
Band Structure of Organic-Ion-Intercalated (EMIM)xFeSe Superconductor



Abstract
:1. Introduction
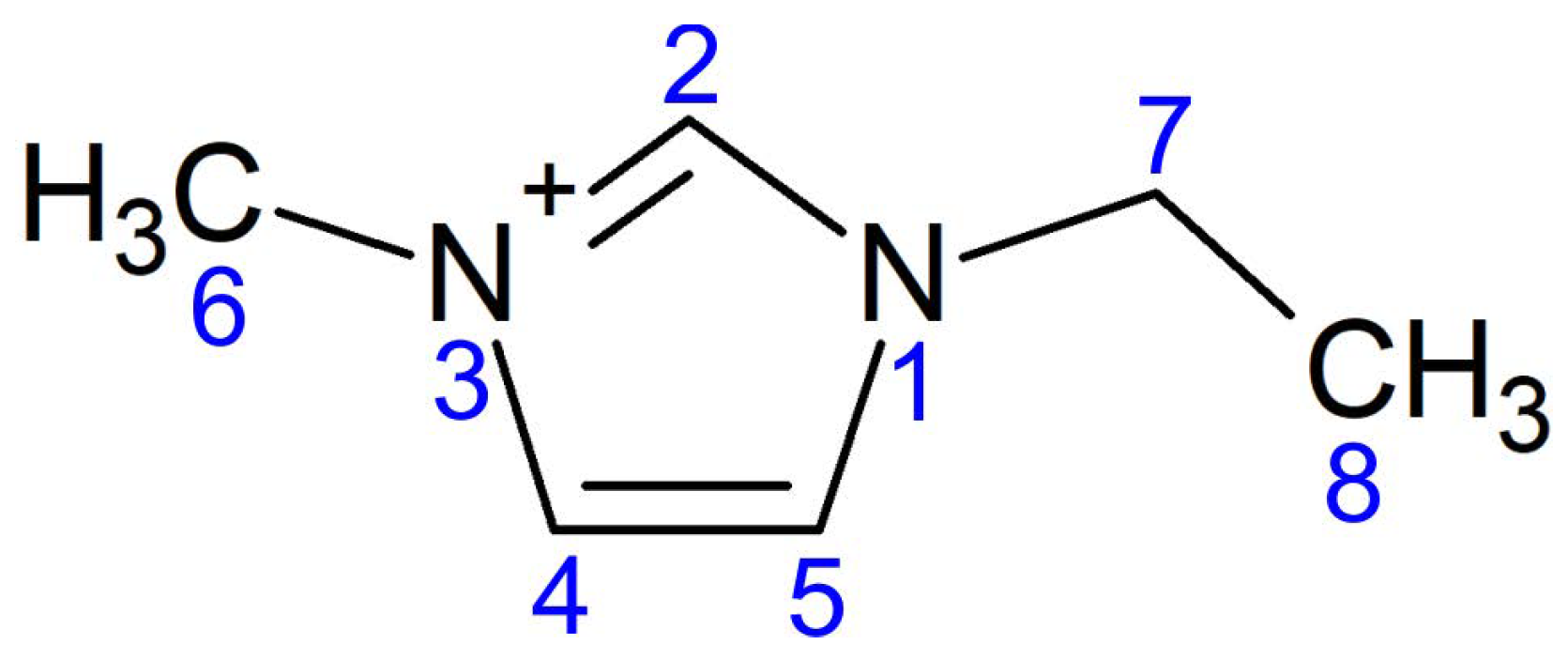
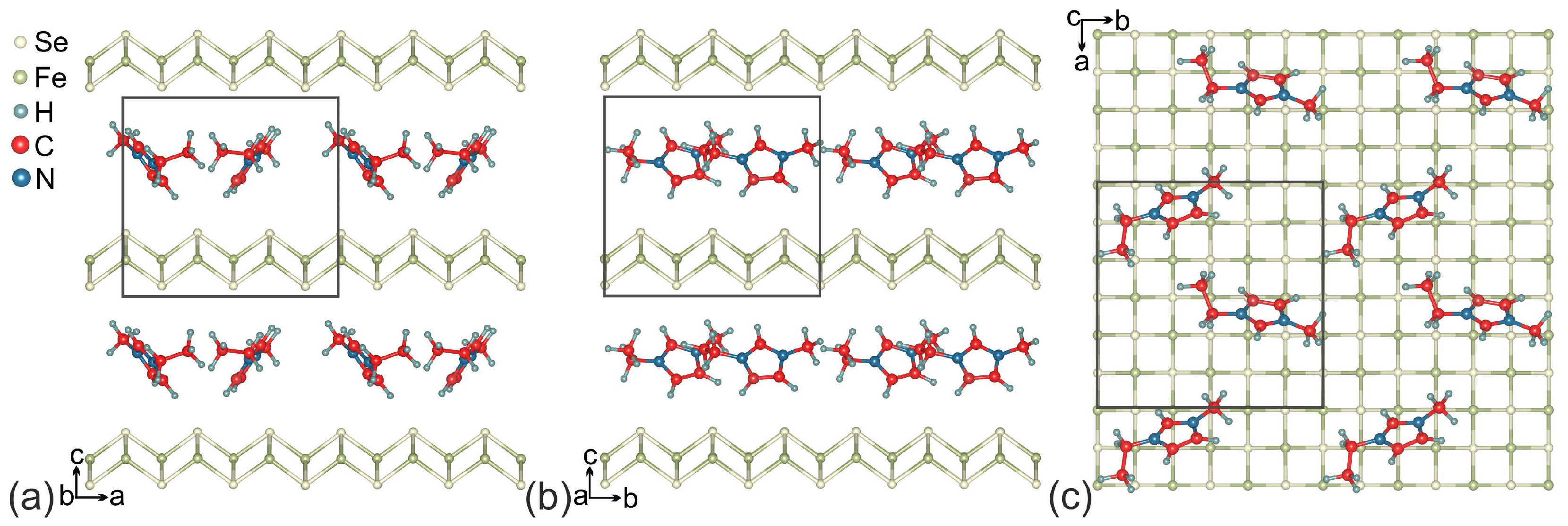
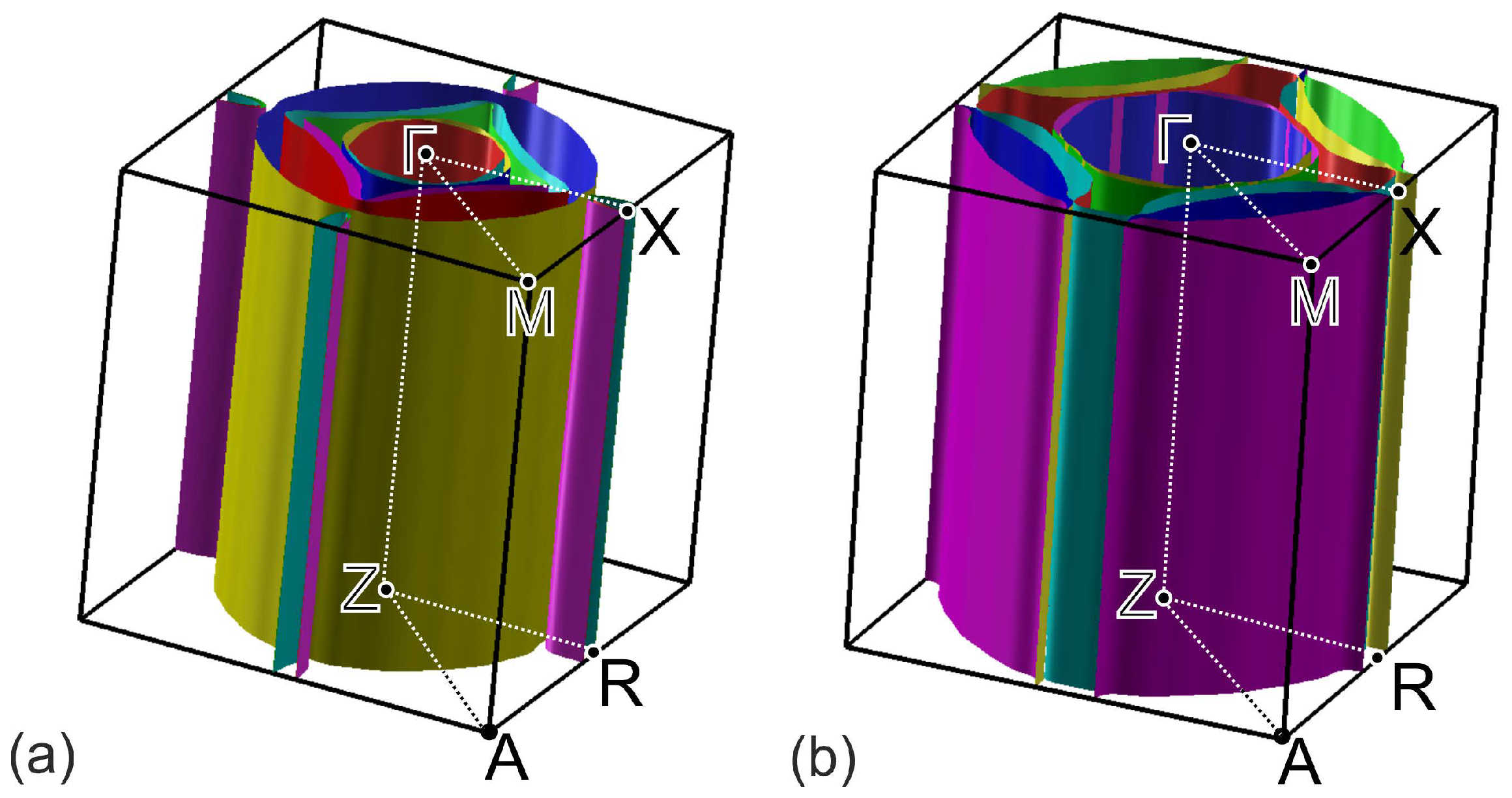
2. Computation Details and Crystal Structure
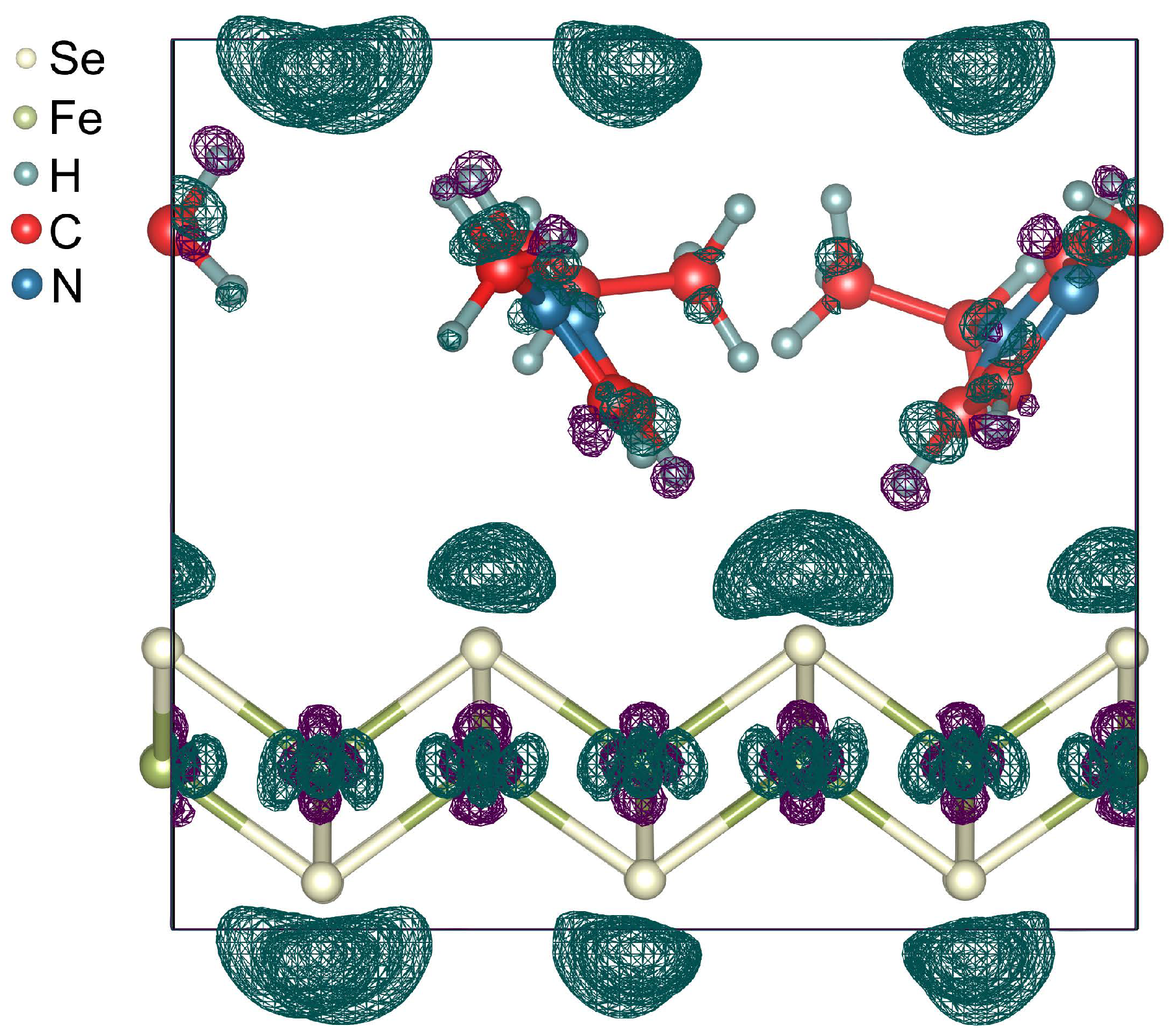
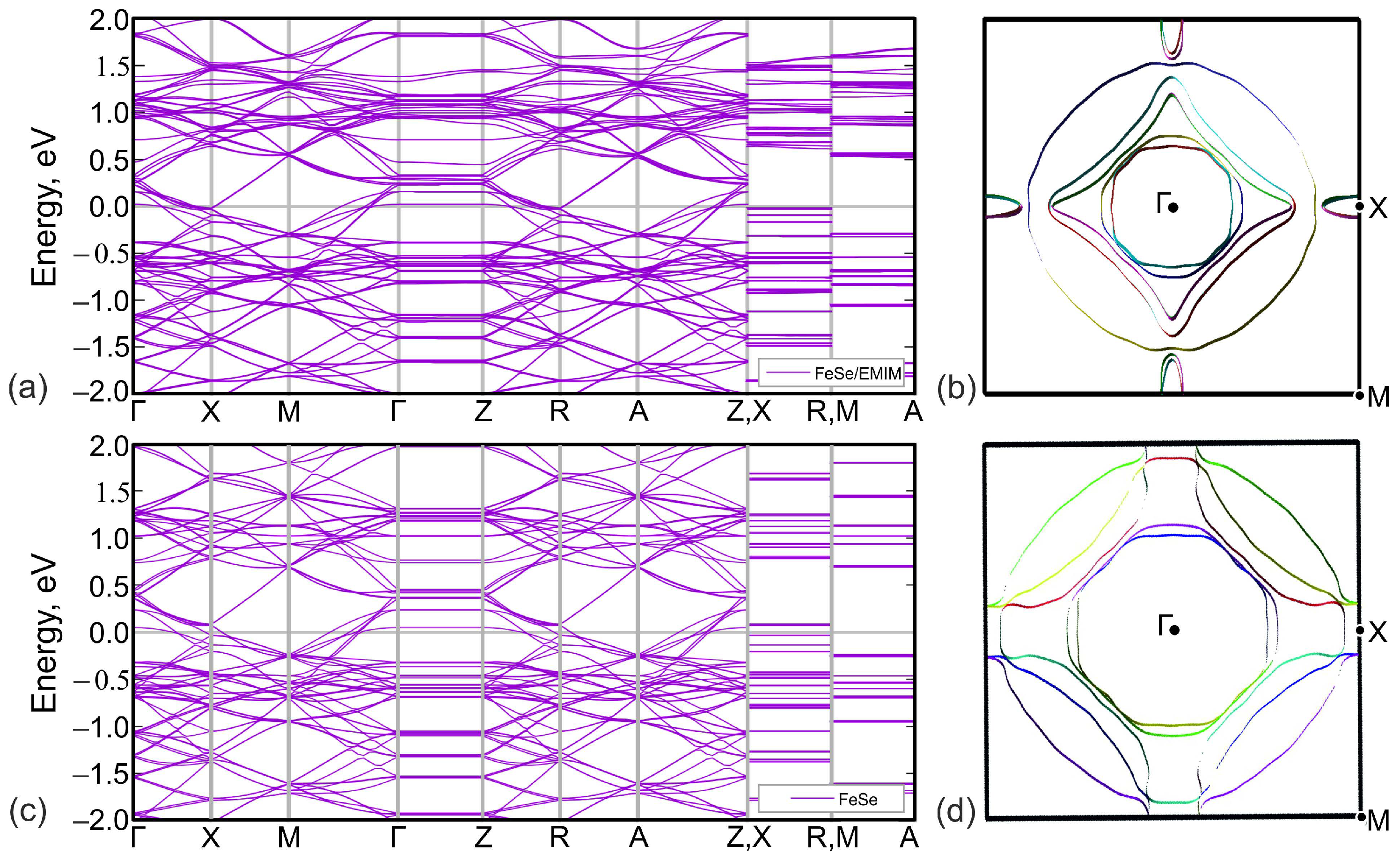
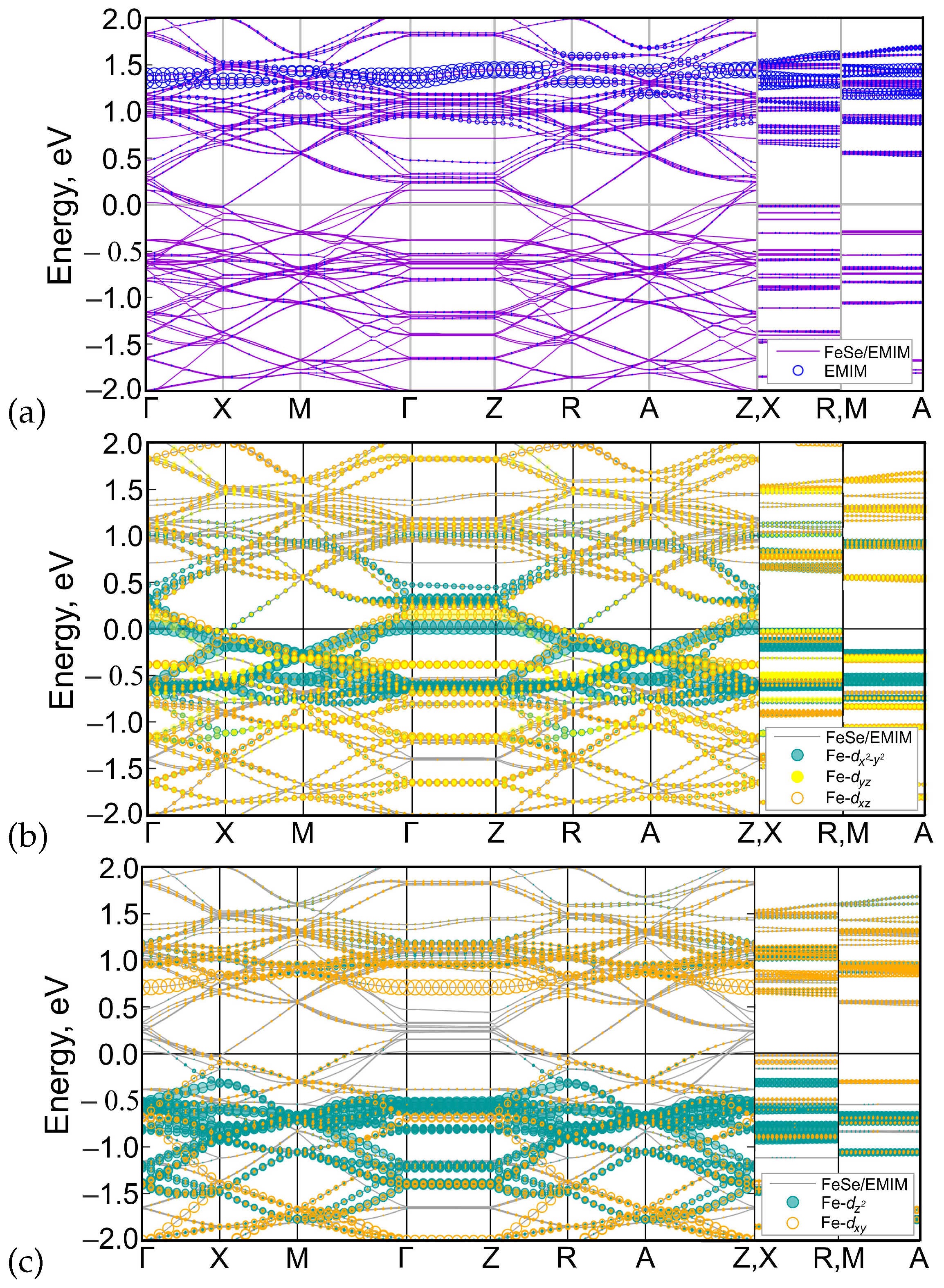
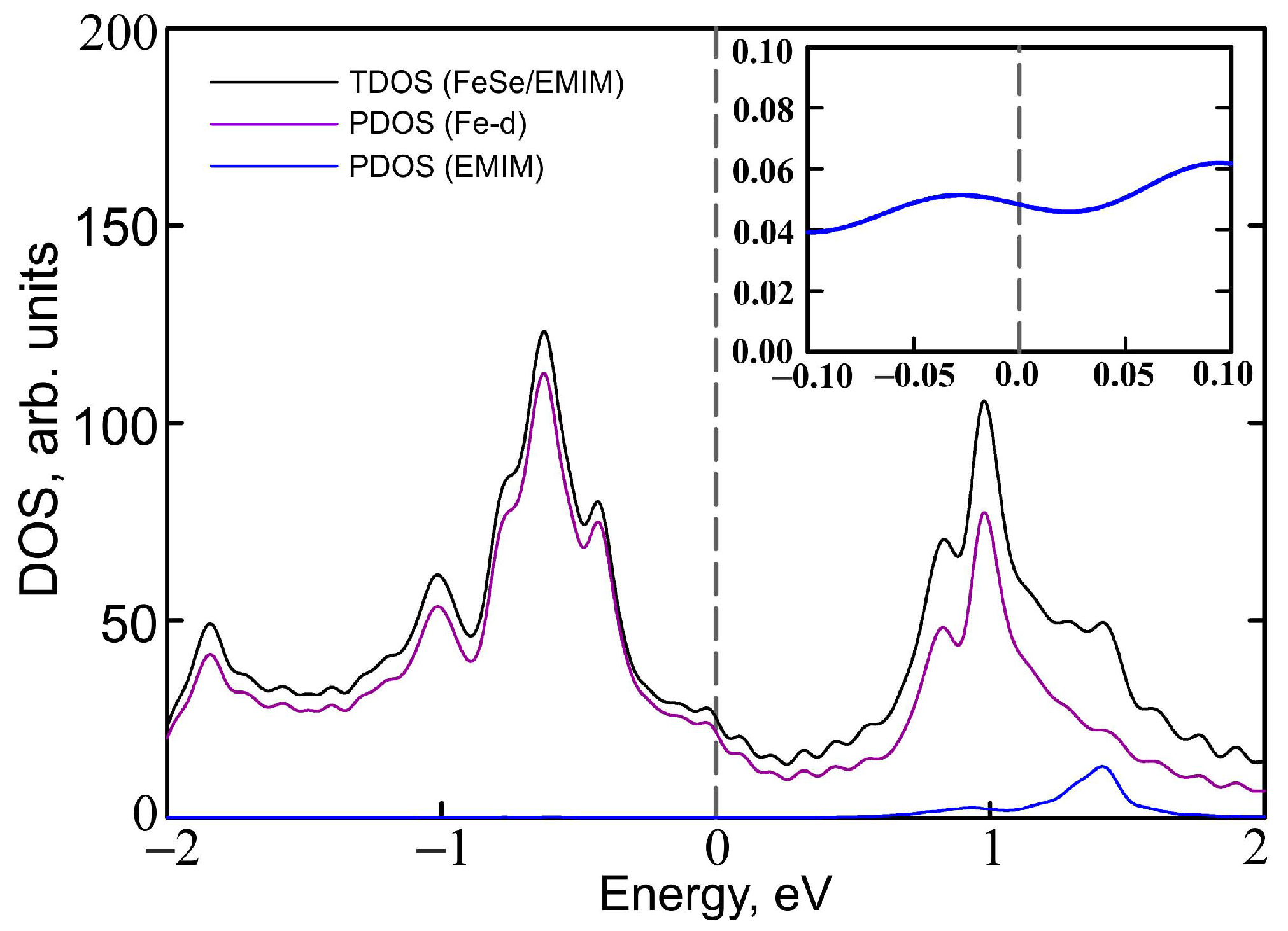
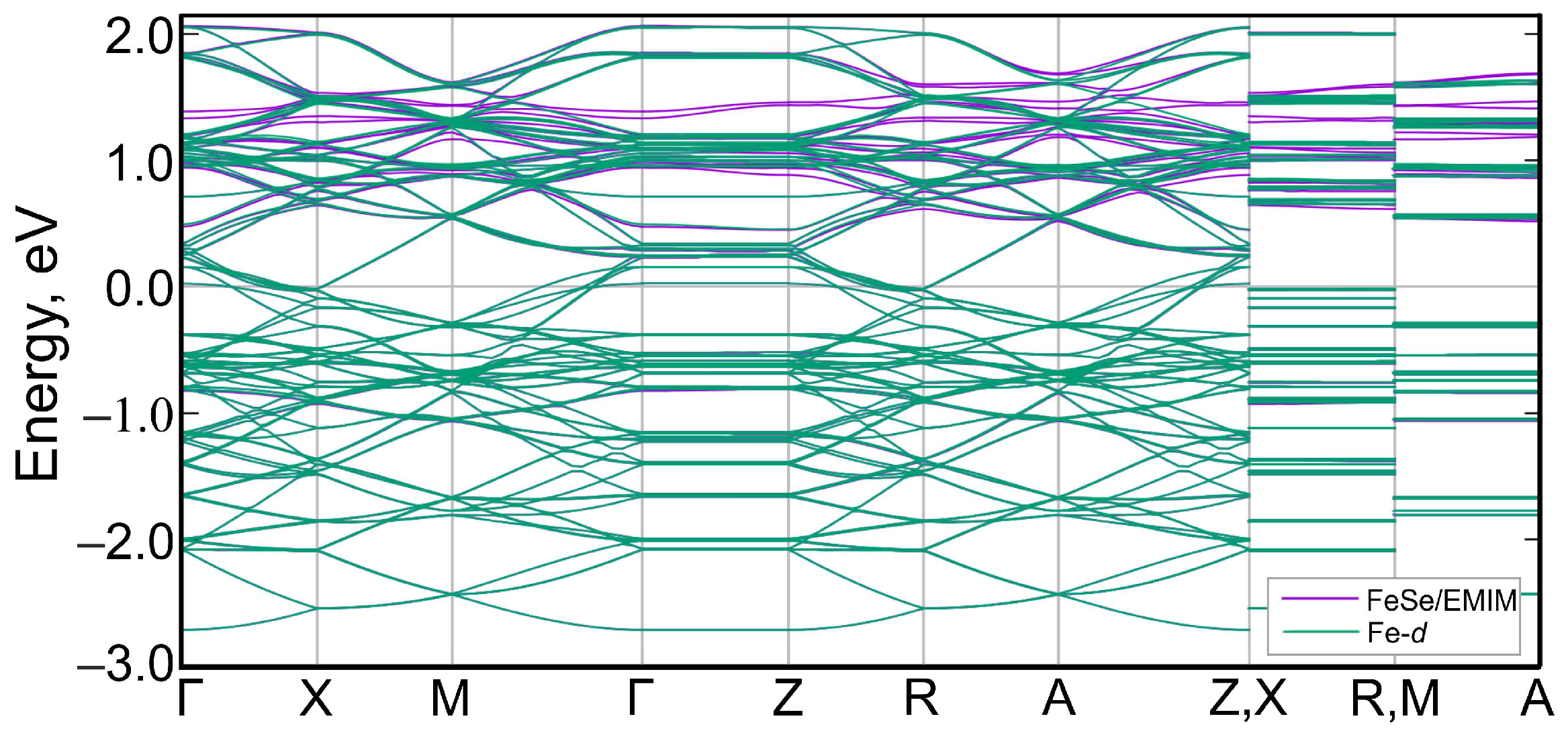
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yu, J.; Sun, Q.; Kawazoe, Y.; Jena, P. Stability and properties of 2D porous nanosheets based on tetraoxa[8]circulene analogues. Nanoscale 2014, 6, 14962–14970. [Google Scholar] [CrossRef] [PubMed]
- Baryshnikov, G.V.; Minaev, B.F.; Karaush, N.N.; Minaeva, V.A. The art of the possible: Computational design of the 1D and 2D materials based on the tetraoxa[8]circulene monomer. RSC Adv. 2014, 4, 25843–25851. [Google Scholar] [CrossRef]
- Kuklin, A.V.; Baryshnikov, G.V.; Minaev, B.F.; Ignatova, N.; Ågren, H. Strong Topological States and High Charge Carrier Mobility in Tetraoxa[8]circulene Nanosheets. J. Phys. Chem. C 2018, 122, 22216–22222. [Google Scholar] [CrossRef]
- Begunovich, L.V.; Kuklin, A.V.; Baryshnikov, G.V.; Valiev, R.R.; Ågren, H. Single-layer polymeric tetraoxa[8]circulene modified by s-block metals: Toward stable spin qubits and novel superconductors. Nanoscale 2021, 13, 4799–4811. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Gao, Y.; Huang, Z.; Chen, X. Superconductivity in p-Terphenyl. arXiv 2017, arXiv:1703.05803. [Google Scholar]
- Wang, R.; Gao, Y.; Huang, Z.; Chen, X. Superconductivity at 43 K in a single C-C bond linked terphenyl. arXiv 2017, arXiv:1703.05804. [Google Scholar]
- Wang, R.; Gao, Y.; Huang, Z.; Chen, X. Superconductivity above 120 kelvin in a chain link molecule. arXiv 2017, arXiv:1703.06641. [Google Scholar]
- Li, H.; Zhou, X.; Parham, S.; Nummy, T.; Griffith, J.; Gordon, K.; Chronister, E.; Dessau, D. Spectroscopic evidence of low-energy gaps persisting up to 120 K in surface-doped p-terphenyl crystals. Phys. Rev. B 2019, 100, 064511. [Google Scholar] [CrossRef] [Green Version]
- Piatti, E. Ionic gating in metallic superconductors: A brief review. Nano Express 2021, 2, 024003. [Google Scholar] [CrossRef]
- Cui, Y.; Hu, Z.; Zhang, J.S.; Ma, W.L.; Ma, M.W.; Ma, Z.; Wang, C.; Yan, J.Q.; Sun, J.P.; Cheng, J.G.; et al. Ionic-Liquid-Gating Induced Protonation and Superconductivity in FeSe, FeSe0.93S0.07, ZrNCl, 1T-TaS2 and Bi2Se3. Chin. Phys. Lett. 2019, 36, 077401. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Li, Q.; Xie, W.; Chen, G.; Zhu, X.; Wen, H.H. Superconductivity at 44.4 K achieved by intercalating EMIM+ into FeSe. Chin. Phys. B 2021, 30, 107402. [Google Scholar] [CrossRef]
- Kamihara, Y.; Watanabe, T.; Hirano, M.; Hosono, H. Iron-Based Layered Superconductor La[O1−xFx]FeAs (x = 0.05–0.12) with Tc = 26 K. J. Am. Chem. Soc. 2008, 130, 3296–3297. [Google Scholar] [CrossRef] [PubMed]
- Sadovskii, M.V. High-temperature superconductivity in iron-based layered compounds. Physics-Uspekhi 2008, 51, 1201–1227. [Google Scholar] [CrossRef] [Green Version]
- Izyumov, Y.A.; Kurmaev, E.Z. FeAs systems: A new class of high-temperature superconductors. Physics-Uspekhi 2008, 51, 1261–1286. [Google Scholar] [CrossRef]
- Ivanovskii, A.L. New high-temperature superconductors based on rare-earth and transition metal oxyarsenides and related phases: Synthesis, properties, and simulations. Physics-Uspekhi 2008, 51, 1229–1260. [Google Scholar] [CrossRef]
- Johnston, D.C. The puzzle of high temperature superconductivity in layered iron pnictides and chalcogenides. Adv. Phys. 2010, 59, 803–1061. [Google Scholar] [CrossRef] [Green Version]
- Paglione, J.; Greene, R.L. High-temperature superconductivity in iron-based materials. Nat. Phys. 2010, 6, 645–658. [Google Scholar] [CrossRef] [Green Version]
- Lumsden, M.D.; Christianson, A.D. Magnetism in Fe-based superconductors. J. Phys. Condens. Matter 2010, 22, 203203. [Google Scholar] [CrossRef]
- Stewart, G.R. Superconductivity in iron compounds. Rev. Mod. Phys. 2011, 83, 1589–1652. [Google Scholar] [CrossRef]
- Hirschfeld, P.J.; Korshunov, M.M.; Mazin, I.I. Gap symmetry and structure of Fe-based superconductors. Rep. Prog. Phys. 2011, 74, 124508. [Google Scholar] [CrossRef] [Green Version]
- Inosov, D.S. Spin fluctuations in iron pnictides and chalcogenides: From antiferromagnetism to superconductivity. Comptes Rendus Phys. 2016, 17, 60–89. [Google Scholar] [CrossRef]
- Qing-Yan, W.; Zhi, L.; Wen-Hao, Z.; Zuo-Cheng, Z.; Jin-Song, Z.; Wei, L.; Hao, D.; Yun-Bo, O.; Peng, D.; Kai, C.; et al. Interface-Induced High-Temperature Superconductivity in Single Unit-Cell FeSe Films on SrTiO3. Chin. Phys. Lett. 2012, 29, 037402. [Google Scholar]
- Wang, Q.-Y.; Li, Z.; Zhang, W.-H.; Zhang, Z.-C.; Zhang, J.-S.; Li, W.; Ding, H.; Ou, Y.-B.; Deng, P.; Chang, K. Onset of the Meissner effect at 65 K in FeSe thin film grown on Nb-doped SrTiO3 substrate. Sci. Bull. 2015, 60, 1301–1304. [Google Scholar] [CrossRef] [Green Version]
- Ge, J.F.; Liu, Z.L.; Liu, C.; Gao, C.L.; Qian, D.; Xue, Q.K.; Liu, Y.; Jia, J.F. Superconductivity above 100 K in single-layer FeSe films on doped SrTiO3. Nat. Mater. 2015, 14, 285–289. [Google Scholar] [CrossRef]
- Zhao, L.; Liang, A.; Yuan, D.; Hu, Y.; Liu, D.; Huang, J.; He, S.; Shen, B.; Xu, Y.; Liu, X.; et al. Common electronic origin of superconductivity in (Li,Fe)OHFeSe bulk superconductor and single-layer FeSe/SrTiO3 films. Nat. Commun. 2016, 7, 10608. [Google Scholar] [CrossRef]
- Sadovskii, M.V. High-temperature superconductivity in monolayers FeSe. Physics-Uspekhi. 2016, 59, 947–967. [Google Scholar] [CrossRef] [Green Version]
- Du, Z.; Yang, X.; Altenfeld, D.; Gu, Q.; Yang, H.; Eremin, I.; Hirschfeld, P.J.; Mazin, I.I.; Lin, H.; Zhu, X.; et al. Sign reversal of the order parameter in (Li1−xFex)OHFe1−yZnySe. Nat. Phys. 2017, 14, 134. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Wang, Z.; Gao, Y.; Liu, X.; Liu, Y.; Wang, Q.H.; Wang, J. Spectroscopic Imaging of Quasiparticle Bound States Induced by Strong Nonmagnetic Scatterings in One-Unit-Cell FeSe/SrTiO3. Phys. Rev. Lett. 2019, 123, 036801. [Google Scholar] [CrossRef]
- Jandke, J.; Yang, F.; Hlobil, P.; Engelhardt, T.; Rau, D.; Zakeri, K.; Gao, C.; Schmalian, J.; Wulfhekel, W. Unconventional pairing in single FeSe layers. Phys. Rev. B 2019, 100, 020503. [Google Scholar] [CrossRef] [Green Version]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef] [Green Version]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, 1133–1138. [Google Scholar] [CrossRef] [Green Version]
- Boker, S.; Neale, M.; Maes, H.; Wilde, M.; Spiegel, M.; Brick, T.; Spies, J.; Estabrook, R.; Kenny, S.; Bates, T.; et al. OpenMX: An open source extended structural equation modeling framework. Psychometrika 2011, 76, 306–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozaki, T. Variationally optimized atomic orbitals for large-scale electronic structures. Phys. Rev. B 2003, 67, 155108. [Google Scholar] [CrossRef]
- Ozaki, T.; Kino, H. Numerical atomic basis orbitals from H to Kr. Phys. Rev. B 2004, 69, 195113. [Google Scholar] [CrossRef]
- Ozaki, T.; Kino, H. Efficient projector expansion for the ab initio LCAO method. Phys. Rev. B 2005, 72, 045121. [Google Scholar] [CrossRef]
- Lejaeghere, K.; Bihlmayer, G.; Björkman, T.; Blaha, P.; Blügel, S.; Blum, V.; Caliste, D.; Castelli, I.E.; Clark, S.J.; Dal Corso, A.; et al. Reproducibility in density functional theory calculations of solids. Science 2016, 351, aad3000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bachelet, G.B.; Hamann, D.R.; Schlüter, M. Pseudopotentials that work: From H to Pu. Phys. Rev. B 1982, 26, 4199–4228. [Google Scholar] [CrossRef]
- Kleinman, L.; Bylander, D.M. Efficacious Form for Model Pseudopotentials. Phys. Rev. Lett. 1982, 48, 1425–1428. [Google Scholar] [CrossRef]
- Blöchl, P.E. Generalized separable potentials for electronic-structure calculations. Phys. Rev. B 1990, 41, 5414–5416. [Google Scholar] [CrossRef]
- Troullier, N.; Martins, J.L. Efficient pseudopotentials for plane-wave calculations. Phys. Rev. B 1991, 43, 1993–2006. [Google Scholar] [CrossRef]
- Morrison, I.; Bylander, D.M.; Kleinman, L. Nonlocal Hermitian norm-conserving Vanderbilt pseudopotential. Phys. Rev. B 1993, 47, 6728–6731. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456. [Google Scholar] [CrossRef] [PubMed]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Mazari, N.; Vanderbilt, D. Maximally localized generalized Wannier functions for composite energy bands. Phys. Rev. B 1997, 56, 12847. [Google Scholar] [CrossRef] [Green Version]
- Souza, I.; Marzari, N.; Vanderbilt, D. Maximally localized Wannier functions for entangled energy bands. Phys. Rev. B 2001, 65, 035109. [Google Scholar] [CrossRef] [Green Version]
- Momma, K.; Izumi, F. VESTA3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]
- Tsuzuki, S.; Honda, K.; Uchimaru, T.; Mikami, M.; Tanabe, K. The Magnitude of the CH/π Interaction between Benzene and Some Model Hydrocarbons. J. Am. Chem. Soc. 2000, 122, 3746–3753. [Google Scholar] [CrossRef]
- Yoshida, Y.; Muroi, K.; Otsuka, A.; Saito, G.; Takahashi, M.; Yoko, T. 1-Ethyl-3-methylimidazolium Based Ionic Liquids Containing Cyano Groups: Synthesis, Characterization, and Crystal Structure. Inorg. Chem. 2004, 43, 1458–1462. [Google Scholar] [CrossRef]
- Wang, Y.; Haoran, L.; Han, S. Structure and conformation properties of 1-alkyl-3-methylimidazolium halide ionic liquids: A density-functional theory study. J. Chem. Phys. 2005, 123, 174501. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.S.; Zhang, Y.; Sinogeikin, S.V.; Xiao, Y.; Kumar, S.; Chow, P.; Cornelius, A.L.; Chen, C. Crystal and Electronic Structure of FeSe at High Pressure and Low Temperature. J. Phys. Chem. B 2010, 114, 12597–12606. [Google Scholar] [CrossRef] [PubMed]
- Kontani, H.; Onari, S. Orbital-Fluctuation-Mediated Superconductivity in Iron Pnictides: Analysis of the Five-Orbital Hubbard-Holstein Model. Phys. Rev. Lett. 2010, 104, 157001. [Google Scholar] [CrossRef] [Green Version]
- Korshunov, M.M. Superconducting state in iron-based materials and spin-fluctuation pairing theory. Phys.-Uspekhi 2014, 57, 813–819. [Google Scholar] [CrossRef] [Green Version]
- Maiti, S.; Korshunov, M.M.; Maier, T.A.; Hirschfeld, P.J.; Chubukov, A.V. Evolution of the Superconducting State of Fe-Based Compounds with Doping. Phys. Rev. Lett. 2011, 107, 147002. [Google Scholar] [CrossRef] [Green Version]
- Maiti, S.; Korshunov, M.M.; Maier, T.A.; Hirschfeld, P.J.; Chubukov, A.V. Evolution of symmetry and structure of the gap in iron-based superconductors with doping and interactions. Phys. Rev. B 2011, 84, 224505. [Google Scholar] [CrossRef] [Green Version]
- Maiti, S.; Korshunov, M.M.; Chubukov, A.V. Gap symmetry in KFe2As2 and the cos4θ gap component in LiFeAs. Phys. Rev. B 2012, 85, 014511. [Google Scholar] [CrossRef] [Green Version]
- Chubukov, A. Pairing Mechanism in Fe-Based Superconductors. Annu. Rev. Condens. Matter Phys. 2012, 3, 57–92. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, R.M.; Chubukov, A.V. Low-energy microscopic models for iron-based superconductors: A review. Rep. Prog. Phys. 2017, 80, 014503. [Google Scholar] [CrossRef]
- Nekrasov, I.A.; Pavlov, N.S.; Sadovskii, M.V.; Slobodchikov, A.A. Electronic structure of FeSe monolayer superconductors. Low Temp. Phys. 2016, 42, 891. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Harriger, L.W.; Luo, H.; Wang, M.; Ewings, R.A.; Guidi, T.; Park, H.; Haule, K.; Kotliar, G.; Hayden, S.M.; et al. Nature of magnetic excitations in superconducting BaFe1.9Ni0.1As2. Nat. Phys. 2012, 8, 376–381. [Google Scholar] [CrossRef] [Green Version]
- Meng, Y.; Xing, X.; Yi, X.; Li, B.; Zhou, N.; Li, M.; Zhang, Y.; Wei, W.; Feng, J.; Terashima, K.; et al. Protonation-induced discrete superconducting phases in bulk FeSe single crystals. arXiv 2021, arXiv:2112.12902. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bond Length, Å | |||||||
|---|---|---|---|---|---|---|---|
| N1–C2 | N1–C5 | N1–C7 | C2–N3 | N3–C4 | N3–C6 | C4–C5 | C7–C8 |
| 1.346 | 1.389 | 1.474 | 1.346 | 1.388 | 1.463 | 1.365 | 1.520 |
| 1.346 | 1.389 | 1.475 | 1.347 | 1.387 | 1.461 | 1.364 | 1.519 |
| Bond Angles in Degrees | |||||||
| N1–C2–N3 | C2–N3–C4 | N3–C4–C5 | C2–N3–C6 | C4–C5–N1 | C2–N1–C5 | C2–N1–C7 | N1–C7–C8 |
| 108.11 | 108.82 | 107.18 | 125.16 | 106.92 | 108.96 | 125.56 | 112.12 |
| 108.01 | 108.88 | 107.21 | 125.61 | 106.92 | 108.97 | 125.40 | 111.96 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Begunovich, L.V.; Korshunov, M.M. Band Structure of Organic-Ion-Intercalated (EMIM)xFeSe Superconductor. Materials 2022, 15, 1856. https://doi.org/10.3390/ma15051856
Begunovich LV, Korshunov MM. Band Structure of Organic-Ion-Intercalated (EMIM)xFeSe Superconductor. Materials. 2022; 15(5):1856. https://doi.org/10.3390/ma15051856
Chicago/Turabian StyleBegunovich, Lyudmila V., and Maxim M. Korshunov. 2022. "Band Structure of Organic-Ion-Intercalated (EMIM)xFeSe Superconductor" Materials 15, no. 5: 1856. https://doi.org/10.3390/ma15051856
APA StyleBegunovich, L. V., & Korshunov, M. M. (2022). Band Structure of Organic-Ion-Intercalated (EMIM)xFeSe Superconductor. Materials, 15(5), 1856. https://doi.org/10.3390/ma15051856