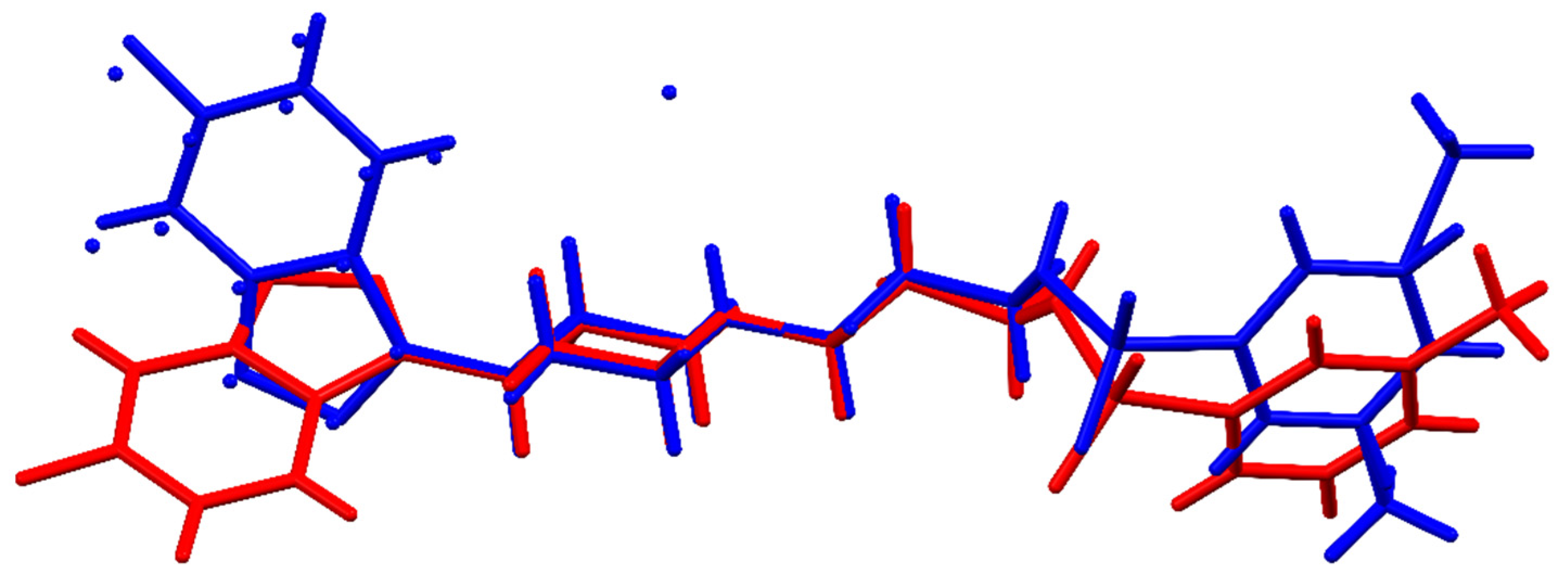
3.1. Single-Crystal X-ray Determination of the MBS and Its Comparison with the MBSHCl Form
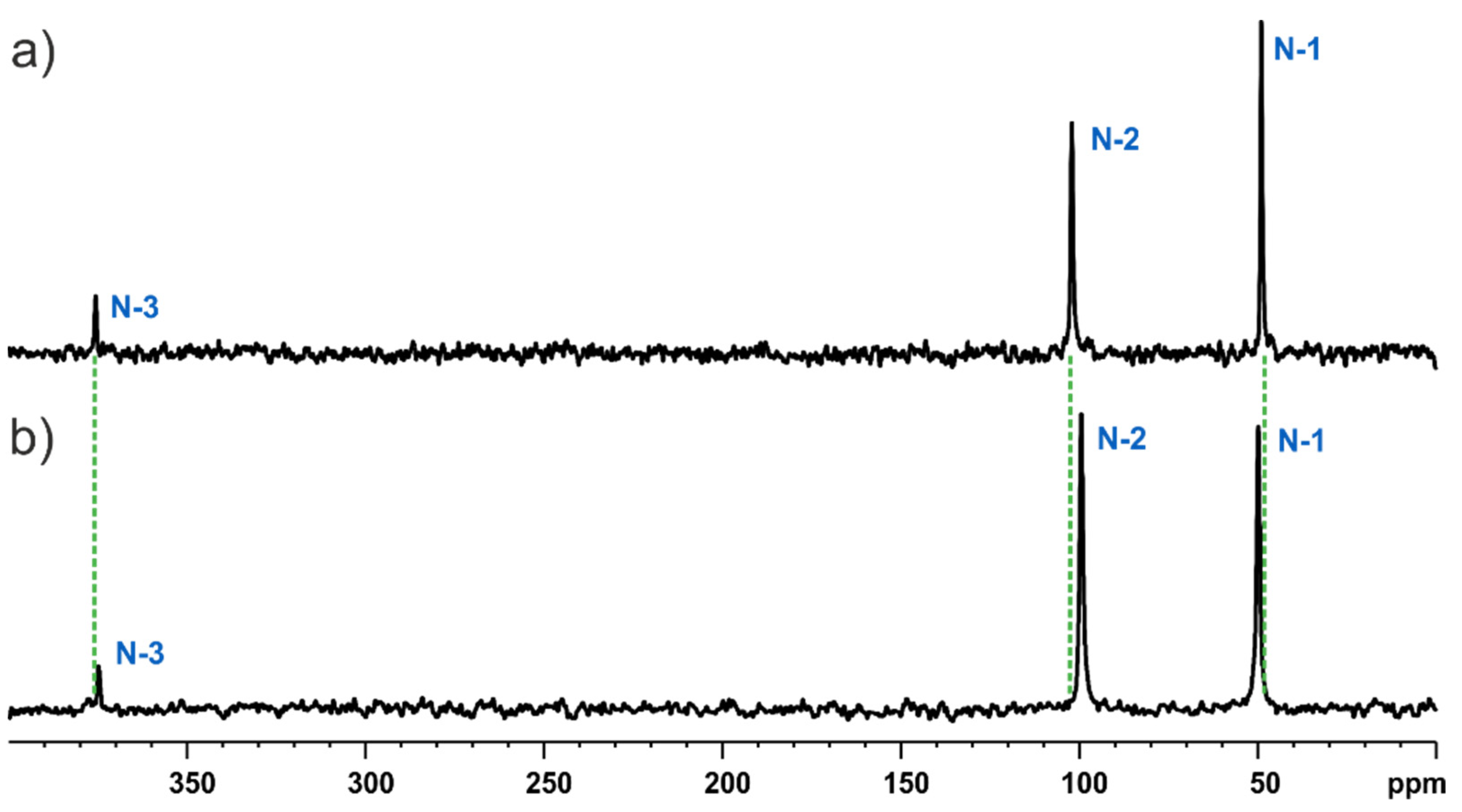
3.2. Solid-State NMR and DFT-D Characterisation of the MBS and MBSHCl
3.3. Fast MAS NMR Final Validation of Structures by Means of 1H Chemical Shifts
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- FDA Today | Generic Drug Approvals Fell in 2020 (and Are likely to Fall again in 2021). Available online: https://www.linkedin.com/pulse/fda-today-generic-drug-approvals-fell-2020-likely-gaffney-ms-rac (accessed on 19 August 2021).
- Braun, D.E.; Nartowski, K.P.; Khimyak, Y.Z.; Morris, K.R.; Byrn, S.R.; Griesser, U.J. Structural Properties, Order–Disorder Phenomena, and Phase Stability of Orotic Acid Crystal Forms. Mol. Pharm. 2016, 13, 1012–1029. [Google Scholar] [CrossRef] [Green Version]
- Beyer, T.; Day, G.M.; Price, S.L. The Prediction, Morphology, and Mechanical Properties of the Polymorphs of Paracetamol. J. Am. Chem. Soc. 2001, 123, 5086–5094. [Google Scholar] [CrossRef]
- Brittain, H.G. Polymorphism in Pharmaceutical Solids; Dekker, M., Ed.; CRC Press: Boca Raton, FL, USA, 1999; ISBN 978-0-585-15829-7. [Google Scholar]
- Raza, K.; Kumar, P.; Ratan, S.; Malik, R.; Arora, S. Polymorphism: The Phenomenon Affecting the Performance of Drugs. SOJ Pharm. Pharm. Sci. 2014, 1, 10. [Google Scholar]
- Yu, L.; Reutzel, S.M.; Stephenson, G.A. Physical Characterization of Polymorphic Drugs: An Integrated Characterization Strategy. Pharm. Sci. Technol. Today 1998, 1, 118–127. [Google Scholar] [CrossRef]
- Duer, M.J. Introduction to Solid-State NMR Spectroscopy; Blackwell: Oxford, UK; Malden, MA, USA, 2004; ISBN 978-1-4051-0914-7. [Google Scholar]
- Ashbrook, S.E.; Griffin, J.M.; Johnston, K.E. Recent Advances in Solid-State Nuclear Magnetic Resonance Spectroscopy. Annu. Rev. Anal. Chem. 2018, 11, 485–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, R.K. Applications of Solid-State NMR to Pharmaceutical Polymorphism and Related Matters. J. Pharm. Pharmacol. 2007, 59, 225–239. [Google Scholar] [CrossRef]
- Schechter, L.E.; Lin, Q.; Smith, D.L.; Zhang, G.; Shan, Q.; Platt, B.; Brandt, M.R.; Dawson, L.A.; Cole, D.; Bernotas, R.; et al. Neuropharmacological Profile of Novel and Selective 5-HT6 Receptor Agonists: WAY-181187 and WAY-208466. Neuropsychopharmacology 2008, 33, 1323–1335. [Google Scholar] [CrossRef] [PubMed]
- Liperoti, R.; Pedone, C.; Corsonello, A. Antipsychotics for the Treatment of Behavioral and Psychological Symptoms of Dementia (BPSD). Curr. Neuropharmacol. 2008, 6, 117–124. [Google Scholar] [CrossRef] [Green Version]
- Hersch, E. Management of the Behavioral and Psychological Symptoms of Dementia. Clin. Interv. Aging 2008, 2, 611–621. [Google Scholar] [CrossRef] [Green Version]
- Jeste, D.V.; Blazer, D.; Casey, D.; Meeks, T.; Salzman, C.; Schneider, L.; Tariot, P.; Yaffe, K. ACNP White Paper: Update on Use of Antipsychotic Drugs in Elderly Persons with Dementia. Neuropsychopharmacol 2008, 33, 957–970. [Google Scholar] [CrossRef]
- Carson, S.; McDonagh, M.S.; Peterson, K. A Systematic Review of the Efficacy and Safety of Atypical Antipsychotics in Patients with Psychological and Behavioral Symptoms of Dementia: Atypical Antipsychotics in Dementia. J. Am. Geriatr. Soc. 2006, 54, 354–361. [Google Scholar] [CrossRef] [PubMed]
- CrysAlisPro. Rigaku Oxford Diffraction; Agilent Technologies UK Ltd.: Oxford, UK, 2017. [Google Scholar]
- Sheldrick, G.M. A Short History of SHELX. Acta Cryst A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Cryst C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury CSD 2.0—New Features for the Visualization and Investigation of Crystal Structures. J. Appl. Cryst. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Spek, A.L. PLATON SQUEEZE: A Tool for the Calculation of the Disordered Solvent Contribution to the Calculated Structure Factors. Acta Cryst. C Struct. Chem. 2015, 71, 9–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fung, B.M.; Khitrin, A.K.; Ermolaev, K. An Improved Broadband Decoupling Sequence for Liquid Crystals and Solids. J. Magn. Reson. 2000, 142, 97–101. [Google Scholar] [CrossRef]
- Dvinskikh, S.V.; Zimmermann, H.; Maliniak, A.; Sandström, D. Heteronuclear Dipolar Recoupling in Liquid Crystals and Solids by PISEMA-Type Pulse Sequences. J. Magn. Reson. 2003, 164, 165–170. [Google Scholar] [CrossRef]
- Dvinskikh, S.V.; Sandström, D. Frequency Offset Refocused PISEMA-Type Sequences. J. Magn. Reson. 2005, 175, 163–169. [Google Scholar] [CrossRef]
- Ramamoorthy, A.; Opella, S.J. Two-Dimensional Chemical Shift/Heteronuclear Dipolar Coupling Spectra Obtained with Polarization Inversion Spin Exchange at the Magic Angle and Magic-Angle Sample Spinning (PISEMAMAS). Solid State Nucl. Magn. Reson. 1995, 4, 387–392. [Google Scholar] [CrossRef]
- Bruker Biospin Gmbh. In Topspin Software, Version 3.5; Bruker Biospin Gmbh: Karlsruhe, German, 2017.
- Mao, K.; Wiench, J.W.; Lin, V.S.-Y.; Pruski, M. Indirectly Detected Through-Bond Chemical Shift Correlation NMR Spectroscopy in Solids under Fast MAS: Studies of Organic–Inorganic Hybrid Materials. J. Magn. Reson. 2009, 196, 92–95. [Google Scholar] [CrossRef]
- Ishii, Y.; Tycko, R. Sensitivity Enhancement in Solid State 15N NMR by Indirect Detection with High-Speed Magic Angle Spinning. J. Magn. Reson. 2000, 142, 199–204. [Google Scholar] [CrossRef] [Green Version]
- Wiench, J.W.; Bronnimann, C.E.; Lin, V.S.-Y.; Pruski, M. Chemical Shift Correlation NMR Spectroscopy with Indirect Detection in Fast Rotating Solids: Studies of Organically Functionalized Mesoporous Silicas. J. Am. Chem. Soc. 2007, 129, 12076–12077. [Google Scholar] [CrossRef] [PubMed]
- Thakur, R.S.; Kurur, N.D.; Madhu, P.K. Swept-Frequency Two-Pulse Phase Modulation for Heteronuclear Dipolar Decoupling in Solid-State NMR. Chem. Phys. Lett. 2006, 426, 459–463. [Google Scholar] [CrossRef]
- Vinod Chandran, C.; Madhu, P.K.; Kurur, N.D.; Bräuniger, T. Swept-Frequency Two-Pulse Phase Modulation (SW f -TPPM) Sequences with Linear Sweep Profile for Heteronuclear Decoupling in Solid-State NMR. Magn. Reson. Chem. 2008, 46, 943–947. [Google Scholar] [CrossRef] [PubMed]
- Marion, D.; Ikura, M.; Tschudin, R.; Bax, A. Rapid Recording of 2D NMR Spectra without Phase Cycling. Application to the Study of Hydrogen Exchange in Proteins. J. Magn. Reson. (1969) 1989, 85, 393–399. [Google Scholar] [CrossRef]
- Morcombe, C.R.; Zilm, K.W. Chemical Shift Referencing in MAS Solid State NMR. J. Magn. Reson. 2003, 162, 479–486. [Google Scholar] [CrossRef]
- Harris, R.K.; Becker, E.D.; Cabral de Menezes, S.M.; Goodfellow, R.; Granger, P. NMR Nomenclature. Nuclear Spin Properties and Conventions for Chemical Shifts(IUPAC Recommendations 2001). Pure Appl. Chem. 2001, 73, 1795–1818. [Google Scholar] [CrossRef]
- Bertani, P.; Raya, J.; Bechinger, B. 15N Chemical Shift Referencing in Solid State NMR. Solid State Nucl. Magn. Reson. 2014, 61–62, 15–18. [Google Scholar] [CrossRef]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.J.; Refson, K.; Payne, M.C. First Principles Methods Using CASTEP. Z. Kristall. 2005, 220, 567–570. [Google Scholar] [CrossRef] [Green Version]
- McNellis, E.R.; Meyer, J.; Reuter, K. Azobenzene at Coinage Metal Surfaces: Role of Dispersive van Der Waals Interactions. Phys. Rev. B 2009, 80, 205414. [Google Scholar] [CrossRef] [Green Version]
- Ambrosetti, A.; Reilly, A.M.; DiStasio, R.A.; Tkatchenko, A. Long-Range Correlation Energy Calculated from Coupled Atomic Response Functions. J. Chem. Phys. 2014, 140, 18A508. [Google Scholar] [CrossRef] [Green Version]
- Vanderbilt, D. Soft Self-Consistent Pseudopotentials in a Generalized Eigenvalue Formalism. Phys. Rev. B 1990, 41, 7892–7895. [Google Scholar] [CrossRef] [PubMed]
- Nocedal, J.; Wright, S.J. Numerical Optimization; Springer Science & Business Media: New York, NY, USA, 2006; ISBN 978-0-387-40065-5. [Google Scholar]
- Monkhorst, H.J.; Pack, J.D. Special Points for Brillouin-Zone Integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Pickard, C.J.; Mauri, F. All-Electron Magnetic Response with Pseudopotentials: NMR Chemical Shifts. Phys. Rev. B 2001, 63, 245101. [Google Scholar] [CrossRef] [Green Version]
- Yates, J.R.; Pickard, C.J.; Mauri, F. Calculation of NMR Chemical Shifts for Extended Systems Using Ultrasoft Pseudopotentials. Phys. Rev. B 2007, 76, 024401. [Google Scholar] [CrossRef]
- Bernstein, J.; Davis, R.E.; Shimoni, L.; Chang, N.-L. Patterns in Hydrogen Bonding: Functionality and Graph Set Analysis in Crystals. Angew. Chem. Int. Ed. Engl. 1995, 34, 1555–1573. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 171–179. [Google Scholar] [CrossRef]
- Kalinowska-Tłuścik, J.; Piaskowska, A.; Kołaczkowski, M. Multifunctional Aryl sulfonamide Derivatives with 5-HT6/5-HT7 Receptor Antagonistic Activity: A Structural Study. Acta Crystallogr. Sect. C Struct. Chem. 2018, 74, 1477–1486. [Google Scholar] [CrossRef]
- Opella, S.J.; Frey, M.H. Selection of Nonprotonated Carbon Resonances in Solid-State Nuclear Magnetic Resonance. J. Am. Chem. Soc. 1979, 101, 5854–5856. [Google Scholar] [CrossRef]
- Semenov, V.A.; Samultsev, D.O.; Krivdin, L.B. Theoretical and Experimental Study of 15 N NMR Protonation Shifts: 15 N NMR Protonation Shifts. Magn. Reson. Chem. 2015, 53, 433–441. [Google Scholar] [CrossRef]
- Pinon, A.C.; Rossini, A.J.; Widdifield, C.M.; Gajan, D.; Emsley, L. Polymorphs of Theophylline Characterized by DNP Enhanced Solid-State NMR. Mol. Pharm. 2015, 12, 4146–4153. [Google Scholar] [CrossRef]
- Pawlak, T.; Potrzebowski, M.J. Fine Refinement of Solid-State Molecular Structures of Leu- and Met-Enkephalins by NMR Crystallography. J. Phys. Chem. B 2014, 118, 3298–3309. [Google Scholar] [CrossRef]
- Webber, A.L.; Emsley, L.; Claramunt, R.M.; Brown, S.P. NMR Crystallography of Campho[2,3-c]Pyrazole ( Z ′ = 6): Combining High-Resolution 1 H- 13 C Solid-State MAS NMR Spectroscopy and GIPAW Chemical-Shift Calculations. J. Phys. Chem. A 2010, 114, 10435–10442. [Google Scholar] [CrossRef]
- Kerr, H.E.; Mason, H.E.; Sparkes, H.A.; Hodgkinson, P. Testing the Limits of NMR Crystallography: The Case of Caffeine–Citric Acid Hydrate. CrystEngComm 2016, 18, 6700–6707. [Google Scholar] [CrossRef] [Green Version]
- Dudenko, D.V.; Yates, J.R.; Harris, K.D.M.; Brown, S.P. An NMR Crystallography DFT-D Approach to Analyse the Role of Intermolecular Hydrogen Bonding and π–π Interactions in Driving Cocrystallisation of Indomethacin and Nicotinamide. CrystEngComm 2013, 15, 8797. [Google Scholar] [CrossRef]
- Tatton, A.S.; Blade, H.; Brown, S.P.; Hodgkinson, P.; Hughes, L.P.; Lill, S.O.N.; Yates, J.R. Improving Confidence in Crystal Structure Solutions Using NMR Crystallography: The Case of β-Piroxicam. Cryst. Growth Des. 2018, 18, 3339–3351. [Google Scholar] [CrossRef]
- Paruzzo, F.M.; Hofstetter, A.; Musil, F.; De, S.; Ceriotti, M.; Emsley, L. Chemical Shifts in Molecular Solids by Machine Learning. Nat. Commun. 2018, 9, 4501. [Google Scholar] [CrossRef] [Green Version]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Crystal Data | |
|---|---|
| Chemical formula | C22H26FN3O3S |
| Mr | 431.52 |
| Crystal system, space group | Monoclinic, P21/c |
| Temperature (K) | 100 |
| a, b, c (Å) | 13.9487 (1), 8.4474 (1), 17.5121 (2) |
| β (°) | 92.163 (1) |
| V (Å3) | 2061.98 (4) |
| Z | 4 |
| Radiation type | Cu Kα |
| Diffractometer | XtaLAB Synergy, Dualflex, Pilatus 300 K |
| No. of measured, independent, and observed [I > 2σ(I)] reflections | 59,889, 4409, 3970 |
| Rint | 0.060 |
| (sin θ/λ)max (Å−1) | 0.637 |
| R[F2 > 2σ(F2)], wR(F2), S | 0.035, 0.091, 1.04 |
| No. of reflections | 4409 |
| No. of parameters | 276 |
| Δ〉max, Δ〉min (e Å−3) | 0.51, −0.48 |
| D—H···A | D—H | H···A | D···A | D—H···A |
|---|---|---|---|---|
| N1—H1···N2 i | 0.87 (2) | 2.13 (2) | 2.9863 (18) | 168.4 (19) |
| C5—H5···O1 ii | 0.93 | 2.49 | 3.3683 (18) | 159 |
| C19—H19···O2 iii | 0.93 | 2.59 | 3.2589 (17) | 130 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pawlak, T.; Szczesio, M.; Potrzebowski, M.J. Single-Crystal X-ray and Solid-State NMR Characterisation of AND-1184 and Its Hydrochloride Form. Materials 2021, 14, 7175. https://doi.org/10.3390/ma14237175
Pawlak T, Szczesio M, Potrzebowski MJ. Single-Crystal X-ray and Solid-State NMR Characterisation of AND-1184 and Its Hydrochloride Form. Materials. 2021; 14(23):7175. https://doi.org/10.3390/ma14237175
Chicago/Turabian StylePawlak, Tomasz, Małgorzata Szczesio, and Marek J. Potrzebowski. 2021. "Single-Crystal X-ray and Solid-State NMR Characterisation of AND-1184 and Its Hydrochloride Form" Materials 14, no. 23: 7175. https://doi.org/10.3390/ma14237175
APA StylePawlak, T., Szczesio, M., & Potrzebowski, M. J. (2021). Single-Crystal X-ray and Solid-State NMR Characterisation of AND-1184 and Its Hydrochloride Form. Materials, 14(23), 7175. https://doi.org/10.3390/ma14237175