

Development of High Temperature Water Sorbents Based on Zeolites, Dolomite, Lanthanum Oxide and Coke



Abstract
:
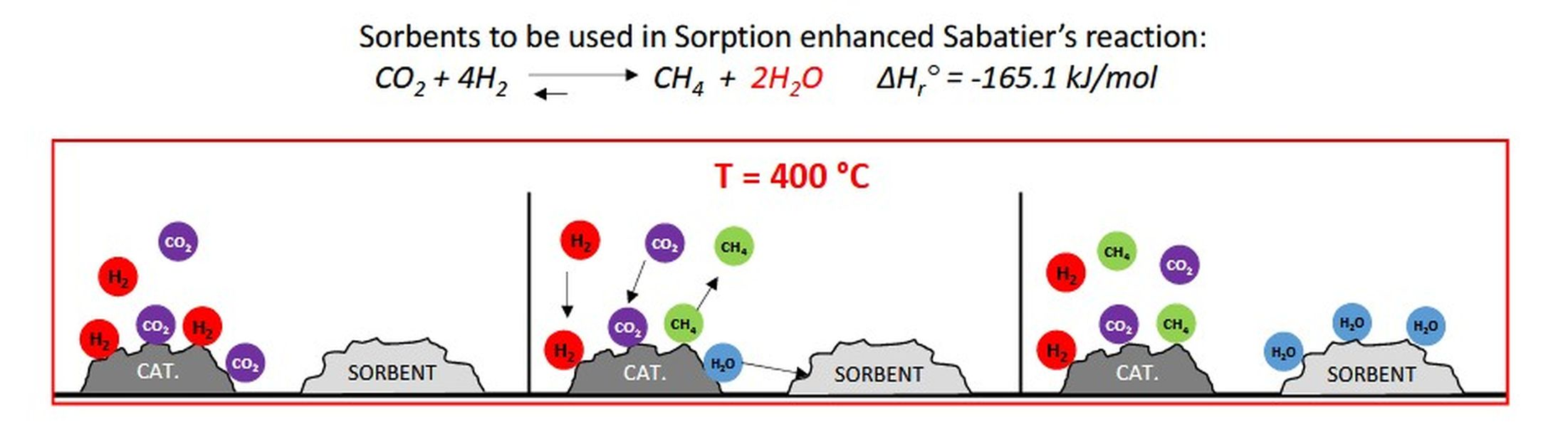
1. Introduction
2. Material and Methods
2.1. Preparation of the Adsorbents
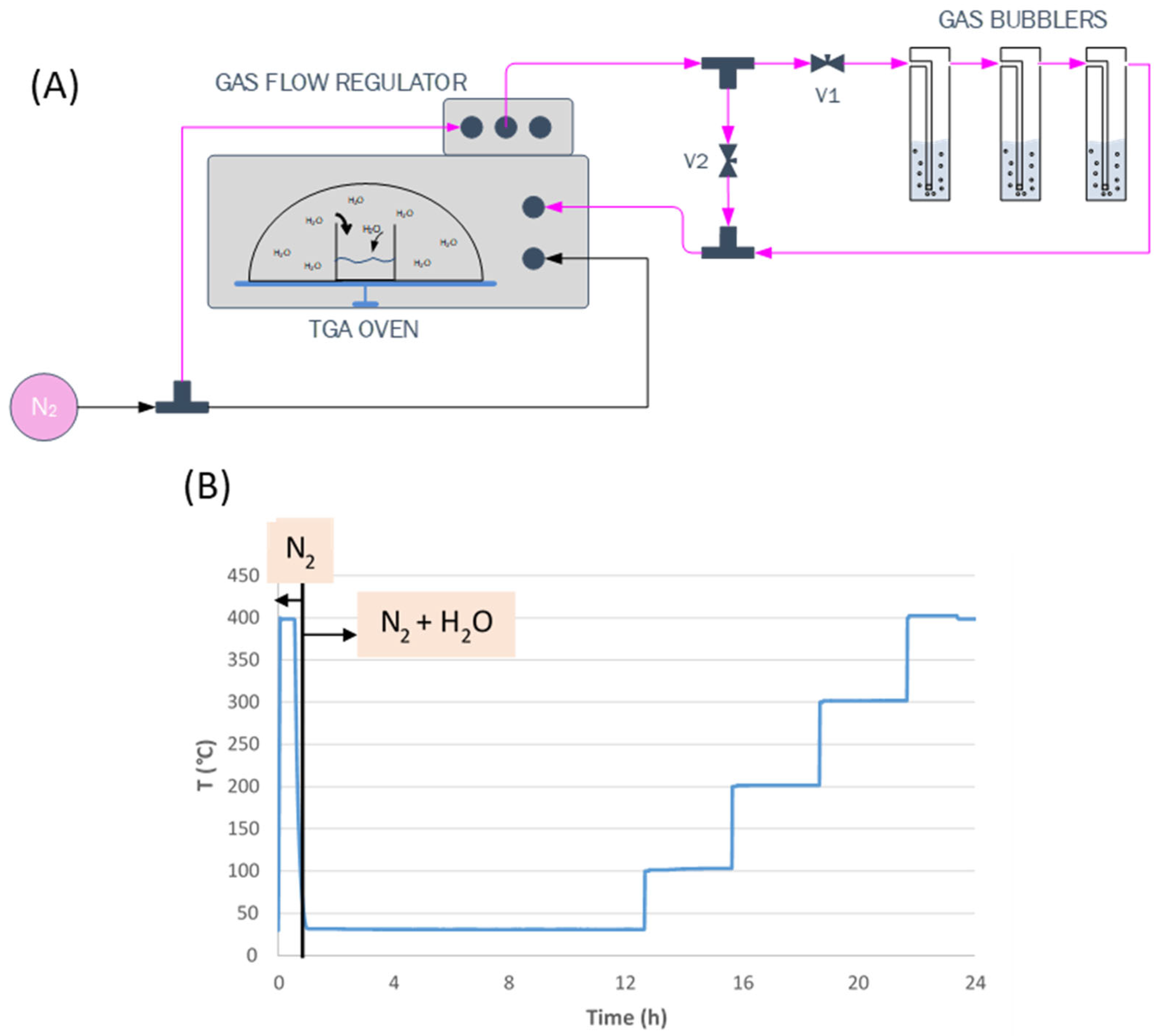
2.2. Measurement of Sorption Capacity of the Sorbents
2.3. Characterization of the Sorbents
3. Results and Discussion
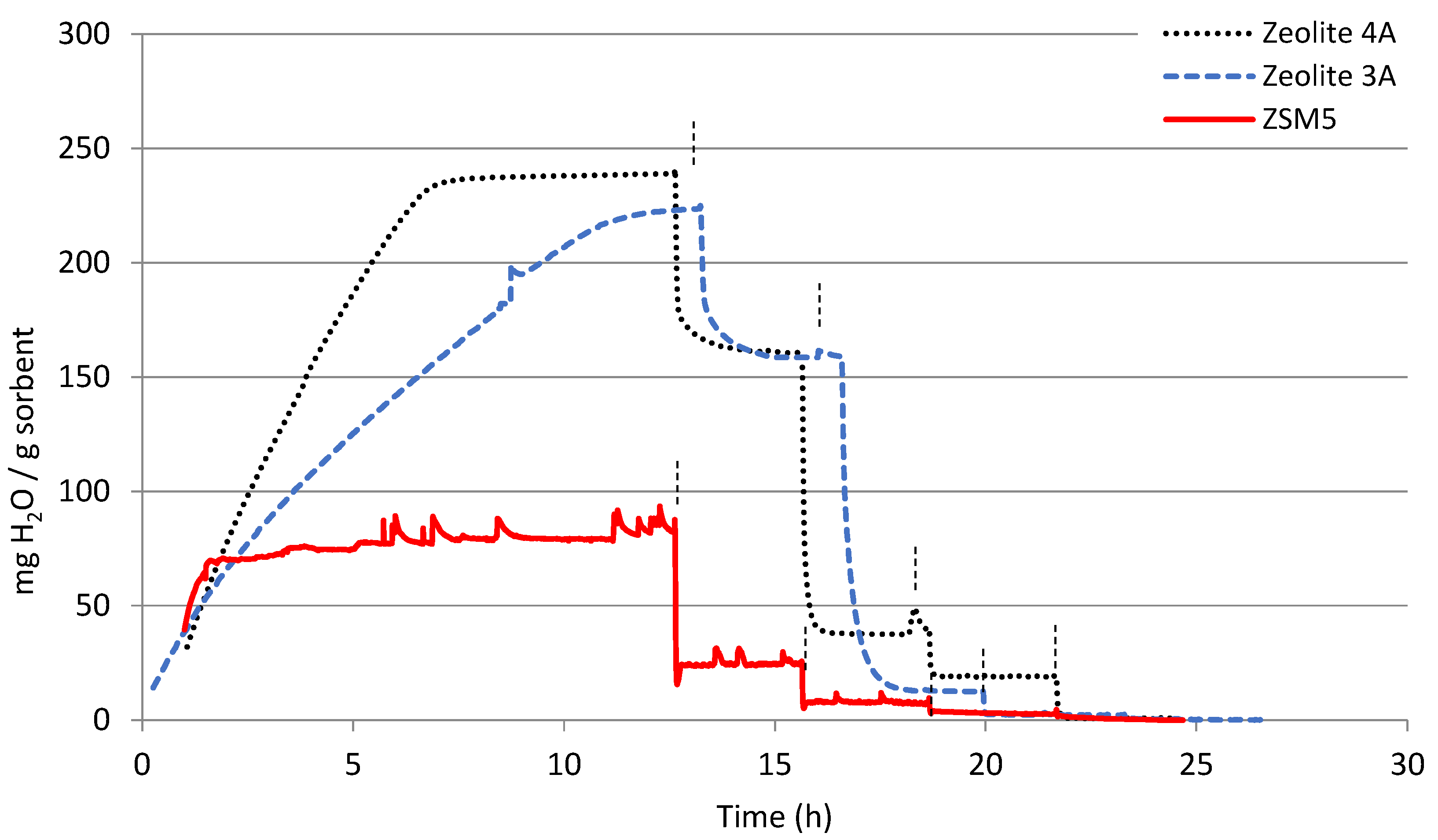
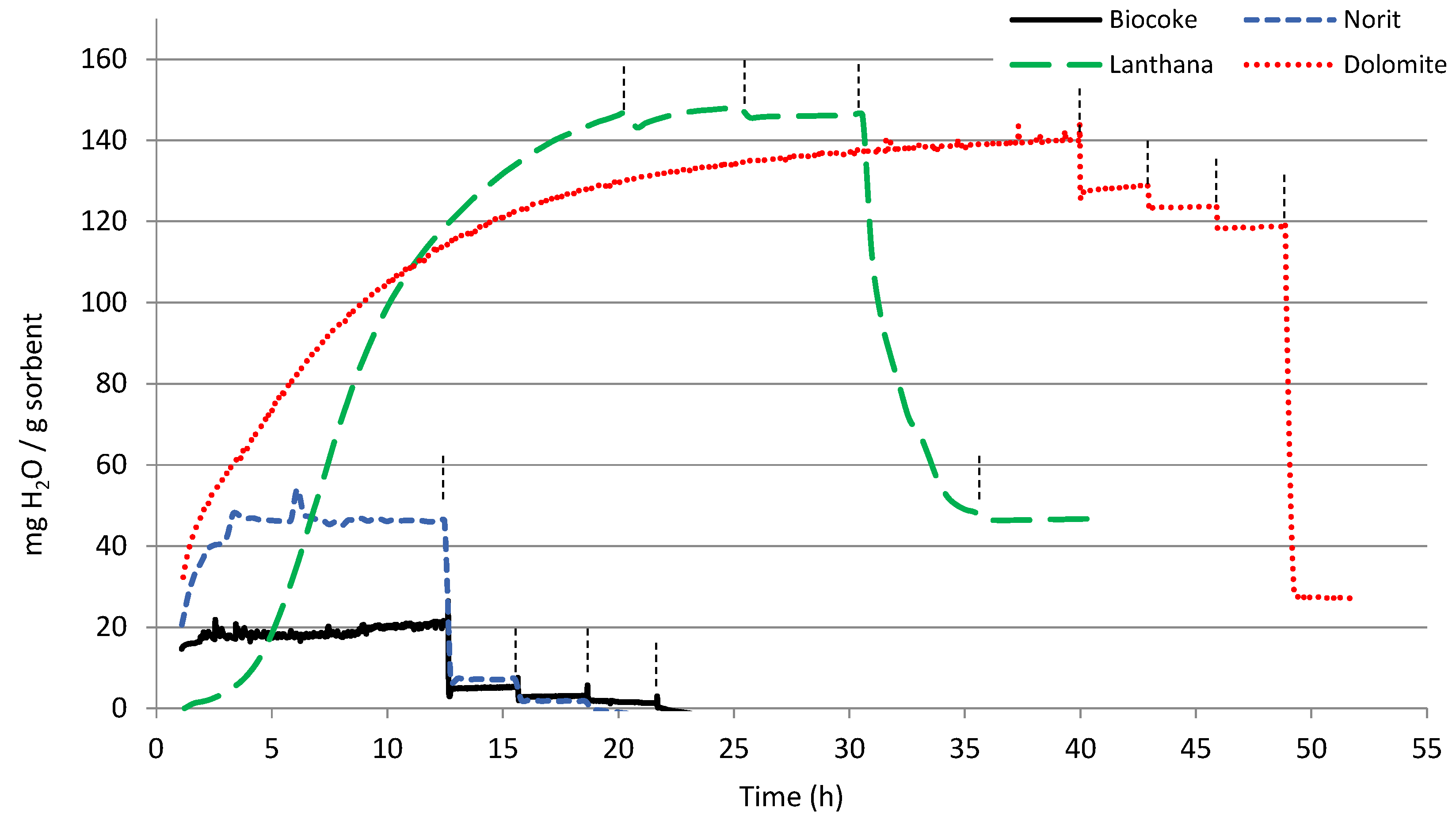
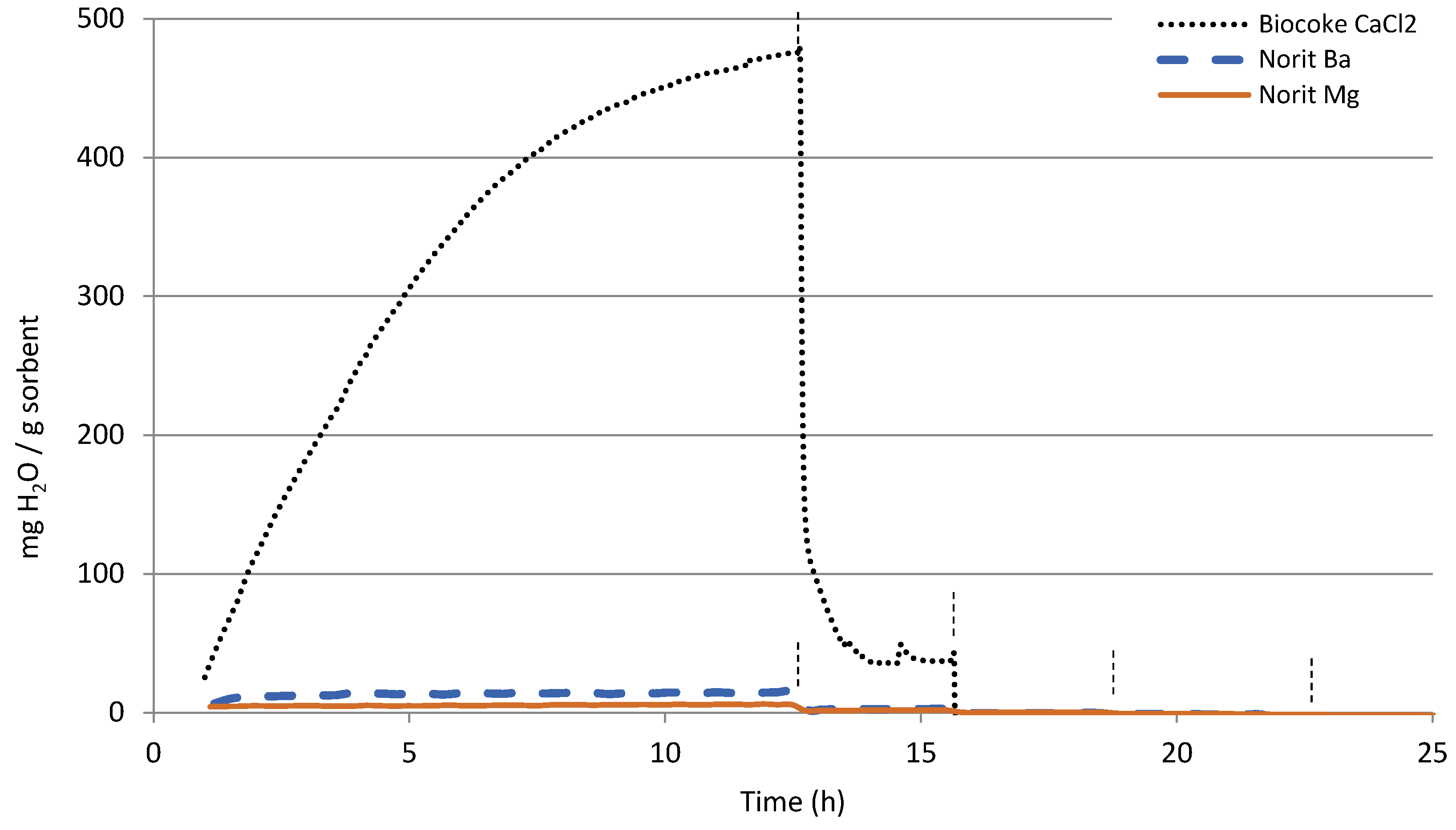
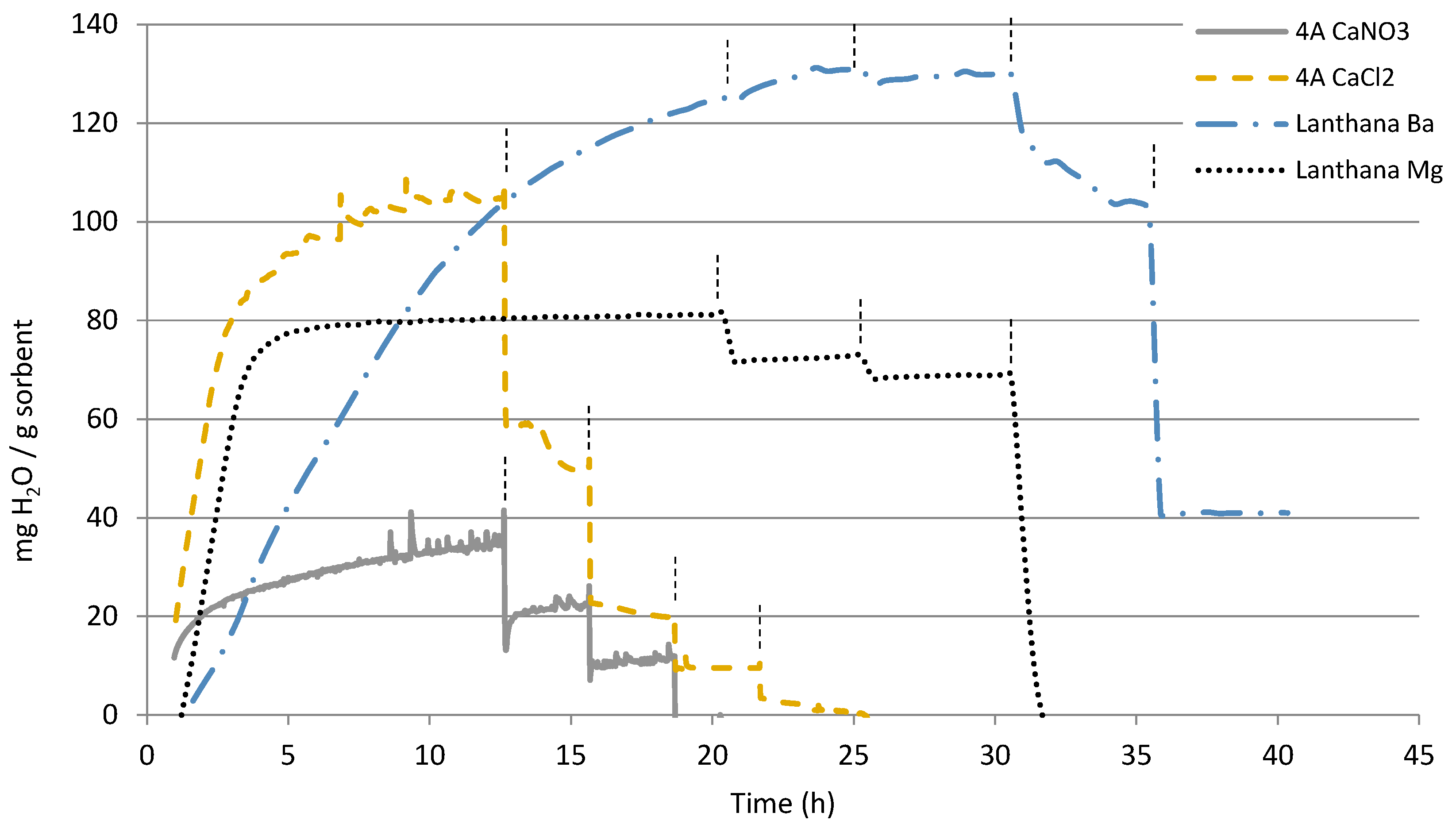
3.1. TGA
3.2. Characterization
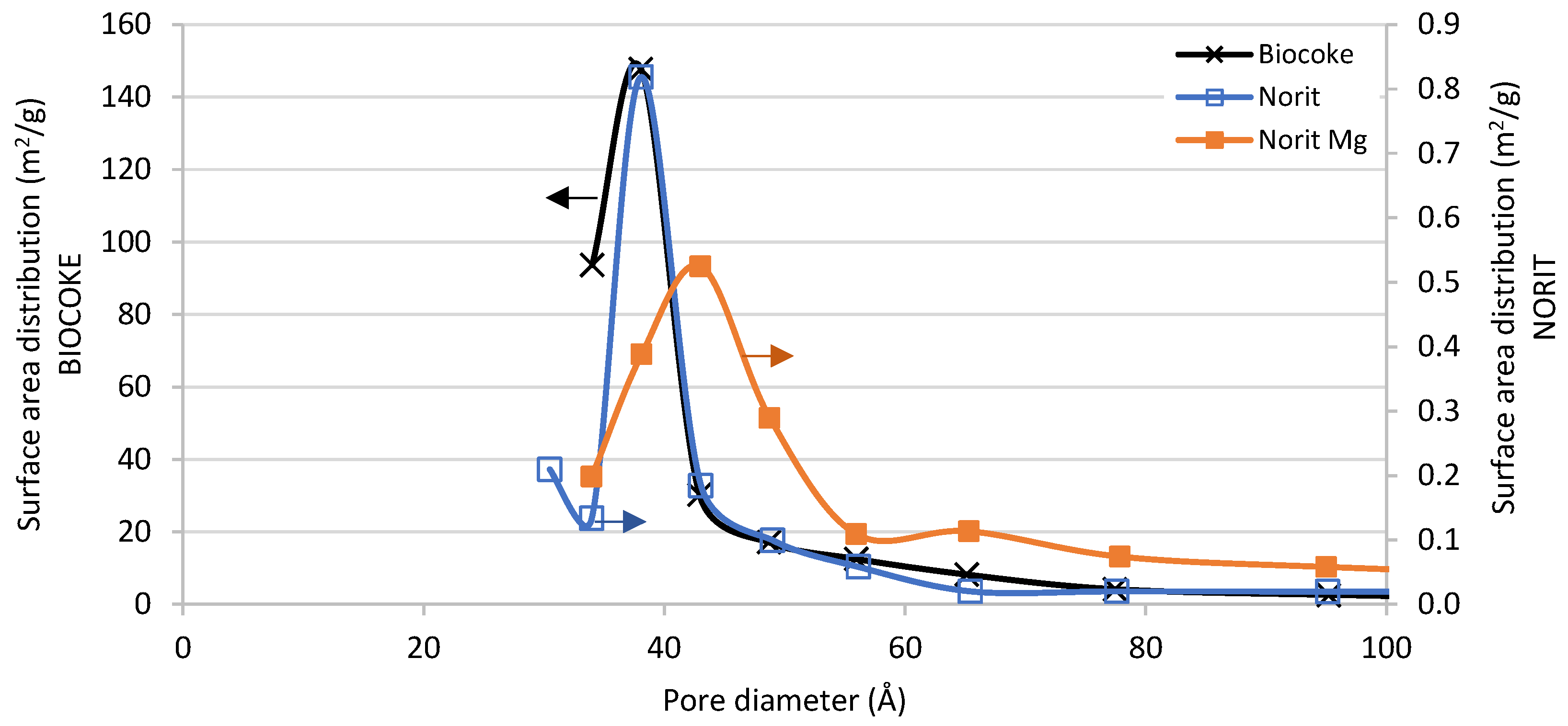
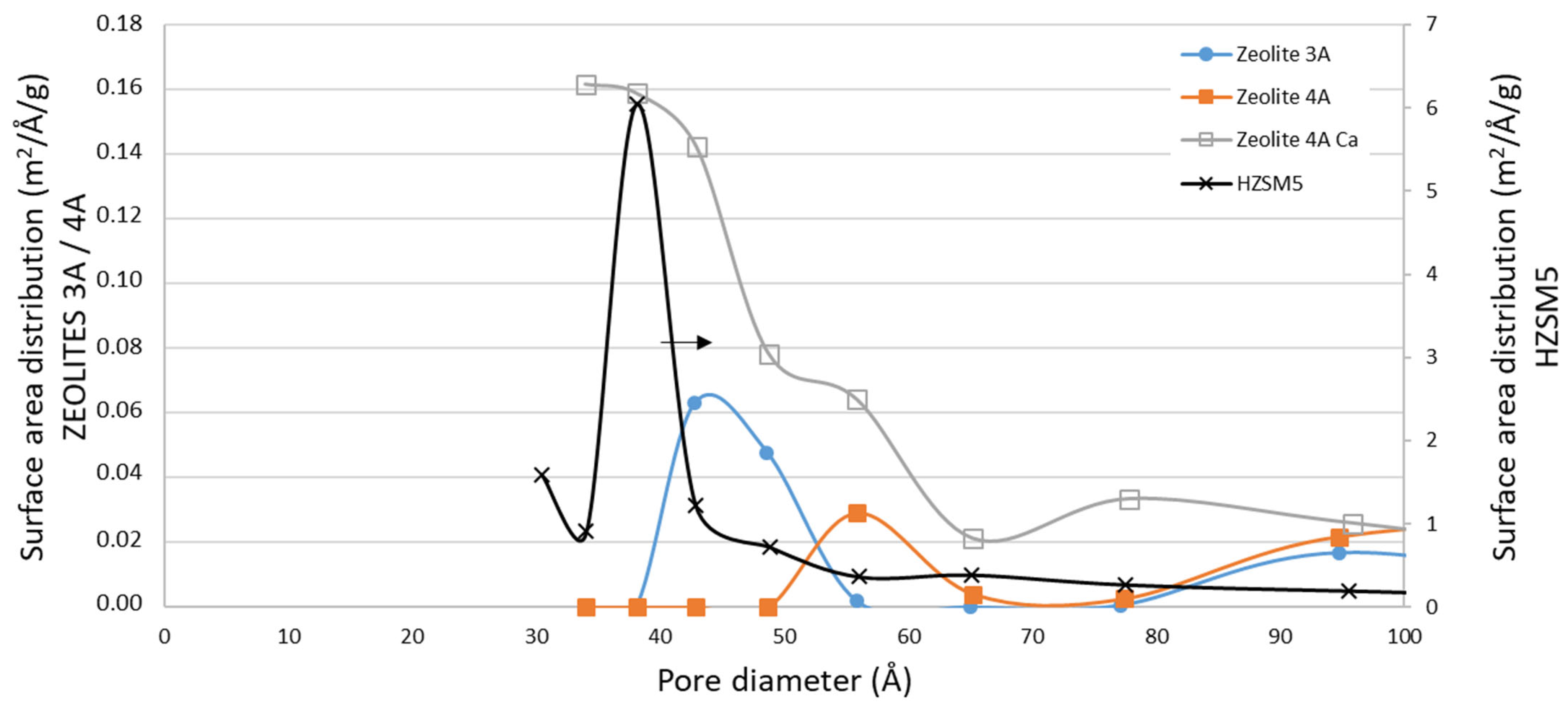
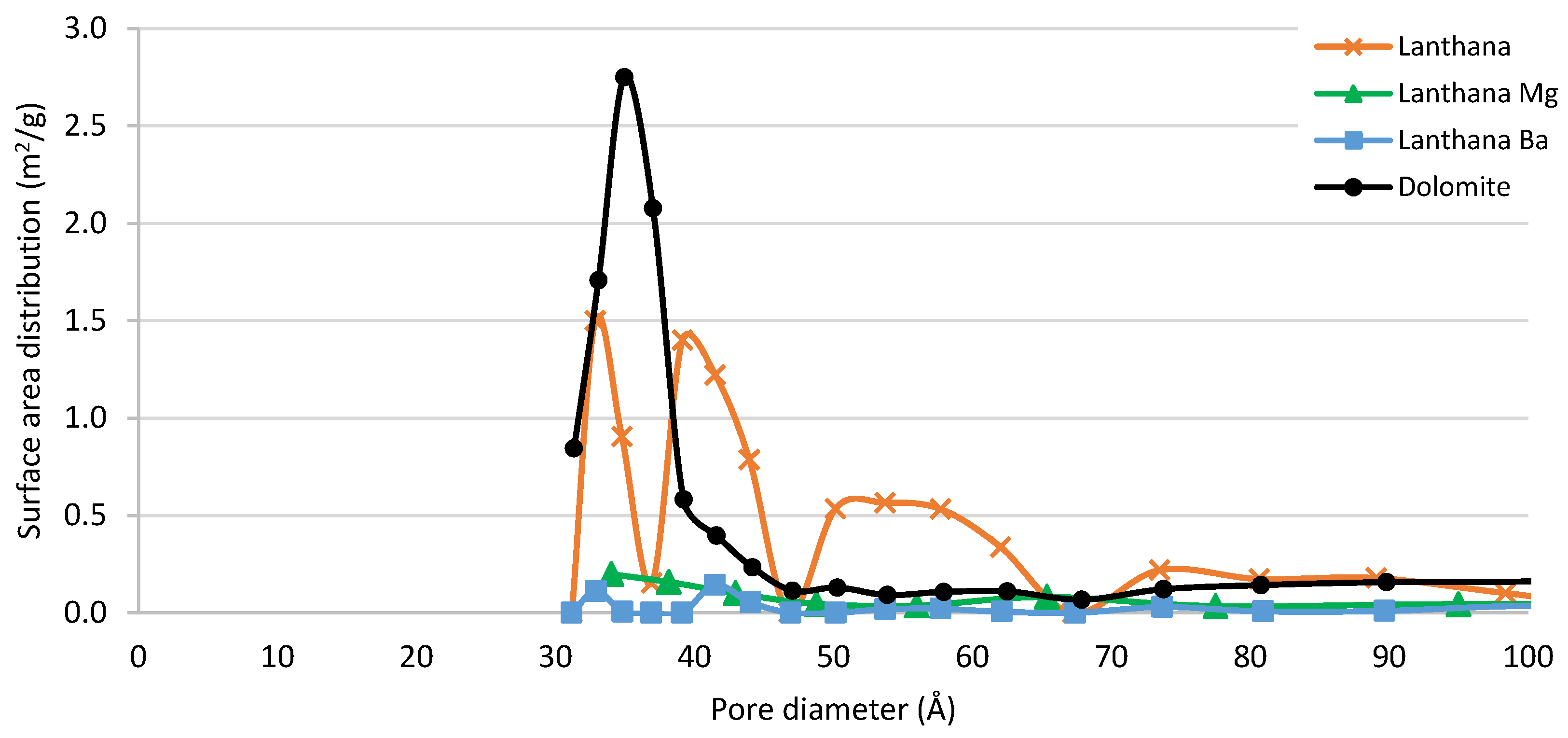
3.2.1. Textural Properties
3.2.2. Temperature-Programmed Desorption of Ammonia, NH3-TPD
3.2.3. CO Chemisorption
3.2.4. Temperature-Programmed Reduction of H2, TPR-H2
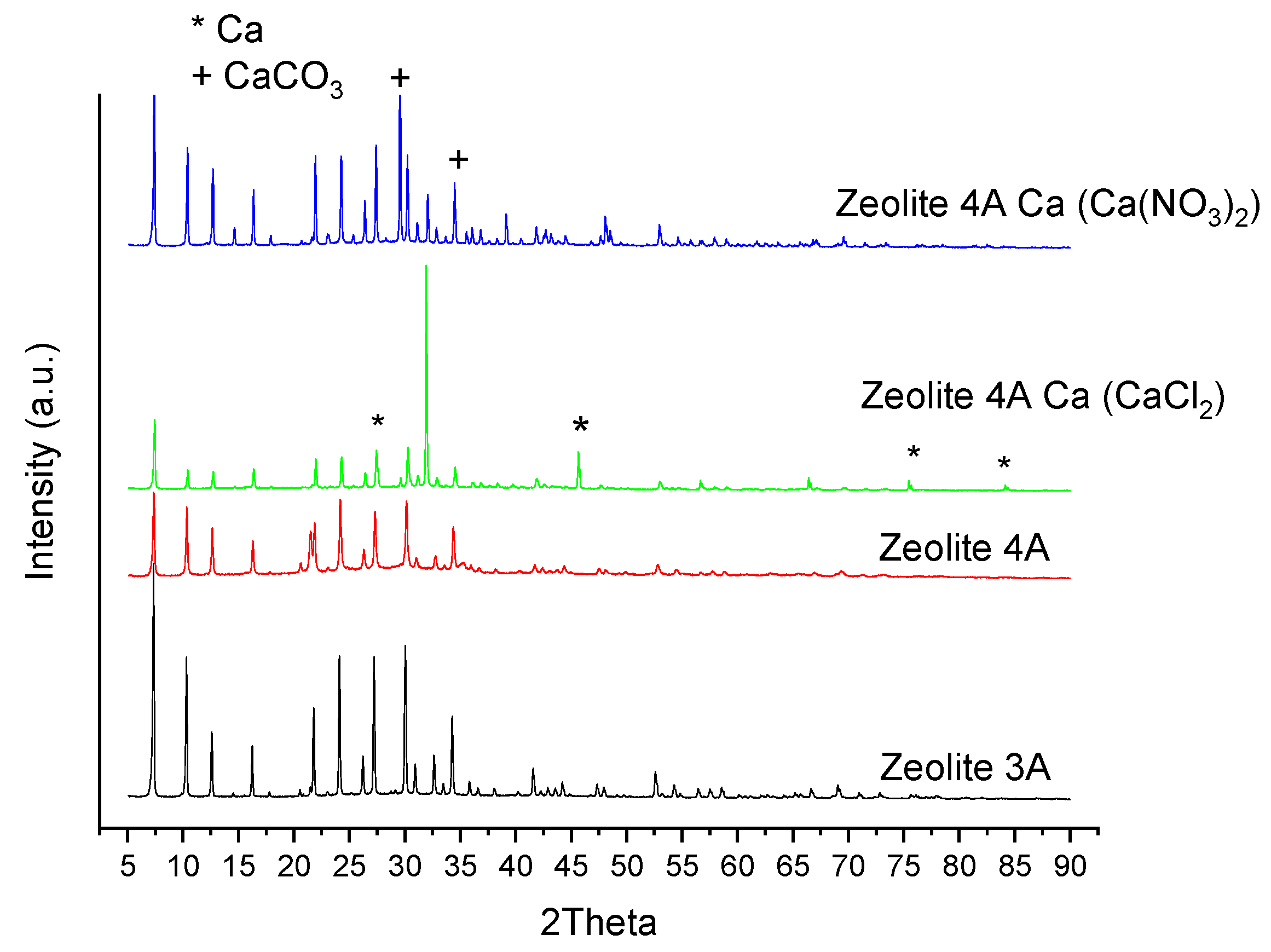
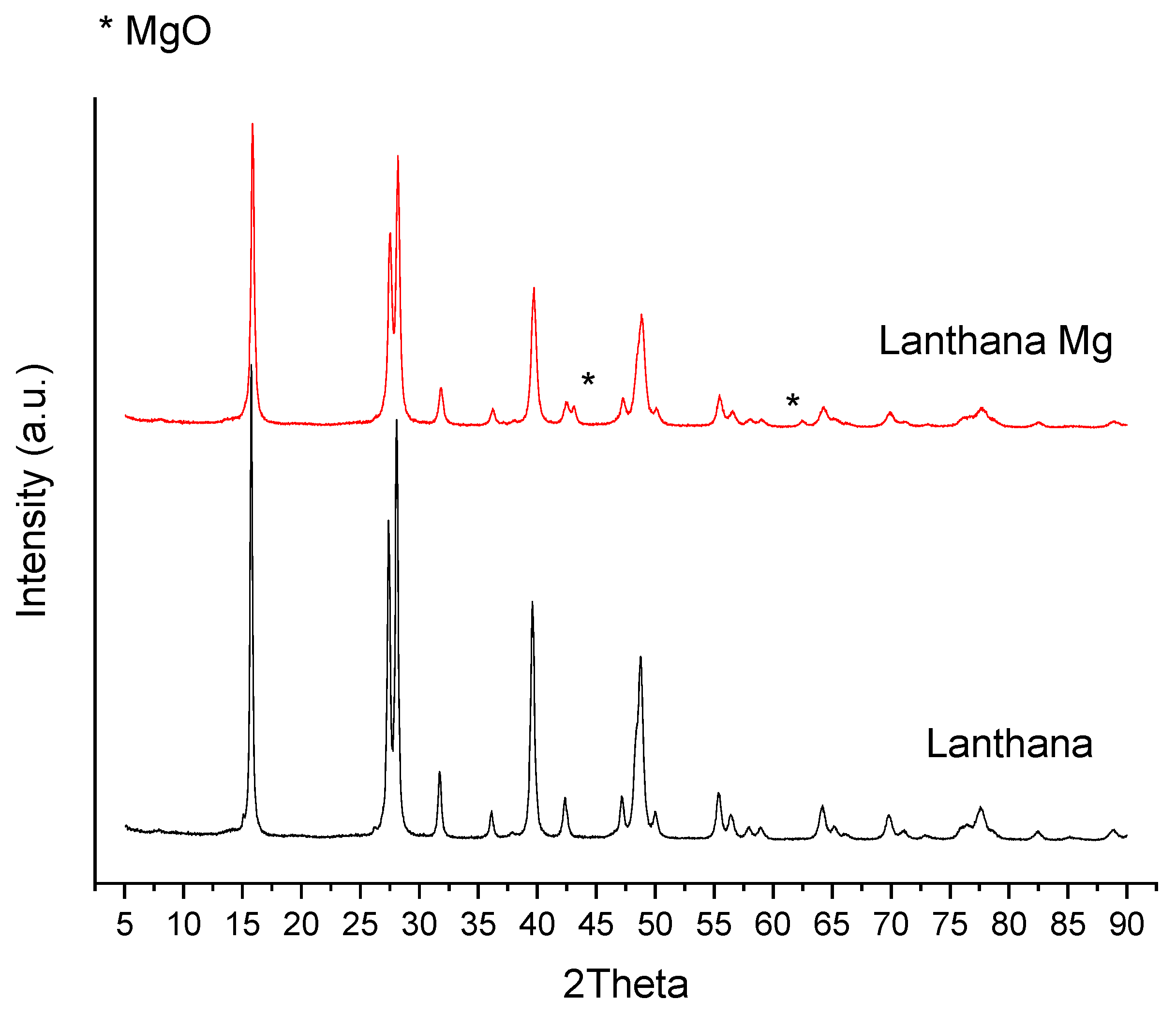
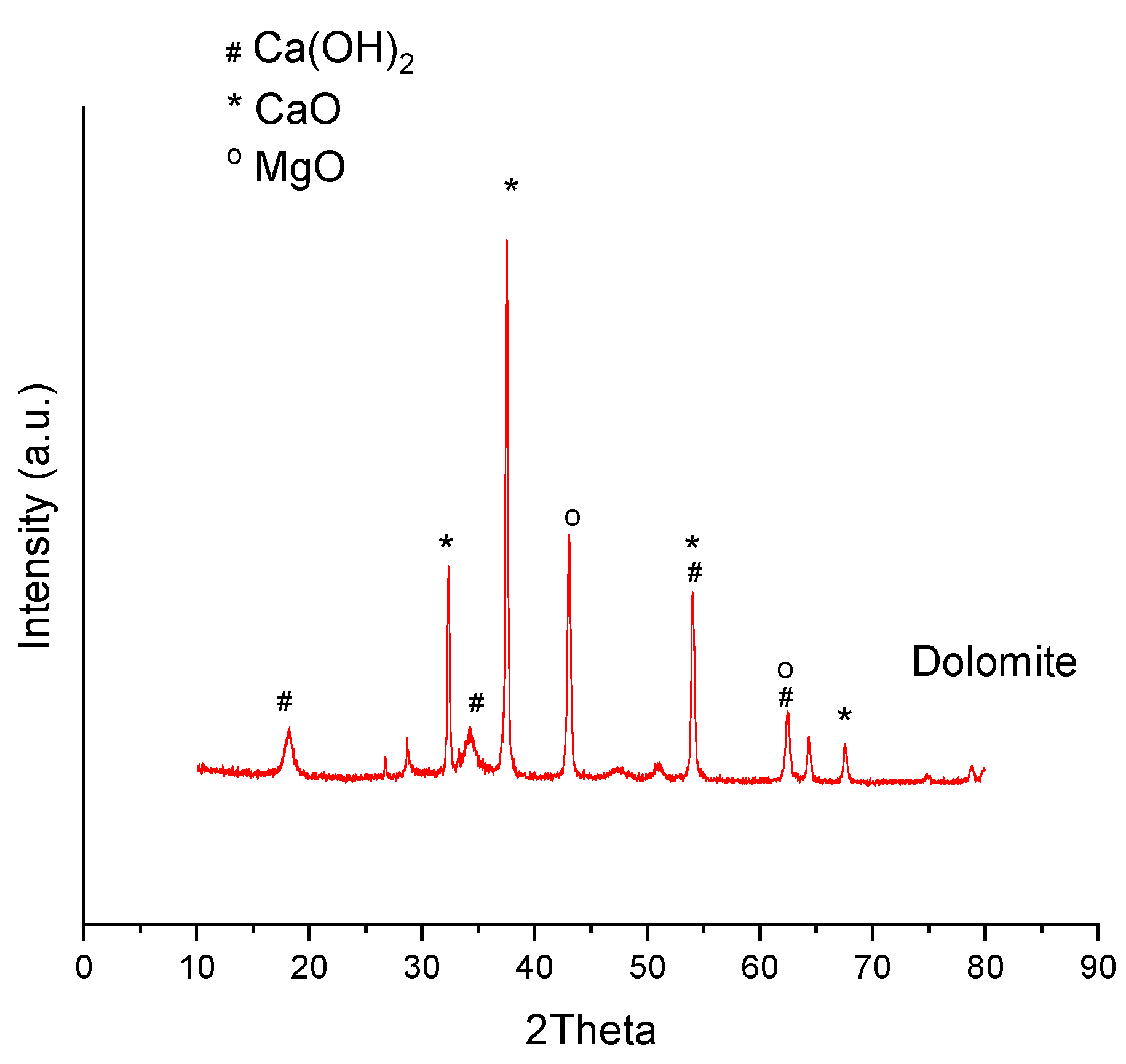
3.2.5. X-ray Diffraction
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Nomenclature
| BET | Brunauer-Emmett-Teller |
| BJH | Barrett-Joyner-Halenda |
| PtG | Power to Gas |
| TGA | thermo-gravimetric analysis |
| TPD | temperature-programmed desorption |
| TPR | temperature-programmed reduction |
| XRD | X-ray diffraction |
References
- Aydin, G. Modeling of Energy Consumption Based on Economic and Demographic Factors: The Case of Turkey with Projections. Renew. Sustain. Energy Rev. 2014, 35, 382–389. [Google Scholar] [CrossRef]
- Aydin, G. Regression Models for Forecasting Global Oil Production. Pet. Sci. Technol. 2015, 33, 1822–1828. [Google Scholar] [CrossRef]
- Xia, A.; Zhu, X.; Liao, Q. Hydrogen Production from Biological Sources. In Encyclopedia of Sustainability Science and Technology; Meyers, R.A., Ed.; Springer: New York, NY, USA, 2017; pp. 833–863. ISBN 978-1-4939-2493-6. [Google Scholar]
- Safari, F.; Dincer, I. Assessment and Optimization of an Integrated Wind Power System for Hydrogen and Methane Production. Energy Convers. Manag. 2018, 177, 693–703. [Google Scholar] [CrossRef]
- European Biogas Association (EBA) EBA’s Biomethane Fact Sheet; European Biogas Association: Brussels, Belgium, 2014.
- García–García, I.; Izquierdo, U.; Barrio, V.L.; Arias, P.L.; Cambra, J.F. Power-to-Gas: Storing Surplus Electrical Energy. Study of Al2O3 Support Modification. Int. J. Hydrog. Energy 2016, 41, 19587–19594. [Google Scholar] [CrossRef]
- Skorek-Osikowska, A.; Martín-Gamboa, M.; Dufour, J. Thermodynamic, Economic and Environmental Assessment of Renewable Natural Gas Production Systems. Energy Convers. Manag. X 2020, 7, 100046. [Google Scholar] [CrossRef]
- Castellani, B.; Rinaldi, S.; Morini, E.; Nastasi, B.; Rossi, F. Flue Gas Treatment by Power-to-Gas Integration for Methane and Ammonia Synthesis—Energy and Environmental Analysis. Energy Convers. Manag. 2018, 171, 626–634. [Google Scholar] [CrossRef]
- Sarić, M.; Dijkstra, J.W.; Walspurger, S.; Haije, W.G. The Potential of “Power to Gas” Technology Integrated with Biomethane Production; ECN: Petten, The Netherlands, 2014. [Google Scholar]
- Walker, S.B.; Van Lanen, D.; Fowler, M.; Mukherjee, U. Economic Analysis with Respect to Power-to-Gas Energy Storage with Consideration of Various Market Mechanisms. Int. J. Hydrog. Energy 2016, 41, 7754–7765. [Google Scholar] [CrossRef]
- Baraj, E.; Vagaský, S.; Hlinčik, T.; Ciahotný, K.; Tekáč, V. Reaction Mechanisms of Carbon Dioxide Methanation. Chem. Pap. 2016, 70, 395–403. [Google Scholar] [CrossRef]
- Lehner, M.; Tichler, R.; Steinmüller, H.; Koppe, M. Methanation. In Power-to-Gas: Technology and Business Models “Methanation”; Springer: Berlin/Heidelberg, Germany, 2014; pp. 41–61. ISBN 978-3-319-03994-7. [Google Scholar]
- Aziz, M.A.A.; Jalil, A.A.; Triwahyono, S.; Sidik, S.M. Methanation of Carbon Dioxide on Metal-Promoted Mesostructured Silica Nanoparticles. Appl. Catal. A Gen. 2014, 486, 115–122. [Google Scholar] [CrossRef]
- Beierlein, D.; Häussermann, D.; Pfeifer, M.; Schwarz, T.; Stöwe, K.; Traa, Y.; Klemm, E. Is the CO2 Methanation on Highly Loaded Ni-Al 2 O 3 Catalysts Really Structure-Sensitive? Appl. Catal. B Environ. 2019, 247, 200–219. [Google Scholar] [CrossRef]
- De Leitenburg, C.; Trovarelli, A.; Kašpar, J. A Temperature-Programmed and Transient Kinetic Study of CO2 Activation and Methanation over CeO2 Supported Noble Metals. J. Catal. 1997, 166, 98–107. [Google Scholar] [CrossRef]
- Panagiotopoulou, P.; Kondarides, D.I.; Verykios, X.E. Selective Methanation of CO over Supported Noble Metal Catalysts: Effects of the Nature of the Metallic Phase on Catalytic Performance. Appl. Catal. A Gen. 2008, 344, 45–54. [Google Scholar] [CrossRef]
- Borgschulte, A.; Gallandat, N.; Probst, B.; Suter, R.; Callini, E.; Ferri, D.; Arroyo, Y.; Erni, R.; Geerlings, H.; Züttel, A. Sorption Enhanced CO2 Methanation. Phys. Chem. Chem. Phys. 2013, 15, 9620–9625. [Google Scholar] [CrossRef] [Green Version]
- Walspurger, S.; Elzinga, G.D.; Dijkstra, J.W.; Sarić, M.; Haije, W.G. Sorption Enhanced Methanation for Substitute Natural Gas Production: Experimental Results and Thermodynamic Considerations. Chem. Eng. J. 2014, 242, 379–386. [Google Scholar] [CrossRef] [Green Version]
- Catarina Faria, A.; Miguel, C.V.; Madeira, L.M. Thermodynamic Analysis of the CO2 Methanation Reaction with in Situ Water Removal for Biogas Upgrading. J. CO2 Util. 2018, 26, 271–280. [Google Scholar] [CrossRef]
- Agirre, I.; Acha, E.; Cambra, J.F.; Barrio, V.L. Water Sorption Enhanced CO2 Methanation Process: Optimization of Reaction Conditions and Study of Various Sorbents. Chem. Eng. Sci. 2021, 237, 116546–116556. [Google Scholar] [CrossRef]
- Gómez, L.; Martínez, I.; Navarro, M.V.; García, T.; Murillo, R. Sorption-Enhanced CO and CO2 Methanation (SEM) for the Production of High Purity Methane. Chem. Eng. J. 2022, 440, 135842. [Google Scholar] [CrossRef]
- Coppola, A.; Massa, F.; Salatino, P.; Scala, F. Evaluation of Two Sorbents for the Sorption-Enhanced Methanation in a Dual Fluidized Bed System. Biomass Convers. Biorefin. 2021, 11, 111–119. [Google Scholar] [CrossRef]
- Pieterse, J.A.Z.; Elzinga, G.D.; Booneveld, S.; van Kampen, J.; Boon, J. Reactive Water Sorbents for the Sorption-Enhanced Reverse Water–Gas Shift. Catal. Lett. 2022, 152, 460–466. [Google Scholar] [CrossRef]
- Lin, H.; Liu, Y.; Deng, J.; Jing, L.; Dai, H. Methane Combustion over the Porous Oxides and Supported Noble Metal Catalysts. Catalysts 2023, 13, 427. [Google Scholar] [CrossRef]
- Mikšík, F.; Miyazaki, T.; Thu, K.; Miyawaki, J.; Nakabayashi, K.; Wijayanta, A.T.; Rahmawati, F. Enhancing water adsorption capacity of acorn nutshell based activated carbon for adsorption thermal energy storage application. Energy Rep. 2020, 6, 255–263. [Google Scholar]
- Tang, Y.; Kourtellaris, A.; Tasiopoulos, A.J.; Teat, S.J.; Dubbeldam, D.; Rothenberg, G.; Tanase, S. Selective CO2 adsorption in water-stable alkaline-earth based metal–organic frameworks. Inorg. Chem. Front. 2018, 5, 541–549. [Google Scholar] [CrossRef]
- Da Silva, A.L.; Wu, L.; Caliman, L.B.; Castro, R.H.; Navrotsky, A.; Gouvêa, D. Energetics of CO2 and H2O adsorption on alkaline earth metal doped TiO2. Phys. Chem. Chem. Phys. 2020, 22, 15600–15607. [Google Scholar] [CrossRef] [PubMed]
- Aristov, Y.I. Selective Water Sorbents, a New Family of Materials for Adsorption Cooling/Heating: State of the Art; V Minsk International Seminar “Hear Pipes, Heat Pumps, Refrigerators”: Minsk, Belarus, 2003. [Google Scholar]
- Glasser, L. Thermodynamics of Inorganic Hydration and of Humidity Control, with an Extensive Database of Salt Hydrate Pairs. J. Chem. Eng. Data 2014, 59, 526–530. [Google Scholar] [CrossRef]
- Ropp, R.C.C. Chapter 3—Group 16 (O, S, Se, Te) Alkaline Earth Compounds. In Encyclopedia of the Alkaline Earth Compounds; Elsevier: Amsterdam, The Netherlands, 2013; pp. 105–197. ISBN 978-0-444-59550-8. [Google Scholar]
- Ng, E.-P.; Mintova, S. Nanoporous Materials with Enhanced Hydrophilicity and High Water Sorption Capacity. Microporous Mesoporous Mater. 2008, 114, 1–26. [Google Scholar] [CrossRef]
- Munthali, M.W.; Elsheikh, M.A.; Johan, E.; Matsue, N. Proton Adsorption Selectivity of Zeolites in Aqueous Media: Effect of Si/Al Ratio of Zeolites. Molecules 2014, 19, 20468–20481. [Google Scholar] [CrossRef]
- Olszak-Humienik, M.; Jablonski, M. Thermal behavior of natural dolomite. J. Therm. Anal. Calorim. 2015, 119, 2239–2248. [Google Scholar] [CrossRef] [Green Version]
- Acha, E.; Chen, D.; Cambra, J.F. Comparison of novel olivine supported catalysts for high purity hydrogen production by CO2 sorption enhanced steam reforming. J. CO2 Util. 2020, 42, 101295. [Google Scholar] [CrossRef]
- Adrados, A.; De Marco, I.; Lopez-Urionabarrenechea, A.; Solar, J.; Caballero, B.M.; Gastelu, N. Biomass Pyrolysis Solids as Reducing Agents: Comparison with Commercial Reducing Agents. Materials 2016, 9, 3. [Google Scholar] [CrossRef]
- Rouquerol, J.; Rouquerol, F.; Llewellyn, P.; Maurin, G.; Sing, K.S.W. (Eds.) 11—Adsorption by Metal Oxides. In Adsorption by Powders and Porous Solids: Principle, Methodology and Applications, 2nd ed.; Academic Press: Oxford, UK, 2014; pp. 393–465. ISBN 978-0-08-097035-6. [Google Scholar]
- Mansir, N.; Hwa Teo, S.; Lokman Ibrahim, M.; Yun Hin, T.-Y. Synthesis and Application of Waste Egg Shell Derived CaO Supported W-Mo Mixed Oxide Catalysts for FAME Production from Waste Cooking Oil: Effect of Stoichiometry. Energy Convers. Manag. 2017, 151, 216–226. [Google Scholar] [CrossRef]
- Antzara, A.; Heracleous, E.; Lemonidou, A.A. Development of CaO-Based Mixed Oxides as Stable Sorbents for Post-Combustion CO2 Capture Via Carbonate Looping. Energy Procedia 2014, 63, 2160–2169. [Google Scholar] [CrossRef] [Green Version]
- Thommes, M.; Kaneko, K.; Neimark, A.V.; Olivier, J.P.; Rodriguez-Reinoso, F.; Rouquerol, J.; Sing, K.S.W. Physisorption of Gases, with Special Reference to the Evaluation of Surface Area and Pore Size Distribution (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1051–1069. [Google Scholar] [CrossRef] [Green Version]
- Pandey, A.; Jain, G.; Vyas, D.; Irusta, S.; Sharma, S. Nonreducible, Basic La2O3 to Reducible, Acidic La2–XSbxO3 with Significant Oxygen Storage Capacity, Lower Band Gap, and Effect on the Catalytic Activity. J. Phys. Chem. C 2017, 121, 481–489. [Google Scholar] [CrossRef]
- Bajdich, M.; Nørskov, J.K.; Vojvodic, A. Surface Energetics of Alkaline-Earth Metal Oxides: Trends in Stability and Adsorption of Small Molecules. Phys. Rev. B 2015, 91, 155401. [Google Scholar] [CrossRef] [Green Version]
- Karge, H.G.; Weitkamp, J. (Eds.) Post-Synthesis Modification I:3 (Molecular Sieves); Springer: Berlin/Heidelberg, Germany, 2002. [Google Scholar]
- Mouat, A.R.; Lohr, T.L.; Wegener, E.C.; Miller, J.T.; Delferro, M.; Stair, P.C.; Marks, T.J. Reactivity of a Carbon-Supported Single-Site Molybdenum Dioxo Catalyst for Biodiesel Synthesis. ACS Catal. 2016, 6, 6762–6769. [Google Scholar] [CrossRef]
- Carabineiro, S.A.C.; Bogdanchikova, N.; Pestryakov, A.; Tavares, P.B.; Fernandes, L.S.G.; Figueiredo, J.L. Gold Nanoparticles Supported on Magnesium Oxide for CO Oxidation. Nanoscale Res. Lett. 2011, 6, 435. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.J.; Hur, Y.G.; Kim, D.H.; Lee, S.H.; Lee, K.-Y. Non-Oxidative Aromatization and Ethylene Formation over Ga/HZSM-5 Catalysts Using a Mixed Feed of Methane and Ethane. Fuel 2019, 253, 449–459. [Google Scholar] [CrossRef]
- Paul, B.; Khatun, R.; Sharma, S.K.; Adak, S.; Singh, G.; Das, D.; Siddiqui, N.; Bhandari, S.; Joshi, V.; Sasaki, T.; et al. Fabrication of Au Nanoparticles Supported on One-Dimensional La2O Nanorods for Selective Esterification of Methacrolein to Methyl Methacrylate with Molecular Oxygen. ACS Sustain. Chem. Eng. 2019, 7, 3982–3994. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| mg H2O/g Sorbent | 25 °C | 100 °C | 200 °C | 300 °C | 400 °C |
|---|---|---|---|---|---|
| Biocoke | 19 | 6 | 4 | <1 | <1 |
| Biocoke Ca (CaCl2) | 473 | 37 | <1 | <1 | <1 |
| Norit | 46 | 7 | 8 | <1 | <1 |
| Norit Mg | 6 | 2 | <1 | <1 | <1 |
| Norit Ba | 15 | 3 | <1 | <1 | <1 |
| ZSM5 | 80 | 24 | 8 | 3 | <1 |
| Zeolite 3A | 222 | 160 | 12 | 3 | <1 |
| Zeolite 4A | 163 | 83 | 26 | 11 | <1 |
| Zeolite 4A Ca (CaCl2) | 105 | 44 | 20 | 10 | 2 |
| Zeolite 4A Ca (Ca(NO3)2) | 46 | 33 | 22 | 7 | <1 |
| Lanthana | 147 | 148 | 146 | 48 | 47 |
| Lanthana Mg | 81 | 73 | 69 | <1 | <1 |
| Lanthana Ba | 125 | 131 | 130 | 102 | 41 |
| Dolomite | 140 | 128 | 124 | 118 | 27 |
| BET Surface Area (m2/g) | BJH Pore Volume (cm3/g) | BJH Pore Size Diameter (Å) | |
|---|---|---|---|
| Biocoke | 624 | 0.22 | 38 |
| Norit | 21 | 0.02 | 38 |
| Norit Mg | 22 | 0.05 | 43 |
| ZSM5 | 251 | 0.08 | 19 |
| Zeolite 3A | 3 | 0.01 | 43 |
| Zeolite 4A | 7 | 0.03 | 31 |
| Zeolite 4A Ca (CaCl2) | 7 | 0.02 | 43 |
| Lanthana | 14 | 0.05 | 39 |
| Lanthana Mg | 15 | 0.04 | 34 |
| Lanthana Ba | 15 | 0.04 | 38 |
| Dolomite | 39 | 0.14 | 35 |
| Total Acidity mmol NH3/gsorbent | |
|---|---|
| Biocoke | 0.214 |
| Biocoke Ca (CaCl2) | 0.059 |
| Norit | 0.162 |
| Norit Mg | 0.191 |
| Norit Ba | 0.198 |
| ZSM5 | 0.595 |
| Zeolite 3A | 0.494 |
| Zeolite 4A | 0.644 |
| Zeolite 4A Ca (CaCl2) | 0.197 |
| Lanthana | 0.232 |
| Lanthana Mg | 0.346 |
| Lanthana Ba | 0.713 |
| Dolomite | 0.336 |
| CO Cumulative Quantity µmol/g | |
|---|---|
| Biocoke | 1.01 |
| Biocoke Ca (CaCl2) | 0.24 |
| Norit | 1.01 |
| Norit Mg | 0.67 |
| Norit Ba | 0.83 |
| Zeolite 3A | 0.42 |
| Zeolite 4A | 1.03 |
| Zeolite 4A Ca (CaCl2) | 0.90 |
| ZSM5 | 1.93 |
| Lanthana | 0.04 |
| Lanthana Ba | 2.05 |
| Lanthana Mg | 0.18 |
| Dolomite | 3.20 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Acha, E.; Agirre, I.; Barrio, V.L. Development of High Temperature Water Sorbents Based on Zeolites, Dolomite, Lanthanum Oxide and Coke. Materials 2023, 16, 2933. https://doi.org/10.3390/ma16072933
Acha E, Agirre I, Barrio VL. Development of High Temperature Water Sorbents Based on Zeolites, Dolomite, Lanthanum Oxide and Coke. Materials. 2023; 16(7):2933. https://doi.org/10.3390/ma16072933
Chicago/Turabian StyleAcha, Esther, Ion Agirre, and V. Laura Barrio. 2023. "Development of High Temperature Water Sorbents Based on Zeolites, Dolomite, Lanthanum Oxide and Coke" Materials 16, no. 7: 2933. https://doi.org/10.3390/ma16072933
APA StyleAcha, E., Agirre, I., & Barrio, V. L. (2023). Development of High Temperature Water Sorbents Based on Zeolites, Dolomite, Lanthanum Oxide and Coke. Materials, 16(7), 2933. https://doi.org/10.3390/ma16072933