

Hyperbranched Polyelectrolyte Copolymers as Novel Candidate Delivery Systems for Bio-Relevant Compounds



Abstract
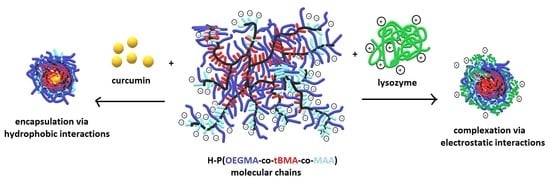
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of H-[P(OEGMA-co-tBMA-co-MAA) Copolymers
2.2.1. Preparation of H-[P(OEGMA-co-tBMA)] Copolymers via RAFT Polymerization
2.2.2. Preparation of H-[P(OEGMA-co-tBMA-co-MAA)] Copolymers via Hydrolysis Reaction
2.3. Preparation of Self-Assembled PNPs in Aqueous Solutions
2.4. Preparation of HC–Lys Complexes
2.5. Preparation of CUR Loaded Aggregates
2.6. Experimental Techniques
2.6.1. Size Exclusion Chromatography
2.6.2. Proton Nuclear Magnetic Resonance Spectroscopy (1H-NMR)
2.6.3. Attenuated Total Reflectance-Fourier Transform Infrared (ATR–FTIR) Spectroscopy
2.6.4. Dynamic Light Scattering (DLS)
2.6.5. Electrophoretic Light Scattering (ELS)
2.6.6. Fluorescence Spectroscopy
2.6.7. UV-Vis Spectroscopy
3. Results and Discussion
3.1. HC Synthesis and Characterization
3.2. Self-Assembly Studies of Amphiphilic Polyelectrolyte Hyperbranched Copolymers in Aqueous Media
3.2.1. CAC determination
3.2.2. Physicochemical Characterization of Self-Assembled Aggregates
3.3. Protein Complexation Studies
3.4. Curcumin Drug Loading Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Souery, W.N.; Bishop, C.J. Clinically Advancing and Promising Polymer-Based Therapeutics. Acta Biomater. 2018, 67, 1–20. [Google Scholar] [CrossRef]
- Begines, B.; Ortiz, T.; Pérez-Aranda, M.; Martínez, G.; Merinero, M.; Argüelles-Arias, F.; Alcudia, A. Polymeric Nanoparticles for Drug Delivery: Recent Developments and Future Prospects. Nanomaterials 2020, 10, 1403. [Google Scholar] [CrossRef] [PubMed]
- Lotocki, V.; Kakkar, A. Miktoarm Star Polymers: Branched Architectures in Drug Delivery. Pharmaceutics 2020, 12, 827. [Google Scholar] [CrossRef] [PubMed]
- Panday, R.; Poudel, A.J.; Li, X.; Adhikari, M.; Ullah, M.W.; Yang, G. Amphiphilic Core-Shell Nanoparticles: Synthesis, Biophysical Properties, and Applications. Colloids Surf. B Biointerfaces 2018, 172, 68–81. [Google Scholar] [CrossRef]
- Sur, S.; Rathore, A.; Dave, V.; Reddy, K.R.; Chouhan, R.S.; Sadhu, V. Recent Developments in Functionalized Polymer Nanoparticles for Efficient Drug Delivery System. Nano-Struct. Nano-Objects 2019, 20, 100397. [Google Scholar] [CrossRef]
- Osorno, L.L.; Brandley, A.N.; Maldonado, D.E.; Yiantsos, A.; Mosley, R.J.; Byrne, M.E. Review of Contemporary Self-Assembled Systems for the Controlled Delivery of Therapeutics in Medicine. Nanomaterials 2021, 11, 278. [Google Scholar] [CrossRef]
- Yasen, W.; Dong, R.; Aini, A.; Zhu, X. Recent Advances in Supramolecular Block Copolymers for Biomedical Applications. J. Mater. Chem. B 2020, 8, 8219–8231. [Google Scholar] [CrossRef]
- Pham, D.T.; Chokamonsirikun, A.; Phattaravorakarn, V.; Tiyaboonchai, W. Polymeric Micelles for Pulmonary Drug Delivery: A Comprehensive Review. J. Mater. Sci. 2021, 56, 2016–2036. [Google Scholar] [CrossRef]
- Corrigan, N.; Jung, K.; Moad, G.; Hawker, C.J.; Matyjaszewski, K.; Boyer, C. Reversible-Deactivation Radical Polymerization (Controlled/Living Radical Polymerization): From Discovery to Materials Design and Applications. Prog. Polym. Sci. 2020, 111, 101311. [Google Scholar]
- Zhang, B.; Zhang, H.; Li, Y.; Hoskins, J.N.; Grayson, S.M. Exploring the Effect of Amphiphilic Polymer Architecture: Synthesis, Characterization, and Self-Assembly of Both Cyclic and Linear Poly(Ethylene Gylcol)- B -Polycaprolactone. ACS Macro Lett. 2013, 2, 845–848. [Google Scholar] [CrossRef]
- Lonsdale, D.E.; Whittaker, M.R.; Monteiro, M.J. Self-Assembly of Well-Defined Amphiphilic Polymeric Miktoarm Stars, Dendrons, and Dendrimers in Water: The Effect of Architecture. J. Polym. Sci. A Polym. Chem. 2009, 47, 6292–6303. [Google Scholar] [CrossRef]
- Zhao, L.; Lin, Z. Self-Assembly of Non-Linear Polymers at the Air/Water Interface: The Effect of Molecular Architecture. Soft. Matter 2011, 7, 10520–10535. [Google Scholar] [CrossRef]
- Cook, A.B.; Perrier, S. Branched and Dendritic Polymer Architectures: Functional Nanomaterials for Therapeutic Delivery. Adv. Funct. Mater. 2020, 30, 1901001. [Google Scholar] [CrossRef] [Green Version]
- Pearce, A.K.; Anane-Adjei, A.B.; Cavanagh, R.J.; Monteiro, P.F.; Bennett, T.M.; Taresco, V.; Clarke, P.A.; Ritchie, A.A.; Alexander, M.R.; Grabowska, A.M.; et al. Effects of Polymer 3D Architecture, Size, and Chemistry on Biological Transport and Drug Delivery In Vitro and in Orthotopic Triple Negative Breast Cancer Models. Adv. Healthc. Mater. 2020, 9, 2000892. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Dai, Y.; Dai, G. Advances in Amphiphilic Hyperbranched Copolymers with an Aliphatic Hyperbranched 2,2-Bis(Methylol)Propionic Acid-Based Polyester Core. Polym. Chem. 2020, 11, 964–973. [Google Scholar] [CrossRef]
- Bera, S.; Barman, R.; Ghosh, S. Hyperbranched vs. Linear Poly(Disulfide) for Intracellular Drug Delivery. Polym. Chem. 2022, 13, 5188–5192. [Google Scholar] [CrossRef]
- Tang, Q.; Cheng, F.; Lou, X.L.; Liu, H.J.; Chen, Y. Comparative Study of Thiol-Free Amphiphilic Hyperbranched and Linear Polymers for the Stabilization of Large Gold Nanoparticles in Organic Solvent. J. Colloid. Interface Sci. 2009, 337, 485–491. [Google Scholar] [CrossRef]
- Liu, J.; Huang, W.; Pang, Y.; Zhu, X.; Zhou, Y.; Yan, D. Self-Assembled Micelles from an Amphiphilic Hyperbranched Copolymer with Polyphosphate Arms for Drug Delivery. Langmuir 2010, 26, 10585–10592. [Google Scholar] [CrossRef]
- Konkolewicz, D.; Monteiro, M.J.; Perrier, S. Dendritic and Hyperbranched Polymers from Macromolecular Units: Elegant Approaches to the Synthesis of Functional Polymers. Macromolecules 2011, 44, 7067–7087. [Google Scholar] [CrossRef]
- Luzon, M.; Boyer, C.; Peinado, C.; Corrales, T.; Whittaker, M.; Tao, L.; Davis, T.P. Water-Soluble, Thermoresponsive, Hyperbranched Copolymers Based on PEG-Methacrylates: Synthesis, Characterization, and LCST Behavior. J. Polym. Sci. A Polym. Chem. 2010, 48, 2783–2792. [Google Scholar] [CrossRef]
- Rikkou-Kalourkoti, M.; Elladiou, M.; Patrickios, C.S. Synthesis and Characterization of Hyperbranched Amphiphilic Block Copolymers Prepared via Self-Condensing RAFT Polymerization. J. Polym. Sci. A Polym. Chem. 2015, 53, 1310–1319. [Google Scholar] [CrossRef]
- Zhukova, O.V.; Sergeeva, T.F.; Gavrina, A.I. Modified Poly(T-Butyl Methacrylate) as a Doxorubicin Carrier for Targeted Delivery. Pharm. Chem. J. 2018, 52, 539–544. [Google Scholar] [CrossRef]
- Porsch, C.; Zhang, Y.; Östlund, Å.; Damberg, P.; Ducani, C.; Malmström, E.; Nyström, A.M. In Vitro Evaluation of Non-Protein Adsorbing Breast Cancer Theranostics Based on 19F-Polymer Containing Nanoparticles. Part. Part. Syst. Charact. 2013, 30, 381–390. [Google Scholar] [CrossRef] [Green Version]
- Carr, D.A.; Gómez-Burgaz, M.; Boudes, M.C.; Peppas, N.A. Complexation Hydrogels for the Oral Delivery of Growth Hormone and Salmon Calcitonin. Ind. Eng. Chem. Res. 2010, 49, 11991–11995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arya, A.; Pathak, D.P.; Majumdar, D.K.; Manchanda, S. Methacrylic Acid-Co-Butylmethacrylate Copolymers: Design, Characterization and Evaluation as Encapsulating Material for Colon Targeted Formulations. Des. Monomers Polym. 2016, 19, 34–46. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Wannasarit, S.; Figueiredo, P.; Molinaro, G.; Ding, Y.; Correia, A.; Casettari, L.; Wiwattanapatapee, R.; Hirvonen, J.; Liu, D.; et al. Intracellular Delivery of Budesonide and Polydopamine Co-Loaded in Endosomolytic Poly(Butyl Methacrylate-Co-Methacrylic Acid) Grafted Acetalated Dextran for Macrophage Phenotype Switch from M1 to M2. Adv. Ther. 2021, 4, 2000058. [Google Scholar] [CrossRef]
- Pan, X.; Guo, X.; Choi, B.; Feng, A.; Wei, X.; Thang, S.H. A Facile Synthesis of PH Stimuli Biocompatible Block Copolymer Poly(Methacrylic Acid)-: Block -Poly(N -Vinylpyrrolidone) Utilizing Switchable RAFT Agents. Polym. Chem. 2019, 10, 2083–2090. [Google Scholar] [CrossRef]
- Sezgin-bayindir, Z.; Ergin, A.D.; Parmaksiz, M.; Elcin, A.E.; Elcin, Y.M.; Yuksel, N. Evaluation of Various Block Copolymers for Micelle Formation and Brain Drug Delivery: In Vitro Characterization and Cellular Uptake Studies. J. Drug. Deliv. Sci. Technol. 2016, 36, 120–129. [Google Scholar] [CrossRef]
- Tripathi, A.K.; Neenan, M.L.; Sundberg, D.C.; Tsavalas, J.G. Influence of N-Alkyl Ester Groups on Efficiency of Crosslinking for Methacrylate Monomers Copolymerized with EGDMA: Experiments and Monte Carlo Simulations of Reaction Kinetics and Sol-Gel Structure. Polymer 2016, 96, 130–145. [Google Scholar] [CrossRef]
- Kurmaz, S.V.; Bubnova, M.L.; Perepelitsina, E.O.; Estrina, G.A. Control of Crosslinking Free-Radical Copolymerization of Ethylene Glycol Dimethacrylate with Alkyl Methacrylates of Various Structures and Macromolecular Design of Copolymers. Polym. Sci.-Ser. A 2006, 48, 696–706. [Google Scholar] [CrossRef]
- Gao, Y.; Zhou, D.; Lyu, J.; Sigen, A.; Xu, Q.; Newland, B.; Matyjaszewski, K.; Tai, H.; Wang, W. Complex Polymer Architectures through Free-Radical Polymerization of Multivinyl Monomers. Nat. Rev. Chem. 2020, 4, 194–212. [Google Scholar]
- Newland, B.; Thomas, L.; Zheng, Y.; Steinhart, M.; Werner, C.; Wang, W. Preparation, Loading, and Cytotoxicity Analysis of Polymer Nanotubes from an Ethylene Glycol Dimethacrylate Homopolymer in Comparison to Multi-walled Carbon Nanotubes. J. Interdiscip. Nanomed. 2016, 1, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Tochwin, A.; El-Betany, A.; Tai, H.; Chan, K.Y.; Blackburn, C.; Wang, W. Thermoresponsive and Reducible Hyperbranched Polymers Synthesized by RAFT Polymerisation. Polymers 2017, 9, 443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogt, A.P.; Gondi, S.R.; Sumerlin, B.S. Hyperbranched Polymers via RAFT Copolymerization of an Acryloyl Trithiocarbonate. Aust. J. Chem. 2007, 60, 396–399. [Google Scholar] [CrossRef]
- Kim, K.-C.; Roh, S.-W.; Ryu, S.-W. Lithium Ion Concentration Dependant Ionic Conductivity and Thermal Properties in Solid Poly(PEGMA-Co-Acrylonitrile) Electrolytes. J. Electrochem. Sci. Technol. 2010, 1, 57–62. [Google Scholar] [CrossRef]
- Ye, M.; Zhang, D.; Han, L.; Tejada, J.; Ortiz, C. Synthesis, Preparation, and Conformation of Stimulus-Responsive End-Grafted Poly(Methacrylic Acid-g-Ethylene Glycol) Layers. Soft Matter 2006, 2, 243–256. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, R.; Heuser, T.; Weyandt, E.; Wang, B.; Walther, A. Polyacid Microgels with Adaptive Hydrophobic Pockets and Ampholytic Character: Synthesis, Solution Properties and Insights into Internal Nanostructure by Cryogenic-TEM. Soft Matter 2015, 11, 8342–8352. [Google Scholar] [CrossRef]
- Mohammad, S.A.; Dolui, S.; Kumar, D.; Mane, S.R.; Banerjee, S. Facile Access to Functional Polyacrylates with Dual Stimuli Response and Tunable Surface Hydrophobicity. Polym. Chem. 2021, 12, 3042–3051. [Google Scholar] [CrossRef]
- Teulère, C.; Ben-Osman, C.; Barry, C.; Nicolaÿ, R. Synthesis and Self-Assembly of Amphiphilic Heterografted Molecular Brushes Prepared by Telomerization. Eur. Polym. J. 2020, 141, 110080. [Google Scholar] [CrossRef]
- Chen, L.; Simpson, J.D.; Fuchs, A.V.; Rolfe, B.E.; Thurecht, K.J. Effects of Surface Charge of Hyperbranched Polymers on Cytotoxicity, Dynamic Cellular Uptake and Localization, Hemotoxicity, and Pharmacokinetics in Mice. Mol. Pharm. 2017, 14, 4485–4497. [Google Scholar] [CrossRef]
- Cazares-Cortes, E.; Baker, B.C.; Nishimori, K.; Ouchi, M.; Tournilhac, F. Polymethacrylic Acid Shows Thermoresponsivity in an Organic Solvent. Macromolecules 2019, 52, 5995–6004. [Google Scholar] [CrossRef] [Green Version]
- Topuzogullari, M.; Bulmus, V.; Dalgakiran, E.; Dincer, S. PH- and Temperature-Responsive Amphiphilic Diblock Copolymers of 4-Vinylpyridine and Oligoethyleneglycol Methacrylate Synthesized by RAFT Polymerization. Polymer 2014, 55, 525–534. [Google Scholar] [CrossRef]
- Hong, C.R.; Park, S.J.; Choi, S.J. Influence of the Hydrophilic Head Size and Hydrophobic Tail Length of Surfactants on the Ability of Micelles to Stabilize Citral. J. Sci. Food Agric. 2016, 96, 3227–3232. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Liu, C.; Zhu, J.; Pochan, D.J.; Jia, X. Hybrid, Elastomeric Hydrogels Crosslinked by Multifunctional Block Copolymer Micelles. Soft Matter 2010, 6, 5293–5297. [Google Scholar] [CrossRef]
- Qu, J.B.; Chapman, R.; Chen, F.; Lu, H.; Stenzel, M.H. Swollen Micelles for the Preparation of Gated, Squeezable, PH-Responsive Drug Carriers. ACS Appl. Mater. Interfaces 2017, 9, 13865–13874. [Google Scholar] [CrossRef]
- Steudle, A.; Pleiss, J. Modelling of Lysozyme Binding to a Cation Exchange Surface at Atomic Detail: The Role of Flexibility. Biophys. J. 2011, 100, 3016–3024. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Zhang, R.; Zhu, Y.; Xu, S.; Liu, X. Influence of Ionic Strength on Gel-like Pickering Emulsions Stabilized by Self-Assembled Colloidal Nanoparticles Containing Lysozyme. Colloid Polym. Sci. 2020, 298, 1249–1262. [Google Scholar] [CrossRef]
- Chernysheva, M.G.; Shnitko, A.V.; Ksenofontov, A.L.; Arutyunyan, A.M.; Petoukhov, M.V.; Badun, G.A. Structural Peculiarities of Lysozyme—PLURONIC Complexes at the Aqueous-Air and Liquid-Liquid Interfaces and in the Bulk of Aqueous Solution. Int. J. Biol. Macromol. 2020, 158, 721–731. [Google Scholar] [CrossRef]
- Sepúlveda-Rivas, S.; Fritz, H.F.; Valenzuela, C.; Santiviago, C.A.; Morales, J.O. Development of Novel EE/Alginate Polyelectrolyte Complex Nanoparticles for Lysozyme Delivery: Physicochemical Properties and in Vitro Safety. Pharmaceutics 2019, 11, 103. [Google Scholar] [CrossRef] [Green Version]
- Karayianni, M.; Gancheva, V.; Pispas, S.; Petrov, P. Complex Formation between Lysozyme and Stabilized Micelles with a Mixed Poly(Ethylene Oxide)/Poly(Acrylic Acid) Shell. J. Phys. Chem. B 2016, 120, 2625–2637. [Google Scholar] [CrossRef]
- Alkudaisi, N.; Russell, B.A.; Jachimska, B.; Birch, D.J.S.; Chen, Y. Detecting Lysozyme Unfolding: Via the Fluorescence of Lysozyme Encapsulated Gold Nanoclusters. J. Mater. Chem. B 2019, 7, 1167–1175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rani, S.; Mishra, S.; Sharma, M.; Nandy, A.; Mozumdar, S. Solubility and Stability Enhancement of Curcumin in Soluplus® Polymeric Micelles: A Spectroscopic Study. J. Dispers. Sci. Technol. 2020, 41, 523–536. [Google Scholar] [CrossRef]
- Yallapu, M.M.; Jaggi, M.; Chauhan, S.C. Curcumin Nanoformulations: A Future Nanomedicine for Cancer. Drug. Discov. Today 2012, 17, 71–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, T.; Trench, D.; Putnam, J.; Stenzel, M.H.; Lord, M.S. Curcumin-Loading-Dependent Stability of PEGMEMA-Based Micelles Affects Endocytosis and Exocytosis in Colon Carcinoma Cells. Mol. Pharm. 2016, 13, 924–932. [Google Scholar] [CrossRef]
- Saxena, S.; Pradeep, A.; Jayakannan, M. Enzyme-Responsive Theranostic FRET Probe Based on l -Aspartic Amphiphilic Polyester Nanoassemblies for Intracellular Bioimaging in Cancer Cells. ACS Appl. Bio. Mater. 2019, 2, 5245–5262. [Google Scholar] [CrossRef]
- Bhatia, N.K.; Kishor, S.; Katyal, N.; Gogoi, P.; Narang, P.; Deep, S. Effect of PH and Temperature on Conformational Equilibria and Aggregation Behaviour of Curcumin in Aqueous Binary Mixtures of Ethanol. RSC Adv. 2016, 6, 103275–103288. [Google Scholar] [CrossRef]
- Kumaraswamy, P.; Sethuraman, S.; Krishnan, U.M. Mechanistic Insights of Curcumin Interactions with the Core-Recognition Motif of β-Amyloid Peptide. J. Agric. Food Chem. 2013, 61, 3278–3285. [Google Scholar] [CrossRef]
- Alam, S.; Panda, J.J.; Chauhan, V.S. Novel Dipeptide Nanoparticles for Effective Curcumin Delivery. Int. J. Nanomed. 2012, 7, 4207–4222. [Google Scholar] [CrossRef] [Green Version]
- Riela, S.; Massaro, M.; Colletti, C.G.; Bommarito, A.; Giordano, C.; Milioto, S.; Noto, R.; Poma, P.; Lazzara, G. Development and Characterization of Co-Loaded Curcumin/Triazole-Halloysite Systems and Evaluation of Their Potential Anticancer Activity. Int. J. Pharm. 2014, 475, 613–623. [Google Scholar] [CrossRef] [Green Version]
- Subhan, M.A.; Alam, K.; Rahaman, M.S.; Rahman, M.A.; Awal, R. Synthesis and Characterization of Metal Complexes Containing Curcumin (C21H20O6) and Study of Their Anti-Microbial Activities and DNA-Binding Properties. J. Sci. Res. 2013, 6, 97–109. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, C.; Maiti, S.; Mustafi, M.; Kuchlyan, J.; Banik, D.; Kundu, N.; Dhara, D.; Sarkar, N. Effect of Encapsulation of Curcumin in Polymeric Nanoparticles: How Efficient to Control ESIPT Process? Langmuir 2014, 30, 10834–10844. [Google Scholar] [CrossRef] [PubMed]
- Sahu, A.; Kasoju, N.; Bora, U. Fluorescence Study of the Curcumin-Casein Micelle Complexation and Its Application as a Drug Nanocarrier to Cancer Cells. Biomacromolecules 2008, 9, 2905–2912. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HC | Initial Feed Ratio c | Mw a (g/mol) ( 104) | Mw/Mn a | %wt tBMA b before Hydrolysis | %wt tBMA b after Hydrolysis |
|---|---|---|---|---|---|
| HC 1 | [7.4]:[21.1]:[2]:[1]:[0.5] | 1.5 | 1.29 | 34 | 32 |
| HC 2 | [5.3]:[35.2]:[2]:[1]:[0.5] | 1.6 | 1.26 | 54 | 39 |
| HC Samples | I90° (a.u.) | Rh (nm) | PDI | ζp (mV) | CAC (g mL−1) | I1/I3 a |
|---|---|---|---|---|---|---|
| HC 1 pH3 | 520 | 14 | 0.55 | −1.6 | -- | -- |
| HC 1 pH7 | 780 | 15 | 0.51 | −5.5 | 2.6 10−6 | 1.2 |
| HC 1 pH10 | 290 | 16 | 0.53 | -- | -- | -- |
| HC 2 pH3 | 500 | 29 | 0.38 | −10.5 | -- | -- |
| HC 2 pH7 | 1200 | 19 | 0.44 | −32.8 | 2.2 10−6 | 1.2 |
| HC 2 pH10 | 110 | 13 | 0.51 | −24.9 | -- | -- |
| Series of Complexes | HC:Lys Ratio | Cpolymer (g/mL) | Clysozyme (g/mL) | I90° (a.u.) | PDI | Rh (nm) | ζp (mV) |
|---|---|---|---|---|---|---|---|
| a | HC Non complexed | 0.43 × 10−4 | - | 48 | 0.44 | 19 | −13.9 |
| a | HC:Lys = 2:1 | 0.43 × 10−4 | 0.69 × 10−4 | 107 | 0.51 | 12 (47%)/99 (53%) | +2.3 |
| a | HC:Lys = 1:1 | 0.43 × 10−4 | 1.38 × 10−4 | 67 | 0.47 | 25 | +0.8 |
| b | HC Non complexed | 0.75 × 10−4 | - | 212 | 0.13 | 24 | −18.9 |
| b | HC:Lys = 2:1 | 0.75 × 10−4 | 1.19 × 10−4 | 235 | 0.16 | 22 | +1.5 |
| b | HC:Lys = 1.5:1 | 0.75 × 10−4 | 1.59 × 10−4 | 230 | 0.17 | 23 | +1.0 |
| b | HC:Lys = 1:1 | 0.75 × 10−4 | 2.39 × 10−4 | 225 | 0.17 | 24 | +0.4 |
| b | HC:Lys = 1:1.5 | 0.75 × 10−4 | 3.58 × 10−4 | 215 | 0.17 | 24 | +1.4 |
| b | HC:Lys = 1:2 | 0.75 × 10−4 | 4.77 × 10−4 | 235 | 0.17 | 24 | +0.7 |
| c | HC Non complexed | 1.87 × 10−4 | - | 136 | 0.32 | 15 | −16.4 |
| c | HC:Lys = 2:1 | 1.87 × 10−4 | 2.97 × 10−4 | 365 | 0.27 | 25 | +1.2 |
| c | HC:Lys = 1.5:1 | 1.87 × 10−4 | 3.96 × 10−4 | 348 | 0.25 | 23 | +6.4 |
| c | HC:Lys = 1:1 | 1.87 × 10−4 | 5.93 × 10−4 | 275 | 0.27 | 22 | +8.8 |
| c | HC:Lys = 1:1.5 | 1.87 × 10−4 | 8.91 × 10−4 | 261 | 0.30 | 23 | +0.7 |
| c | HC:Lys = 1:2 | 1.87 × 10−4 | 11.87 × 10−4 | 276 | 0.30 | 23 | +1.4 |
| HC Samples | I90° (a.u.) | Rh (nm) | PDI | ζp (mV) |
|---|---|---|---|---|
| HC 1 | 780 | 15 | 0.51 | −5.5 |
| HC 1/20% CUR | 380 | 13 | 0.55 | −23.3 |
| HC 2 | 1200 | 19 | 0.44 | −32.8 |
| HC 2/20% CUR | 185 | 16 | 0.56 | −14.8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balafouti, A.; Pispas, S. Hyperbranched Polyelectrolyte Copolymers as Novel Candidate Delivery Systems for Bio-Relevant Compounds. Materials 2023, 16, 1045. https://doi.org/10.3390/ma16031045
Balafouti A, Pispas S. Hyperbranched Polyelectrolyte Copolymers as Novel Candidate Delivery Systems for Bio-Relevant Compounds. Materials. 2023; 16(3):1045. https://doi.org/10.3390/ma16031045
Chicago/Turabian StyleBalafouti, Anastasia, and Stergios Pispas. 2023. "Hyperbranched Polyelectrolyte Copolymers as Novel Candidate Delivery Systems for Bio-Relevant Compounds" Materials 16, no. 3: 1045. https://doi.org/10.3390/ma16031045
APA StyleBalafouti, A., & Pispas, S. (2023). Hyperbranched Polyelectrolyte Copolymers as Novel Candidate Delivery Systems for Bio-Relevant Compounds. Materials, 16(3), 1045. https://doi.org/10.3390/ma16031045