
Construction of Al-Mg-Zn Interatomic Potential and the Prediction of Favored Glass Formation Compositions and Associated Driving Forces


Abstract
:1. Introduction
2. Construction of Al-Mg-Zn Interatomic Potential
3. Metallic Glass Formation for the Al-Mg-Zn System
3.1. Evaluation of Favored Glass-Forming Compositions
3.2. Optimization of Glass-Forming Stoichiometries
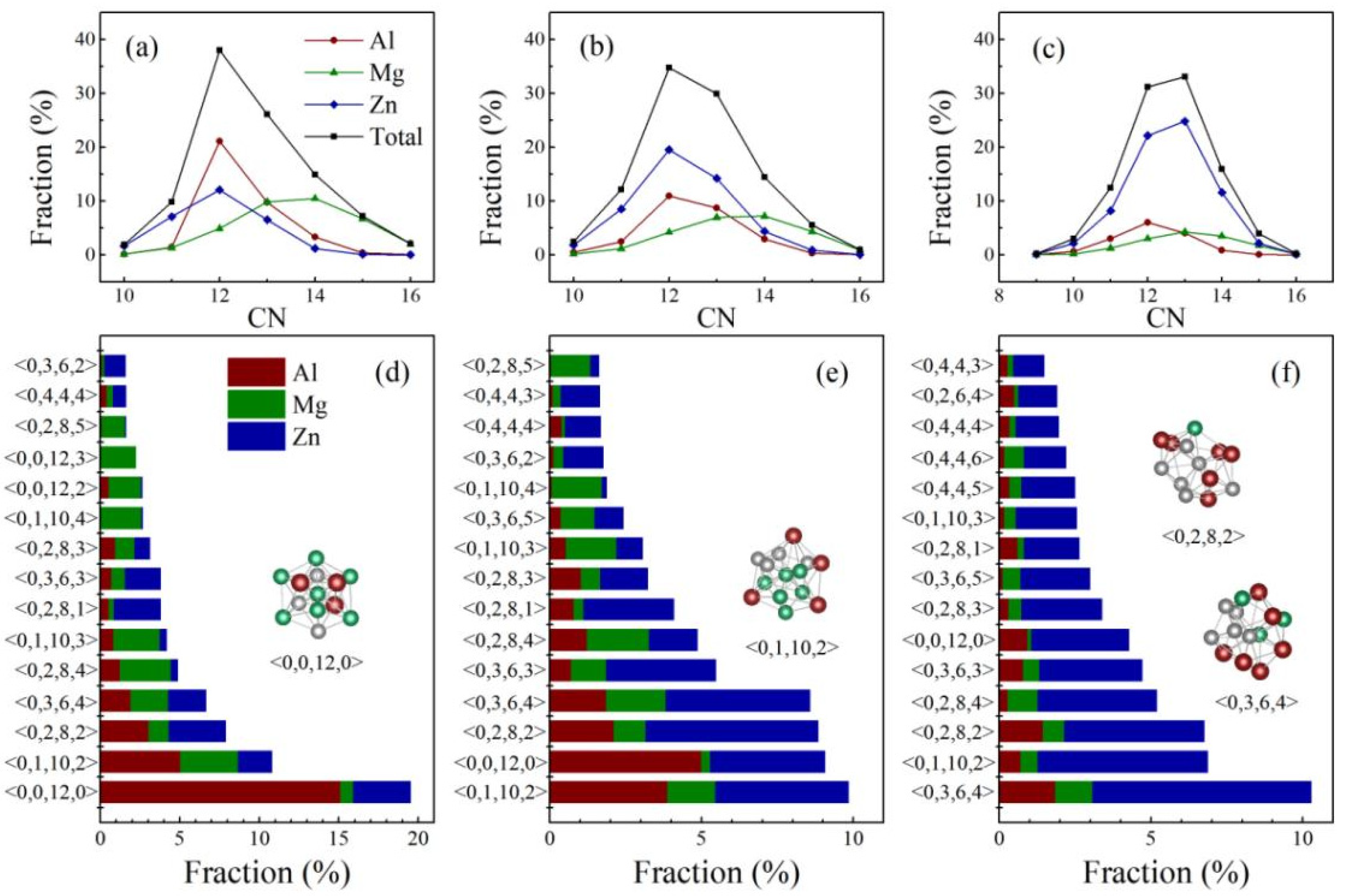
4. Atomic-Level Structure of Al-Mg-Zn Metallic Glasses
4.1. Local Atomic Arrangements in the Short-Range
4.2. Structural Signature of High Glass-Forming Ability
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gutierrez-Urrutia, I. Low density Fe-Mn-Al-C steels: Phase structures, mechanisms and properties. ISIJ Int. 2021, 61, 16–25. [Google Scholar] [CrossRef]
- Clancy, A.J.; Anthony, D.B.; De Luca, F. Metal Mimics: Lightweight, strong, and tough nanocomposites and nanomaterial assemblies. Acs. Appl. Mater. Inter. 2020, 12, 15955–15975. [Google Scholar] [CrossRef] [PubMed]
- Makineni, S.K.; Singh, M.P.; Chattopadhyay, K. Low-density, high-temperature Co base superalloys. Annu. Rev. Mater. Res. 2021, 51, 187–208. [Google Scholar] [CrossRef]
- Pollock, T.M. Weight loss with magnesium alloys. Science 2010, 328, 986–987. [Google Scholar] [CrossRef] [PubMed]
- Brenna, M.; Bucci, V.; Falvo, M.C.; Foiadelli, F.; Ruvio, A.; Sulligoi, G.; Vicenzutti, A. A review on energy efficiency in three transportation sectors: Railways, electrical vehicles and marine. Energies 2020, 13, 2378. [Google Scholar] [CrossRef]
- Yi, J. Fabrication and properties of micro- and nanoscale metallic glassy wires. Adv. Eng. Mater. 2018, 20, 1700875. [Google Scholar] [CrossRef]
- Khan, M.M.; Nemati, A.; Rahman, Z.U.; Shah, U.H.; Asgar, H.; Haider, W. Recent advancements in bulk metallic glasses and their applications. Crit. Rev. Solid State 2018, 43, 233–268. [Google Scholar] [CrossRef]
- Jiang, H.Y.; Shang, T.T.; Xian, H.J.; Sun, B.A.; Zhang, Q.H.; Yu, Q.; Bai, H.Y.; Gu, L.; Wang, W.H. Structures and functional properties of amorphous alloys. Small Struct. 2021, 2, 2000057. [Google Scholar] [CrossRef]
- Gerard, A.Y.; Lutton, K.; Lucente, A.; Frankel, G.S.; Scully, J.R. Progress in understanding the origins of excellent corrosion resistance in metallic alloys: From binary polycrystalline alloys to metallic glasses and high entropy alloys. Corrosion 2020, 76, 485–499. [Google Scholar] [CrossRef]
- Li, H.; Li, Z.; Yang, J.; Ke, H.B.; Sun, B.; Yuan, C.C. Interface design enabled manufacture of giant metallic glasses. Sci. China Mater. 2021, 64, 964–972. [Google Scholar] [CrossRef]
- He, T.; Chen, S.; Lu, T.; Zhao, P.; Chen, W.; Scudino, S. High-strength and ductile ultrafine-grained Al-Y-Ni-Co alloy for high-temperature applications. J. Alloys Compd. 2020, 848, 156655. [Google Scholar] [CrossRef]
- Peng, S.Y.; Zhang, Y.H.; Cui, B.; Ngai, T.L.; Liu, Y.F.; Xiao, Z.Y.; Chen, W. Lamellar-structured Al-based alloys with high strength and plasticity. J. Alloy Compd. 2021, 865, 158927. [Google Scholar] [CrossRef]
- Bai, J.; Xu, Y.; Fan, Q.Z.; Cao, R.H.; Zhou, X.X.; Cheng, Z.J.; Dong, Q.S.; Xue, F. Mechanical properties and degradation behaviors of Zn-xMg alloy fine wires for biomedical applications. Scanning 2021, 12, 4831387. [Google Scholar] [CrossRef] [PubMed]
- Dickel, D.E.; Baskes, M.I.; Aslam, I.; Barrett, C.D. New interatomic potential for Mg-Al-Zn alloys with specific application to dilute Mg-based alloys. Modelling Simul. Mater. Sci. Eng. 2018, 26, 045010. [Google Scholar] [CrossRef]
- Foroughi, A.; Tavakoli, R. Topological and chemical short-range order and their correlation with glass form ability of Mg-Zn metallic glasses: A molecular dynamics study. Comput. Mater. Sci. 2020, 180, 109709. [Google Scholar] [CrossRef]
- Zhao, S.; Li, J.H.; An, S.M.; Li, S.N.; Liu, B.X. Atomistic modeling to investigate the favored composition for metallic glass formation in the Ca-Mg-Ni ternary system. Phys. Chem. Chem. Phys. 2017, 19, 12056–12063. [Google Scholar] [CrossRef] [PubMed]
- Dai, R.; Ashcraft, R.; Gangopadhyay, A.K.; Kelton, K.F. Predicting metallic glass formation from properties of the high temperature liquid. J. Non-Cryst. Solids 2019, 525, 119673. [Google Scholar] [CrossRef]
- Cui, K.Y.; Deng, Y.L.; Zhang, C.; Zhang, B.W.; Liao, S.Z. Prediction of glass forming ranges in Ti-Ni-Zr, Ti-Cu-Zr and Ti-Cu-Hf systems based on miedema and atomic parameter models. Mater. Trans. 2020, 61, 1200–1204. [Google Scholar]
- Niu, X.F.; Yao, G.X.; Qiao, J.W.; Feng, S.D.; Pan, S.P. Effect of Y on the structure-property relationship of Mg65Cu25Y10 metallic glass. Comp. Mater. Sci. 2020, 171, 109285. [Google Scholar] [CrossRef]
- Ramsey, F.P. A mathematical theory of saving. Econ. J. 1928, 38, 543–559. [Google Scholar] [CrossRef]
- Zhang, P.; Maldonis, J.J.; Besse, M.F.; Kramer, M.J.; Voyles, P.M. Medium-range structure and glass forming ability in Zr-Cu-Al bulk metallic glasses. Acta Mater. 2016, 109, 103–114. [Google Scholar] [CrossRef] [Green Version]
- Han, C.Y.; Yang, W.Y.; Lan, Y.K.; Sun, M.H. Al addition on the short and medium range order of CuZrAl metallic glasses. Phys. B 2021, 619, 413237. [Google Scholar] [CrossRef]
- Samavatian, M.; Gholamipour, R.; Samavatian, V.; Farahani, F. Effects of Nb minor addition on atomic structure and glass forming ability of Zr55Cu30Ni5Al10 bulk metallic glass. Mater. Res. Express 2019, 6, 065202. [Google Scholar] [CrossRef]
- Kbirou, M.; Trady, S.; Hasnaoui, A.; Mazroui, M. Short and medium-range orders in Co3Al metallic glass. Chem. Phys. 2018, 513, 58–66. [Google Scholar] [CrossRef]
- Ren, L.; Gao, T.H.; Ma, R.; Xie, Q.; Tian, Z.A.; Chen, Q.; Liang, Y.C.; Hu, X.C. The connection of icosahedral and defective icosahedral clusters in medium range order structures of CuZrAl alloy. J. Non-Cryst. Solids 2019, 521, 119475. [Google Scholar] [CrossRef]
- Davani, F.A.; Hilke, S.; Roesner, H.; Geissler, D.; Gebert, A.; Wilde, G. Correlations between the ductility and medium-range order of bulk metallic glasses. J. Appl. Phys. 2020, 128, 015103. [Google Scholar] [CrossRef]
- Huang, B.; Yuan, C.C.; Wang, Z.Q.; Tong, Y.; Wang, Q.; Yi, J.; Wang, G.; He, Q.F.; Shek, C.H.; Yang, Y. Influence of short- to medium-range electronic and atomic structure on secondary relaxations in metallic glasses. Acta Mater. 2020, 196, 88–100. [Google Scholar] [CrossRef]
- Feng, S.D.; Chan, K.C.; Zhao, L.; Pan, S.P.; Qi, L.; Wang, L.M.; Liu, R.P. Rejuvenation by weakening the medium range order in Zr46Cu46Al8 metallic glass with pressure preloading: A molecular dynamics simulation study. Mater. Des. 2018, 158, 248–255. [Google Scholar] [CrossRef]
- Hilke, S.; Roesner, H.; Geissler, D.; Gebert, A.; Peterlechner, M.; Wilde, G. The influence of deformation on the medium-range order of a Zr-based bulk metallic glass characterized by variable resolution fluctuation electron microscopy. Acta Mater. 2019, 171, 275–281. [Google Scholar] [CrossRef]
- Ghaemi, M.; Tavakoli, R.; Foroughi, A. Comparing short-range and medium-range ordering in Cu-Zr and Ni-Zr metallic glasses—Correlation between structure and glass form ability. J. Non-Cryst. Solids 2018, 499, 227–236. [Google Scholar] [CrossRef]
- Pekin, T.C.; Ding, J.; Gammer, C.; Ozdol, B.; Ophus, C.; Asta, M.; Ritchie, R.O.; Minor, A.M. Direct measurement of nanostructural change during in situ deformation of a bulk metallic glass. Nat. Commun. 2019, 10, 2445. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.H.; Li, J.H.; Liu, B.X. Fractal analysis on the cluster network in metallic liquid and glass. J. Alloy Compd. 2018, 757, 228–232. [Google Scholar] [CrossRef]
- Xie, Z.C.; Gao, T.H.; Guo, X.T.; Qin, X.M.; Xie, Q. Network connectivity in icosahedral medium-range order of metallic glass: A molecular dynamics simulation. J. Non-Cryst. Solids 2014, 406, 31–36. [Google Scholar] [CrossRef]
- Wang, C.C.; Wong, C.H. Interpenetrating networks in Zr-Cu-Al and Zr-Cu metallic glasses. Intermetallics 2012, 22, 13–16. [Google Scholar] [CrossRef]
- Li, J.H.; Dai, X.D.; Wang, T.L.; Liu, B.X. A binomial truncation function proposed for the second-moment approximation of tight-binding potential and application in the ternary Ni-Hf-Ti system. J. Phys. Condens. Matter 2007, 19, 271–296. [Google Scholar] [CrossRef]
- Zhao, S.; Li, J.H.; Liu, J.B.; Li, S.N.; Liu, B.X. Atomistic approach to design favored compositions for the ternary Al-Mg-Ca metallic glass formation. RSC Adv. 2015, 5, 93623–93630. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab-initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef] [PubMed]
- Garland, C.W.; Dalven, R. Elastic constants of zinc from 4.2°K to 77.6°K. Phys. Rev. 1958, 111, 1232. [Google Scholar] [CrossRef]
- Porter, F.C. Zinc Handbook: Properties, Processing, and Use in Design; Marcel Dekker: New York, NY, USA, 1991. [Google Scholar]
- Rose, J.H.; Smith, J.R.; Guinea, F.; Ferrante, J. Universal features of the equation of state of metals. Phys. Rev. B Condens. Matter Mater. Phys. 1984, 29, 2963–2969. [Google Scholar] [CrossRef]
- Wang, W.H.; Dong, C.; Shek, C.H. Bulk metallic glasses. Mater. Sci. Eng. R 2004, 44, 45–89. [Google Scholar] [CrossRef]
- Schroers, J. Processing of bulk metallic glass. Adv. Mater. 2010, 22, 1566–1597. [Google Scholar] [CrossRef] [PubMed]
- Basu, J.; Murty, B.S.; Ranganathan, S.J. Glass forming ability: Miedema approach to (Zr, Ti, Hf)-(Cu, Ni) binary and ternary alloys. J. Alloys Compd. 2008, 465, 163–172. [Google Scholar] [CrossRef]
- Dai, Y.; Li, J.H.; Che, X.L.; Liu, B.X. Proposed long-range empirical potential to study the metallic glasses in the Ni-Nb-Ta system. J. Phys. Chem. B 2009, 113, 7282–7290. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182. [Google Scholar] [CrossRef]
- Allen, M.P.; Tildesley, D.J. Computer Simulation of Liquids; Oxford University Press: New York, NY, USA, 1989. [Google Scholar]
- Dai, Y.; Li, J.H.; Che, X.L. Glass formation region of the Ni-Nb-Ta ternary metal system determined directly from n-body potential through molecular dynamics simulations. J. Mater. Res. 2009, 24, 1815–1819. [Google Scholar] [CrossRef]
- Dai, Y.; Li, J.H.; Liu, B.X. First-principles molecular dynamics simulations to study the crystal-to-amorphous transition in the Mg-Zn system. Intermetallics 2012, 29, 75–79. [Google Scholar] [CrossRef]
- Din, S.U.; Chishti, S.Y. Synthesis and characterization of Al40Mg25Zn35 amorphous powder by rapid solidification. Powder Technol. 2001, 114, 51–54. [Google Scholar] [CrossRef]
- Niikura, A.; Tsai, A.P.; Nishiyama, N.; Inoue, A.; Masumoto, T. Amorphous and quasi-crystalline phases in rapidly solidified Mg-Al-Zn alloys. Mater. Sci. Eng. A 1994, 182, 1387–1391. [Google Scholar] [CrossRef]
- Richter, R.; Baxter, D.V.; Stromolsen, J.O. Quantum corrections to the conductivity in Mg-based metallic glasses. Phys. Rev. B 1988, 38, 10421. [Google Scholar] [CrossRef] [PubMed]
- Calka, A.; Polk, D.E.; Giessen, B.C.; Matyja, H.; Sande, J.V.; Madhava, M. A transition-metal-free amorphous alloy: Mg70Zn30. Scripta Metall. 1977, 11, 65–70. [Google Scholar] [CrossRef]
- Calka, A.; Radlinski, A.P. Amorphization of Mg-Zn alloys by mechanical alloying. Mater. Sci. Eng. A 1989, 11, 131–135. [Google Scholar] [CrossRef]
- Calka, A. The room-temperature stability of amorphous Mg-Zn alloys. J Phys. F Met. Phys. 1986, 16, 1577. [Google Scholar] [CrossRef]
- Richter, R.; Baxter, D.V.; Stromolsen, J.O. Quantum corrections to the conductivity in Mg70Cu30-0Au0, Mg70Cu30-1Au1, Mg70Cu30-3Au3, Mg70Cu30-9Au9 and Mg70Zn30-0Au0, Mg70Zn30-3Au3. Mater. Sci. Eng. 1988, 99, 183. [Google Scholar] [CrossRef]
- Gorsse, S.; Orveillon, G.; Senkov, O.N.; Miracle, D.B. Thermodynamic analysis of glass-forming ability in a Ca-Mg-Zn ternary alloy system. Phys. Rev. B 2006, 73, 224202. [Google Scholar] [CrossRef] [Green Version]
- Lide, D.R.; Bruno, T.J. CRC Handbook of Chemistry and Physics; CRC Press: Boca Raton, FL, USA, 2012. [Google Scholar]
- Dai, X.D.; Li, J.H.; Liu, B.X. Molecular statics calculation of the formation enthalpy for ternary metal systems based on the long-range empirical interatomic potentials. Appl. Phys. Lett. 2007, 90, 131904. [Google Scholar] [CrossRef]
- Finney, J.L. Random packings and the structure of simple liquids. The geometry of random close packing. Proc. R. Soc. Lond. Ser. A 1970, 319, 479–493. [Google Scholar]
- Cheng, Y.Q.; Ma, E.; Sheng, H.W. Atomic level structure in multicomponent bulk metallic glass. Phys. Rev. Lett. 2009, 102, 245501. [Google Scholar] [CrossRef] [PubMed]
- Li, J.H.; Zhao, S.Z.; Dai, Y.; Cui, Y.Y.; Liu, B.X. Formation and structure of Al-Zr metallic glasses studied by Monte Carlo simulations. J. Appl. Phys. 2011, 109, 113538. [Google Scholar] [CrossRef]
- Sheng, H.W.; Luo, W.K.; Alamgir, F.M.; Bai, J.M.; Ma, E. Atomic packing and short-to-medium-range order in metallic glasses. Nature 2006, 439, 419–425. [Google Scholar] [CrossRef]
- Wu, S.Q.; Wang, C.Z.; Hao, S.G.; Zhu, Z.Z.; Ho, K.M. Energetics of local clusters in Cu64.5Zr35.5 metallic liquid and glass. Appl. Phys. Lett. 2010, 97, 21901. [Google Scholar] [CrossRef]
- Guan, P.F.; Fujita, T.; Hirata, A.; Liu, Y.H.; Chen, M.W. Structural origins of the excellent glass forming ability of Pd40Ni40P20. Phys. Rev. Lett. 2012, 108, 175501. [Google Scholar] [CrossRef] [PubMed]
- Zemp, J.; Celino, M.; Schonfeld, B.; Loeffler, J.F. Icosahedral superclusters in Cu64Zr36 metallic glass. Phys. Rev. B Condens. Matter Mater. Phys. 2014, 90, 144108. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Potential Parameters | Al-Al | Mg-Mg | Zn-Zn | Al-Mg | Al-Zn | Mg-Zn |
|---|---|---|---|---|---|---|
| p1 | 8.776460 | 10.373070 | 12.365126 | 10.238284 | 7.906891 | 9.350041 |
| A1 (10−19 J) | 0.402184 | 0.145780 | 0.122940 | 0.190712 | 0.518358 | 0.436637 |
| rm1 (Å) | 2.764394 | 3.522308 | 2.287506 | 2.654430 | 2.667892 | 2.590032 |
| n1 | 4 | 4 | 4 | 4 | 4 | 4 |
| p1m | 2.558558 | 3.850843 | 5.432586 | 3.447699 | 1.328661 | 3.962105 |
| A1m (10−19 J) | 2.917212 | 0.538535 | 3.572668 | 3.457161 | 0.505861 | 2.898628 |
| rc1 (Å) | 4.607023 | 5.487015 | 3.875254 | 4.421071 | 5.375373 | 4.800655 |
| p2 | 5.249466 | 4.375061 | 6.403749 | 3.439822 | 6.359076 | 9.050849 |
| A2 (10−38 J2) | 4.738155 | 0.951887 | 0.600921 | 1.890899 | 3.558540 | 1.873409 |
| rm2 (Å) | 3.786874 | 2.588516 | 3.891120 | 2.636021 | 4.330974 | 3.521581 |
| n2 | 5 | 5 | 5 | 5 | 5 | 5 |
| p2m | 0.000477 | 0.000378 | 0.000389 | 0.000439 | 0.000286 | 0.000486 |
| A2m (10−38 J2) | 1.114067 | 1.130393 | 0.146081 | 0.441638 | 0.376332 | 6.968794 |
| rc2 (Å) | 6.515324 | 6.250000 | 6.039821 | 6.996006 | 6.538981 | 5.166639 |
| r0 (Å) | 2.864321 | 3.203567 | 2.751782 | 2.999131 | 2.808051 | 2.977674 |
| Physical Properties | hcp-Zn | fcc-Zn | bcc-Zn | |||
|---|---|---|---|---|---|---|
| Fitted | Experiments | Fitted | Ab Initio | Fitted | Ab Initio | |
| a or a, c (Å) | 2.651, 4.614 | 2.665, 4.947 | 3.891 | 3.932 | 3.098 | 3.135 |
| Ec (eV/atom) | 1.348 | 1.350 | 1.330 | 1.325 | 1.317 | 1.264 |
| C11 (Mbar) | 1.63 | 1.77 | 1.086 | 1.106 | 0.311 | 0.365 |
| C12 (Mbar) | 0.428 | 0.348 | 0.504 | 0.522 | 0.851 | 0.813 |
| C13 (Mbar) | 0.452 | 0.528 | ||||
| C33 (Mbar) | 0.403 | 0.685 | ||||
| C44 (Mbar) | 0.325 | 0.459 | 0.012 | 0.005 | 0.107 | 0.127 |
| B0 (Mbar) | 0.703 | 0.700 | 0.698 | 0.717 | 0.671 | 0.664 |
| Physical Properties | B2-AlZn | L12-AlZn3 | L12-Al3Zn | |||
|---|---|---|---|---|---|---|
| Fitted | Ab Initio | Fitted | Ab Initio | Fitted | Ab Initio | |
| a (Å) | 3.201 | 3.196 | 3.952 | 3.965 | 4.061 | 4.022 |
| Ec (eV/atom) | 2.249 | 2.396 | 1.934 | 2.000 | 2.851 | 2.846 |
| C11 (Mbar) | 0.564 | 0.407 | 1.186 | 1.354 | 0.915 | 1.048 |
| C12 (Mbar) | 0.728 | 0.767 | 0.501 | 0.437 | 0.597 | 0.617 |
| C44 (Mbar) | 0.235 | 0.216 | 0.105 | 0.023 | 0.241 | 0.257 |
| B0 (Mbar) | 0.673 | 0.647 | 0.729 | 0.743 | 0.703 | 0.761 |
| Physical Properties | MgZn | MgZn3 | Mg3Zn | MgZn2 |
|---|---|---|---|---|
| B2 | L12 | L12 | C14 | |
| a or a, c (Å) | 3.440 | 4.142 | 4.429 | 5.3066, 8.2746 |
| 3.306 | 4.041 | 4.331 | 5.2073, 8.5315 | |
| Ec (eV/atom) | 1.508 | 1.459 | 1.493 | 1.522 |
| 1.510 | 1.438 | 1.492 | 1.541 | |
| B0 (Mbar) | 0.520 | 0.592 | 0.445 | 0.609 |
| 0.501 | 0.595 | 0.413 | 0.653 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cai, B.; Li, J.; Lai, W.; Liu, J.; Liu, B. Construction of Al-Mg-Zn Interatomic Potential and the Prediction of Favored Glass Formation Compositions and Associated Driving Forces. Materials 2022, 15, 2062. https://doi.org/10.3390/ma15062062
Cai B, Li J, Lai W, Liu J, Liu B. Construction of Al-Mg-Zn Interatomic Potential and the Prediction of Favored Glass Formation Compositions and Associated Driving Forces. Materials. 2022; 15(6):2062. https://doi.org/10.3390/ma15062062
Chicago/Turabian StyleCai, Bei, Jiahao Li, Wensheng Lai, Jianbo Liu, and Baixin Liu. 2022. "Construction of Al-Mg-Zn Interatomic Potential and the Prediction of Favored Glass Formation Compositions and Associated Driving Forces" Materials 15, no. 6: 2062. https://doi.org/10.3390/ma15062062
APA StyleCai, B., Li, J., Lai, W., Liu, J., & Liu, B. (2022). Construction of Al-Mg-Zn Interatomic Potential and the Prediction of Favored Glass Formation Compositions and Associated Driving Forces. Materials, 15(6), 2062. https://doi.org/10.3390/ma15062062