
Development of Ni-Sr(V,Ti)O3-δ Fuel Electrodes for Solid Oxide Fuel Cells



Abstract
:1. Introduction
2. Materials and Methods
2.1. Synthesis and Ceramic Processing
2.2. Characterization of Materials
2.3. Fabrication and Electrochemical Characterization of Electrodes
2.4. Electrode Modifications by Infiltration
3. Results and Discussion
3.1. Synthesis and Phase Evolution
3.1.1. Effect of Mechanochemical Treatment
3.1.2. Effect of Calcination Temperature
3.1.3. Phase Evolution in Consecutive Calcination Steps
3.1.4. Final Synthetic Procedure
3.2. Crystal Structure and Microstructure
3.3. Thermomechanical Compatibility of STVN with Solid Electrolyte
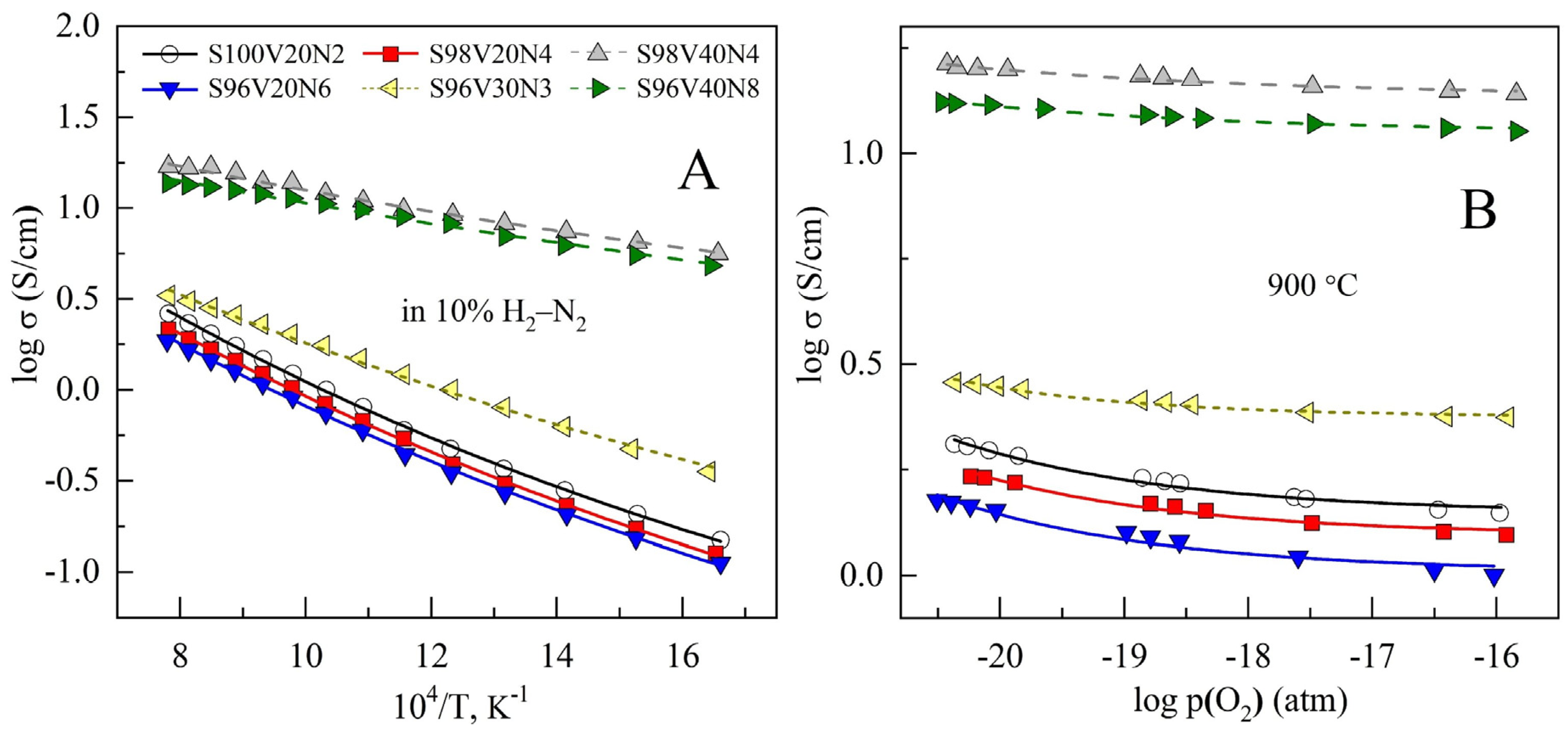
3.4. Electrical Conductivity
3.5. Electrochemical Characterization of STVN Electrodes
3.5.1. As-Prepared STVN Electrodes
3.5.2. Chemical Reactivity between STVN and Solid Electrolytes
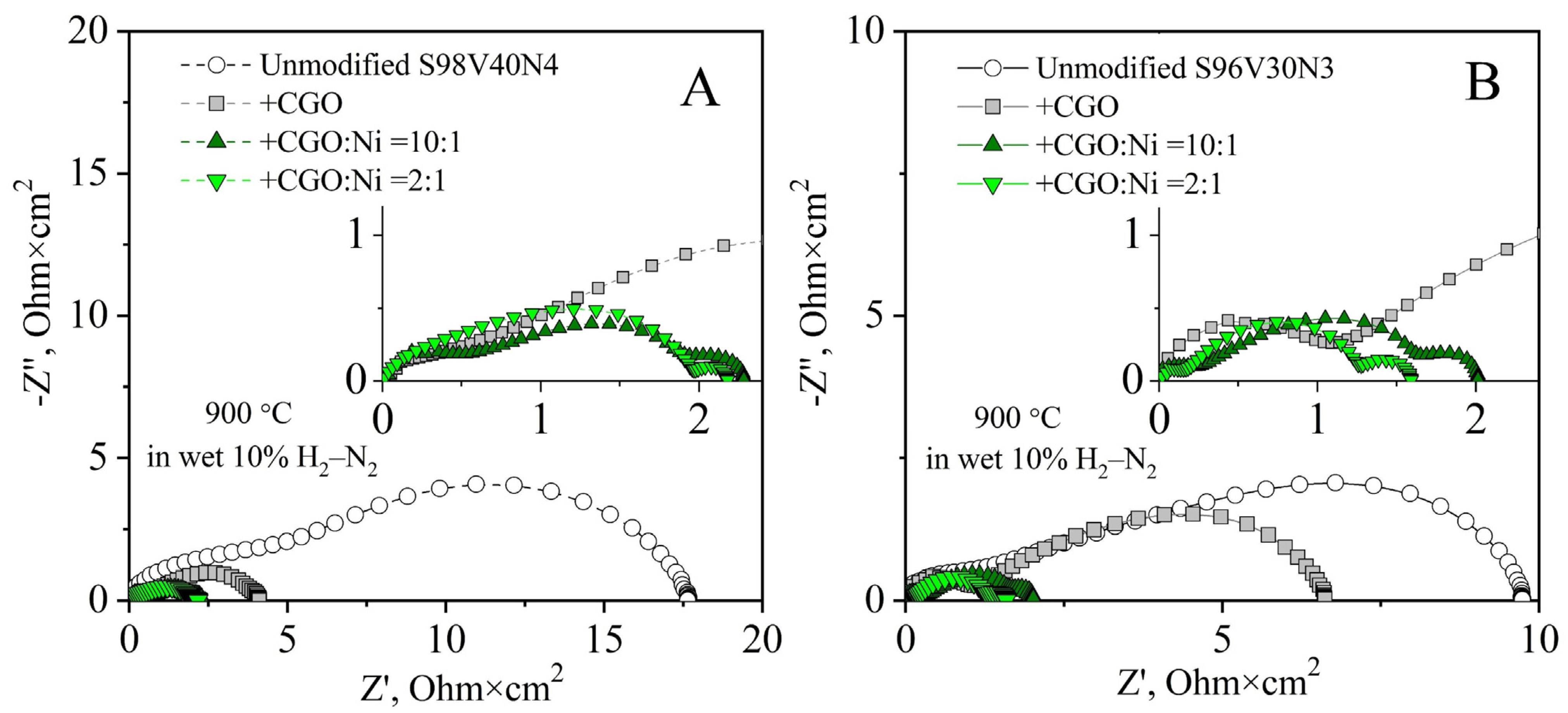
3.5.3. Modification of STVN Electrodes via Infiltration
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shri Prakash, B.; Senthil Kumar, S.; Aruna, S.T. Properties and development of Ni/YSZ as an anode material in solid oxide fuel cell: A review. Renew. Sustain. Energy Rev. 2014, 36, 149–179. [Google Scholar] [CrossRef]
- Shu, L.; Sunarso, J.; Hashim, S.S.; Mao, J.; Zhou, W.; Liang, F. Advanced perovskite anodes for solid oxide fuel cells: A review. Int. J. Hydrogen Energy 2019, 44, 31275–31304. [Google Scholar] [CrossRef]
- Ettler, M.; Timmermann, H.; Malzbender, J.; Weber, A.; Menzler, N.H. Durability of Ni anodes during reoxidation cycles. J. Power Sources 2010, 195, 5452–5467. [Google Scholar] [CrossRef]
- Heo, Y.H.; Lee, J.W.; Lee, S.B.; Lim, T.H.; Park, S.J.; Song, R.H.; Park, C.O.; Shin, D.R. Redox-induced performance degradation of anode-supported tubular solid oxide fuel cells. Int. J. Hydrogen Energy 2011, 36, 797–804. [Google Scholar] [CrossRef]
- Faes, A.; Hessler-Wyser, A.; Zryd, A.; Van Herle, J. A review of RedOx cycling of solid oxide fuel cells anode. Membranes 2012, 2, 585–664. [Google Scholar] [CrossRef] [Green Version]
- Iwanschitz, B.; Holzer, L.; Mai, A.; Schütze, M. Nickel agglomeration in solid oxide fuel cells: The influence of temperature. Solid State Ion. 2012, 211, 69–73. [Google Scholar] [CrossRef]
- Kan, W.H.; Thangadurai, V. Challenges and prospects of anodes for solid oxide fuel cells (SOFCs). Ionics 2015, 21, 301–318. [Google Scholar] [CrossRef]
- Gong, M.; Liu, X.; Trembly, J.; Johnson, C. Sulfur-tolerant anode materials for solid oxide fuel cell application. J. Power Sources 2007, 168, 289–298. [Google Scholar] [CrossRef]
- Gür, T.M. Comprehensive review of methane conversion in solid oxide fuel cells: Prospects for efficient electricity generation from natural gas. Progr. Energy Combust. Sci. 2016, 54, 1–64. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Yan, N.; Chuang, K.T.; Luo, J. Progress in La-doped SrTiO3 (LST)-based anode materials for solid oxide fuel cells. RSC Adv. 2014, 4, 118–131. [Google Scholar] [CrossRef] [Green Version]
- Sadykov, V.A.; Eremeev, N.F.; Sadovskaya, E.M.; Shlyakhtina, A.V.; Pikalova, E.Y.; Osinkin, D.A.; Yaremchenko, A.A. Design of materials for solid oxide fuel cells, permselective membranes, and catalysts for biofuel transformation into syngas and hydrogen based on fundamental studies of their real structure, transport properties, and surface reactivity. Curr. Opin. Green Sustain. Chem. 2022, 33, 100558. [Google Scholar] [CrossRef]
- Hui, S.; Petric, A. Conductivity and stability of SrVO3 and mixed perovskites at low oxygen partial pressures. Solid State Ion. 2001, 143, 275–283. [Google Scholar] [CrossRef]
- Cheng, Z.; Zha, S.; Aguilar, L.; Liu, M. Chemical, electrical, and thermal properties of strontium doped lanthanum vanadate. Solid State Ion. 2005, 176, 1921–1928. [Google Scholar] [CrossRef]
- Yaremchenko, A.A.; Brinkmann, B.; Janssen, R.; Frade, J.R. Electrical conductivity, thermal expansion and stability of Y- and Al-substituted SrVO3 as prospective SOFC anode material. Solid State Ion. 2013, 247–248, 86–93. [Google Scholar] [CrossRef]
- Macías, J.; Yaremchenko, A.A.; Fagg, D.P.; Frade, J.R. Structural and defect chemistry guidelines for Sr(V,Nb)O3-based SOFC anode materials. Phys. Chem. Chem. Phys. 2015, 17, 10749–10758. [Google Scholar] [CrossRef]
- Macías, J.; Yaremchenko, A.A.; Frade, J.R. Redox transitions in strontium vanadates: Electrical conductivity and dimensional changes. J. Alloys Compd. 2014, 601, 186–194. [Google Scholar] [CrossRef]
- Rey, M.J.; Dehaudt, P.; Joubert, J.C.; Lambert-Andron, B.; Cyrot, M.; Cyrot-Lackmann, F. Preparation and structure of the compounds SrVO3 and Sr2VO4. J. Solid State Chem. 1990, 86, 101–108. [Google Scholar] [CrossRef]
- Niu, B.; Jin, F.; Fu, R.; Feng, T.; Shen, Y.; Liu, J.; He, T. Pd-impregnated Sr1.9VMoO6-δ double perovskite as an efficient and stable anode for solid-oxide fuel cells operating on sulfur-containing syngas. Electrochim. Acta 2018, 274, 91–102. [Google Scholar] [CrossRef]
- Peng, C.; Luo, J.; Sanger, A.R.; Chuang, K.T. Sulfur-tolerant anode catalyst for solid oxide fuel cells operating on H2S-containing syngas. Chem. Mater. 2010, 22, 1032–1037. [Google Scholar] [CrossRef]
- Ge, X.; Zhang, L.; Fang, Y.; Zeng, J.; Chan, S.H. Robust solid oxide cells for alternate power generation and carbon conversion. RSC Adv. 2011, 1, 715–724. [Google Scholar] [CrossRef]
- Tamm, K.; Küngas, R.; Gorte, R.J.; Lust, E. Solid oxide fuel cell anodes prepared by infiltration of strontium doped lanthanum vanadate into doped ceria electrolyte. Electrochim. Acta 2013, 106, 398–405. [Google Scholar] [CrossRef]
- Macías, J.; Yaremchenko, A.A.; Frade, J.R. Enhanced stability of perovskite-like SrVO3-based anode materials by donor-type substitutions. J. Mater. Chem. A 2016, 4, 10186–10194. [Google Scholar] [CrossRef]
- Chan, N.; Sharma, R.K.; Smyth, D.M. Nonstoichiometry in SrTiO3. J. Electrochem. Soc. 1981, 128, 1762–1769. [Google Scholar] [CrossRef]
- Abrantes, J.C.C.; Ferreira, A.A.L.; Labrincha, J.A.; Frade, J.R. Electrical conductivity of Sr1-xTiO3-δ materials. Ionics 1997, 3, 436–441. [Google Scholar] [CrossRef]
- Yaremchenko, A.A.; Macías, J.; Kovalevsky, A.V.; Arias-Serrano, B.I.; Frade, J.R. Electrical conductivity and thermal expansion of Ln-substituted SrTiO3 for solid oxide cell electrodes and interconnects: The effect of rare-earth cation size. J. Power Sources 2020, 474, 228531. [Google Scholar] [CrossRef]
- Flores, J.J.A.; Rodríguez, M.L.Á.; Espinosa, G.A.; Vera, J.V.A. Advances in the development of titanates for anodes in SOFC. Int. J. Hydrogen Energy 2019, 44, 12529–12542. [Google Scholar] [CrossRef]
- Kurokawa, H.; Yang, L.; Jacobson, C.P.; De Jonghe, L.C.; Visco, S.J. Y-doped SrTiO3 based sulfur tolerant anode for solid oxide fuel cells. J. Power Sources 2007, 164, 510–518. [Google Scholar] [CrossRef]
- Shao, L.; Si, F.; Fu, X.Z.; Luo, J.L. Archiving high-performance solid oxide fuel cells with titanate anode in sulfur- and carbon-containing fuels. Electrochim. Acta 2018, 280, 9–13. [Google Scholar] [CrossRef]
- Slater, P.R.; Fagg, D.P.; Irvine, J.T.S. Synthesis and electrical characterisation of doped perovskite titanates as potential anode materials for solid oxide fuel cells. J. Mater. Chem. 1997, 7, 2495–2498. [Google Scholar] [CrossRef]
- Marina, O.A.; Canfield, N.L.; Stevenson, J.W. Thermal, electrical, and electrocatalytical properties of lanthanum-doped strontium titanate. Solid State Ion. 2002, 149, 21–28. [Google Scholar] [CrossRef]
- Yaremchenko, A.A.; Patrício, S.G.; Frade, J.R. Thermochemical behavior and transport properties of Pr-substituted SrTiO3 as potential solid oxide fuel cell anode. J. Power Sources 2014, 245, 557–569. [Google Scholar] [CrossRef]
- Neagu, D.; Irvine, J.T.S. Structure and properties of La0.4Sr0.4TiO3 ceramics for use as anode materials in solid oxide fuel cells. Chem. Mater. 2010, 22, 5042–5053. [Google Scholar] [CrossRef]
- Yaremchenko, A.A.; Naumovich, E.N.; Patricio, S.G.; Merkulov, O.V.; Patrakeev, M.V.; Frade, J.R. Rare-earth-substituted strontium titanate: Insight into local oxygen-rich structures and redox kinetics. Inorg. Chem. 2016, 55, 4836–4849. [Google Scholar] [CrossRef] [PubMed]
- Macías, J.; Yaremchenko, A.A.; Rodríguez-Castellón, E.; Starykevich, M.; Frade, J.R. Compromising between phase stability and electrical performance: SrVO3–SrTiO3 solid solutions as solid oxide fuel cell anode components. ChemSusChem 2019, 12, 240–251. [Google Scholar] [CrossRef]
- Fu, Q.X.; Tietz, F. Ceramic-based anode materials for improved redox cycling of solid oxide fuel cells. Fuel Cells 2008, 8, 283–293. [Google Scholar] [CrossRef]
- Ikebe, T.; Muroyama, H.; Matsui, T.; Eguchi, K. Fabrication of redox tolerant anode with an electronic conductive oxide of Y-doped SrTiO3. J. Electrochem. Soc. 2010, 157, B970–B974. [Google Scholar] [CrossRef]
- Ma, Q.; Tietz, F.; Sebold, D.; Stöver, D. Y-substituted SrTiO3–YSZ composites as anode materials for solid oxide fuel cells: Interaction between SYT and YSZ. J. Power Sources 2010, 195, 920–1925. [Google Scholar] [CrossRef]
- Puengjinda, P.; Muroyama, H.; Matsui, T.; Eguchi, K. Optimization of anode material composed of Y-doped SrTiO3 and metal and/or oxide additives for solid oxide fuel cells. J. Power Sources 2012, 204, 67–73. [Google Scholar] [CrossRef]
- Lu, X.C.; Zhu, J.H.; Yang, Z.; Xia, G.; Stevenson, J.W. Pd-impregnated SYT/LDC composite as sulfur-tolerant anode for solid oxide fuel cells. J. Power Sources 2009, 192, 381–384. [Google Scholar] [CrossRef]
- Hussain, A.M.; Høgh, J.V.T.; Jacobsen, T.; Bonanos, N. Nickel-ceria infiltrated Nb-doped SrTiO3 for low temperature SOFC anodes and analysis on gas diffusion impedance. Int. J. Hydrogen Energy 2012, 37, 4309–4318. [Google Scholar] [CrossRef]
- Lee, S.; Kim, G.; Vohs, J.M.; Gorte, R.J. SOFC anodes based on infiltration of La0.3Sr0.7TiO3. J. Electrochem. Soc. 2008, 55, B1179–B1183. [Google Scholar] [CrossRef]
- Gross, M.D.; Carver, K.M.; Deighan, M.A.; Schenkel, A.; Smith, B.M.; Yee, A.Z. Redox stability of SrNbxTi1-xO3-YSZ for use in SOFC anodes. J. Electrochem. Soc. 2009, 156, B540–B545. [Google Scholar] [CrossRef]
- Savaniu, C.D.; Irvine, J.T.S. Reduction studies and evaluation of surface modified A-site deficient La-doped SrTiO3 as anode material for IT-SOFCs. J. Mater. Chem. 2009, 19, 8119–8128. [Google Scholar] [CrossRef]
- Zhu, T.; Troiani, H.E.; Mogni, L.V.; Han, M.; Barnett, S.A. Ni-substituted Sr(Ti,Fe)O3 SOFC anodes: Achieving high performance via metal alloy nanoparticle exsolution. Joule 2008, 2, 478–496. [Google Scholar] [CrossRef] [Green Version]
- Qin, Q.; Wu, G.; Chen, S.; Doherty, W.; Xie, K.; Wu, Y. Perovskite titanate cathode decorated by in-situ grown iron nanocatalyst with enhanced electrocatalytic activity for high-temperature steam electrolysis. Electrochim. Acta 2014, 127, 215–227. [Google Scholar] [CrossRef]
- Glaser, R.; Zhu, T.; Troiani, H.; Caneiro, A.; Mogni, L.; Barnett, S. The enhanced electrochemical response of Sr(Ti0.3Fe0.7Ru0.07)O3-δ anodes due to exsolved Ru–Fe nanoparticles. J. Mater. Chem. A 2018, 6, 5193–5201. [Google Scholar] [CrossRef] [Green Version]
- Mori, T. Taguchi Methods: Benefits, Impacts, Mathematics, Statistics, and Applications; ASME: New York, NY, USA, 2011. [Google Scholar]
- Tsuiki, H.; Kitazawa, K.; Fueki, K. The Donor Level of V4+ and the Metal-Nonmetal Transition in SrTi1-xVxO3. Jpn. J. Appl. Phys. 1983, 22, 590–596. [Google Scholar] [CrossRef]
- Shannon, R.D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chaleogenides. Acta Crystallogr. A 1976, 32, 751–767. [Google Scholar] [CrossRef]
- Moos, R.; Hardtl, K.H. Defect chemistry of donor-doped and undoped strontium titanate ceramics between 1000° and 1400 °C. J. Am. Ceram. Soc. 1997, 80, 2549–2562. [Google Scholar] [CrossRef]
- Akhtar, M.J.; Akhtar, Z.U.N.; Jackson, R.A.; Catlow, C.R.A. Computer simulation studies of strontium titanate. J. Am. Ceram. Soc. 1995, 78, 421–428. [Google Scholar] [CrossRef]
- Borchardt, G.; Gömann, K.; Kilo, M.; Schmidt, H. Diffusion in Ceramics. In Ceramics Science and Technology; Riedel, R., Chen, I.W., Eds.; Wiley-VCH Verlag: Weinheim, Germany, 2008; pp. 105–182. [Google Scholar]
- Krasnenko, T.I.; Tugova, N.P.; Slobodin, B.V.; Fotiev, A.A.; Syrneva, O.N.; Kiyaeva, G.A. Phase composition and sequence of transformations in SrO-V2O5 system. Depos. Doc. VINITI 1983, 2866-83, 1–12. [Google Scholar]
- Fotiev, A.A.; Slobodin, B.V.; Khodos, M.Y.; Pletnev, R.N. Vanadates. Composition, Synthesis, Structure, Properties; Nauka: Moscow, Russia, 1988. [Google Scholar]
- Ardila, D.R.; Andreeta, J.P.; Basso, H.C. Preparation, microstructural and electrical characterization of SrVO3 single crystal fiber. J. Cryst. Growth 2000, 211, 313–317. [Google Scholar] [CrossRef]
- Hossain, K.M.; Rubel, M.H.K.; Rahaman, M.M.; Hossain, M.M.; Hossain, M.I.; Khatun, A.A.; Hossain, J.; Islam, A.K.M.A. A comparative theoretical study on physical properties of synthesized AVO3 (A = Ba, Sr, Ca, Pb) perovskites. arXiv 2019, arXiv:1905.01437. Available online: https://arxiv.org/abs/1905.01437 (accessed on 16 November 2021).
- Natoli, A.; Frade, J.R.; Bamburov, A.; Żurawska, A.; Yaremchenko, A. Impact of praseodymia additions and firing conditions on structural and electrical transport properties of 5 mol.% yttria partially stabilized zirconia (5YSZ). Appl. Sci. 2021, 11, 5939. [Google Scholar] [CrossRef]
- Atkinson, A.; Barnett, S.; Gorte, R.J.; Irvine, J.T.S.; McEvoy, A.J.; Mogensen, M.; Singhal, S.C.; Vohs, J. Advanced anodes for high-temperature fuel cells. Nat. Mater. 2004, 3, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Jamnik, J.; Maier, J. Treatment of the impedance of mixed conductors equivalent circuit model and explicit approximate solutions. J. Electrochem. Soc. 1999, 146, 4183–4188. [Google Scholar] [CrossRef]
- Barsoukov, E.; Macdonald, J.R. Impedance Spectroscopy: Theory, Experiment, and Applications, 2nd ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2005. [Google Scholar]
- Primdahl, S.; Mogensen, M. Oxidation of hydrogen on Ni/yttria-stabilized zirconia cermet anodes. J. Electrochem. Soc. 1997, 144, 3409–3419. [Google Scholar] [CrossRef]
- Zhou, J.; Shin, T.H.; Ni, C.; Chen, G.; Wu, K.; Cheng, Y.; Irvine, J.T.S. In situ growth of nanoparticles in layered perovskite La0.8Sr1.2Fe0.9Co0.1O4−δ as an active and stable electrode for symmetrical solid oxide fuel cells. Chem. Mater. 2016, 28, 2981–2993. [Google Scholar] [CrossRef]
- Jiang, S.P. Development of lanthanum strontium manganite perovskite cathode materials of solid oxide fuel cells: A review. J. Mater. Sci. 2008, 43, 6799–6833. [Google Scholar] [CrossRef]
- Thommy, L.; Joubert, O.; Hamon, J.; Caldes, M.T. Impregnation versus exsolution: Using metal catalysts to improve electrocatalytic properties of LSCM-based anodes operating at 600 C. Int. J. Hydrogen Energy 2016, 41, 14207–14216. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| α | β | γ | Nominal Composition | Notation | Lattice Parameter a, Å | Density ρexp, g/cm3 | Relative Density ρexp/ρtheor, % 1 |
|---|---|---|---|---|---|---|---|
| 0 | 0.2 | 0.1 | SrTi0.78V0.20Ni0.02O3-δ | S100V20N2 | 3.9000(1) | 4.93 | 95.5 |
| 0 | 0.3 | 0.2 | SrTi0.64V0.30Ni0.06O3-δ | S100V30N6 | - | - | - |
| 0 | 0.4 | 0.3 | SrTi0.48V0.40Ni0.12O3-δ | S100V40N12 | - | - | - |
| 0.02 | 0.2 | 0.2 | Sr0.98Ti0.76V0.20Ni0.04O3-δ | S98V20N4 | 3.9006 (1) | 4.91 | 95.9 |
| 0.02 | 0.3 | 0.3 | Sr0.98Ti0.61V0.30Ni0.09O3-δ | S98V30N9 | - | - | - |
| 0.02 | 0.4 | 0.1 | Sr0.98Ti0.56V0.40Ni0.04O3-δ | S98V40N4 | 3.8868 (2) | 4.89 | 94.4 |
| 0.04 | 0.2 | 0.3 | Sr0.96Ti0.74V0.20Ni0.06O3-δ | S96V20N6 | 3.9018 (1) | 4.70 | 92.7 |
| 0.04 | 0.3 | 0.1 | Sr0.96Ti0.67V0.30Ni0.03O3-δ | S96V30N3 | 3.8956 (1) | 4.53 | 88.9 |
| 0.04 | 0.4 | 0.2 | Sr0.96Ti0.52V0.40Ni0.08O3-δ | S96V40N8 | 3.9024 (1) | 4.96 | 97.4 |
| Notation | Perovskite Phase | Fraction of Sr3(VO4)2, mol.% 1 |
|---|---|---|
| S100V20N2 | SrTi0.830V0.170O3 | 2.04 |
| S98V20N4 | SrTi0.826V0.174O3 | 2.04 |
| S98V40N4 | SrTi0.609V0.391O3 | 2.04 |
| S96V20N6 | SrTi0.822V0.178O3 | 2.05 |
| S96V30N3 | Sr0.990Ti0.691V0.309O3 | 0 |
| S96V40N8 | SrTi0.619V0.381O3 | 4.20 |
| Composition | Average TEC ± 0.1, ppm/K (25–1100 °C) | Parameters of Arrhenius Model for σ (500–1000 °C) 1 | |
|---|---|---|---|
| EA, kJ/mol | ln(A0) (S/cm) | ||
| S100V20N2 | 12.5 | 31.4 ± 0.3 | 3.91 ± 0.03 |
| S98V20N4 | 12.6 | 30.8 ± 0.2 | 3.65 ± 0.02 |
| S98V40N4 | 14.0 | 11.3 ± 0.1 | 3.90 ± 0.02 |
| S96V20N6 | 13.0 | 30.6 ± 0.3 | 3.50 ± 0.03 |
| S96V30N3 | 13.3 | 22.4 ± 0.1 | 3.33 ± 0.02 |
| S96V40N8 | 14.3 | 10.0 ± 0.1 | 3.58 ± 0.02 |
| Electrode | S98V40N4 | S96V30N3 | ||
|---|---|---|---|---|
| Load, wt.% | EA, kJ/mol 1 | Load, wt.% | EA, kJ/mol | |
| Unmodified | - | 141 ± 1 | - | 116 ± 3 |
| + CGO | 21 | 97 ± 1 | 21 | 99 ± 1 |
| + CGO:Ni = 9:1 | 30 | 63 ± 1 | 27 | 73 ± 2 |
| + CGO:Ni = 5:1 | 27 | 87 ± 1 | 21 | 64 ± 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Serôdio Costa, B.F.; Arias-Serrano, B.I.; Yaremchenko, A.A. Development of Ni-Sr(V,Ti)O3-δ Fuel Electrodes for Solid Oxide Fuel Cells. Materials 2022, 15, 278. https://doi.org/10.3390/ma15010278
Serôdio Costa BF, Arias-Serrano BI, Yaremchenko AA. Development of Ni-Sr(V,Ti)O3-δ Fuel Electrodes for Solid Oxide Fuel Cells. Materials. 2022; 15(1):278. https://doi.org/10.3390/ma15010278
Chicago/Turabian StyleSerôdio Costa, Bernardo F., Blanca I. Arias-Serrano, and Aleksey A. Yaremchenko. 2022. "Development of Ni-Sr(V,Ti)O3-δ Fuel Electrodes for Solid Oxide Fuel Cells" Materials 15, no. 1: 278. https://doi.org/10.3390/ma15010278
APA StyleSerôdio Costa, B. F., Arias-Serrano, B. I., & Yaremchenko, A. A. (2022). Development of Ni-Sr(V,Ti)O3-δ Fuel Electrodes for Solid Oxide Fuel Cells. Materials, 15(1), 278. https://doi.org/10.3390/ma15010278