
Adsorption Tuning of Polarity and Magnetism in AgCr2S4 Monolayer


Abstract
:1. Introduction
2. Methods
3. Results
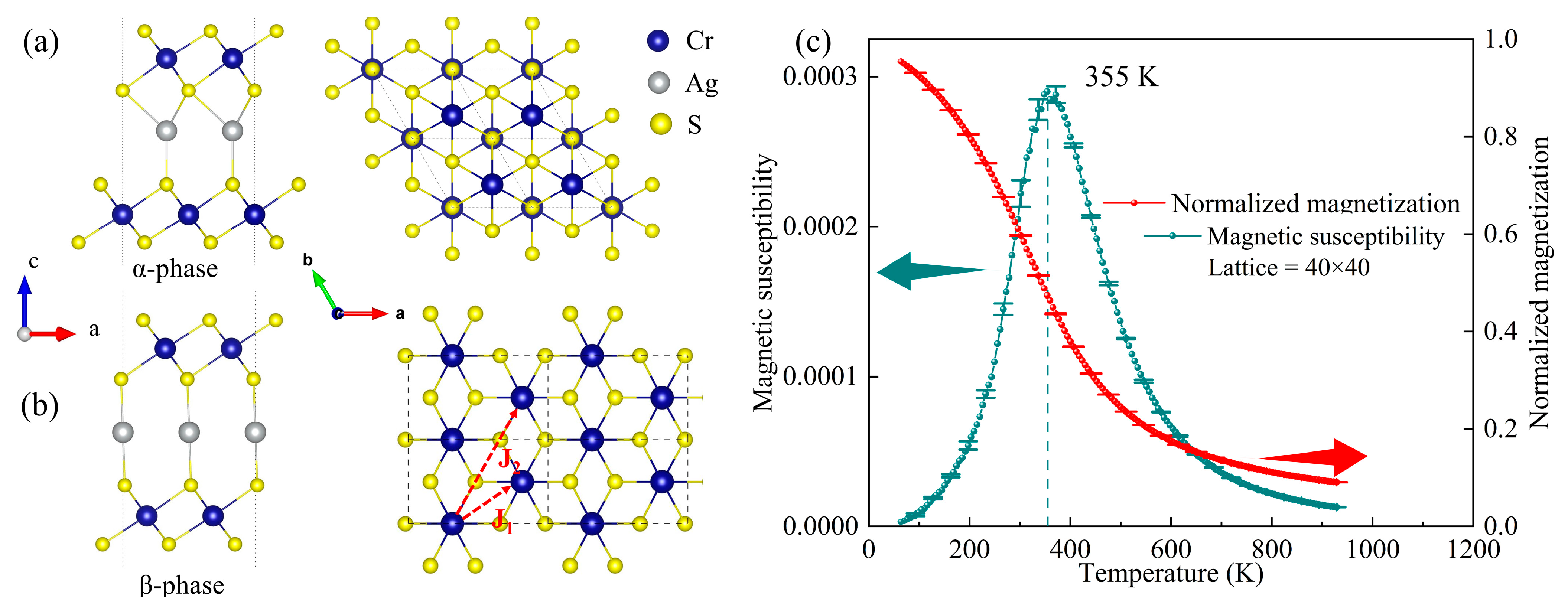
3.1. AgCrS2 Bulk Properties
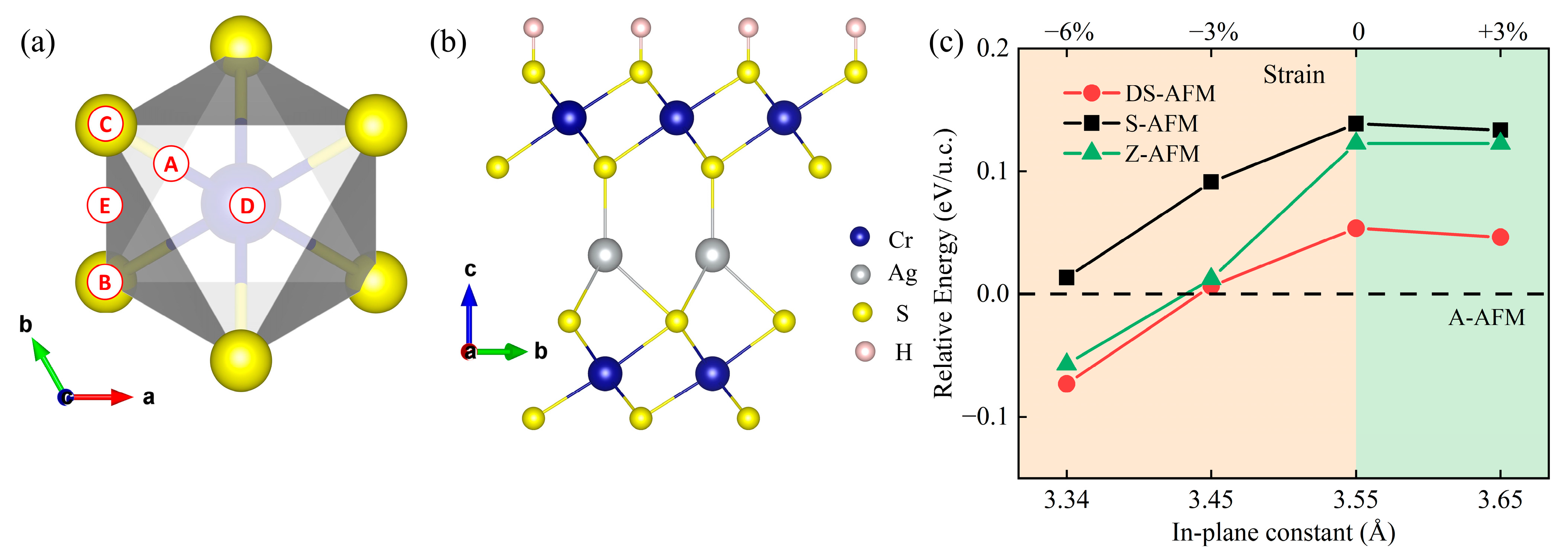
3.2. AgCr2S4 Monolayer
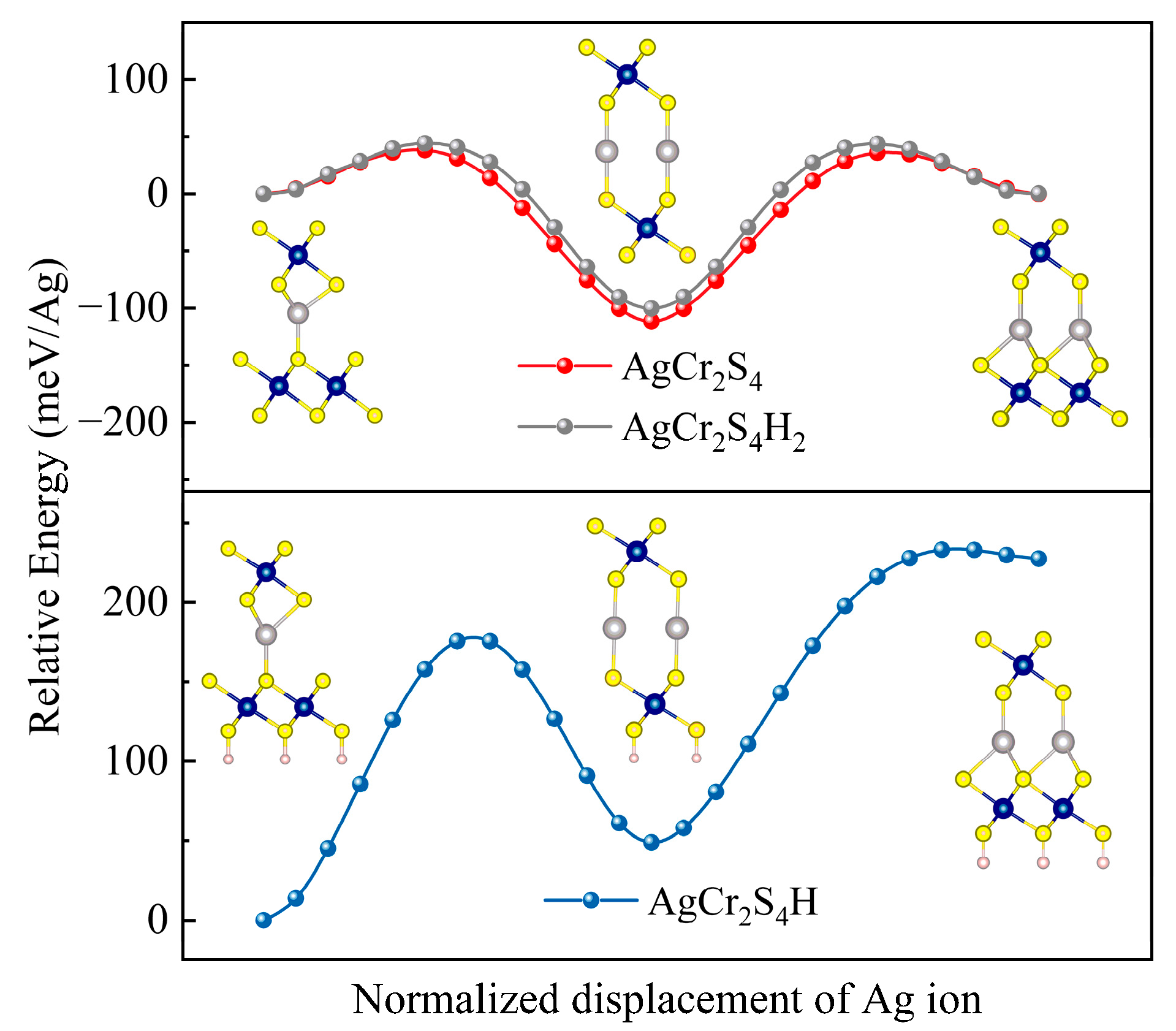
3.3. H Adsorption Effect
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Ueff | Effective Hubbard parameter. In order to deal with the strong correlation of Cr3+, the Hubbard empirical parameter U is added using Dudarev’s approach, Ueff = U − J, where U and J are the on-site Coulomb interaction and the strength of the effective on-site exchange interaction, respectively. |
| Si | The normalized spin vector sitting on lattice site i. |
| J1 and J2 | The in-plane nearest neighbor and the next-nearest neighbor magnetic exchange coupling parameters. |
| kB | The Boltzmann constant. |
| M | The normalized magnetization. |
| χ | The magnetic susceptibility. |
References
- Huang, B.; Clark, G.; Navarro-Moratalla, E.; Klein, D.R.; Cheng, R.; Seyler, K.L.; Zhong, D.; Schmidgall, E.; McGuire, M.A.; Cobden, D.H.; et al. Layer-dependent ferromagnetism in a van der Waals crystal down to the monolayer limit. Nature 2017, 546, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, C.; Li, L.; Li, Z.; Ji, H.; Stern, A.; Xia, Y.; Cao, T.; Bao, W.; Wang, C.; Wang, Y.; et al. Discovery of intrinsic ferromagnetism in two-dimensional van der Waals crystals. Nature 2017, 546, 265–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, W.; Zhu, J.; Wang, Z.; Gao, Y.; Xiao, D.; Gu, Y.; Zhang, Z.; Zhu, W. Prediction of intrinsic two-dimensional ferroelectrics in In2Se3 and other III2-VI3 van der Waals materials. Nat. Commun. 2017, 8, 14956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.; You, L.; Seyler, K.L.; Li, X.; Yu, P.; Lin, J.; Wang, X.; Zhou, J.; Wang, H.; He, H.; et al. Room-temperature ferroelectricity in CuInP2S6 ultrathin flakes. Nat. Commun. 2016, 7, 12357. [Google Scholar] [CrossRef]
- Chang, K.; Liu, J.W.; Lin, H.C.; Wang, N.; Zhao, K.; Zhang, A.M.; Jin, F.; Zhong, Y.; Hu, X.P.; Duan, W.H.; et al. Discovery of robust in-plane ferroelectricity in atomic-thick SnTe. Science 2016, 353, 274–278. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.U.; Lee, S.; Ryoo, J.H.; Kang, S.; Kim, T.Y.; Kim, P.; Park, C.H.; Park, J.G.; Cheong, H. Ising type magnetic ordering in ordering thin FePS3. Nano Lett. 2016, 16, 7433–7438. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; You, L.; Cobden, D.; Wang, J. Towards two-dimensional van der Waals ferroelectrics. Nat. Mater. 2023. [Google Scholar] [CrossRef]
- Dong, S.; Liu, J.-M.; Cheong, S.-W.; Ren, Z. Multiferroic materials and magnetoelectric physics: Symmetry, entanglement, excitation, and topology. Adv. Phys. 2015, 64, 519–626. [Google Scholar] [CrossRef] [Green Version]
- Shao, Z.; Liang, J.; Cui, Q.; Chshiev, M.; Fert, A.; Zhou, T.; Yang, H. Multiferroic materials based on transition-metal dichalcogenides: Potential platform for reversible control of Dzyaloshinskii-Moriya interaction and skyrmion via electric field. Phy. Rev. B 2022, 105, 174404. [Google Scholar] [CrossRef]
- Wang, J.; Neaton, J.B.; Zheng, H.; Nagarajan, V.; Ogale, S.B.; Liu, B.; Viehland, D.; Vaithyanathan, V.; Schlom, D.G.; Waghmare, U.V.; et al. Epitaxial BiFeO3 multiferroic thin film heterostructures. Science 2003, 299, 1719–1722. [Google Scholar] [CrossRef]
- Khan, K.; Tareen, A.K.; Aslam, M.; Wang, R.; Zhang, Y.; Mahmood, A.; Ouyang, Z.; Zhang, H.; Guo, Z. Recent developments in emerging two-dimensional materials and their applications. J. Mater. Chem. C 2020, 8, 387–440. [Google Scholar] [CrossRef]
- An, M.; Zhang, Y.; Chen, J.; Zhang, H.-M.; Guo, Y.; Dong, S. Tuning magnetism in layered magnet VI3: A theoretical study. J. Phys. Chem. C 2019, 123, 30545–30550. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, K.; Taniguchi, T.; Kanda, H. Direct-bandgap properties and evidence for ultraviolet lasing of hexagonal boron nitride single crystal. Nat. Mater. 2004, 3, 404–409. [Google Scholar] [CrossRef] [PubMed]
- Meric, I.; Dean, C.R.; Petrone, N.; Wang, L.; Hone, J.; Kim, P.; Shepard, K.L. Graphene field-effect transistors based on Boron–Nitride dielectrics. Proc. IEEE 2013, 101, 1609–1619. [Google Scholar] [CrossRef]
- Shirodkar, S.N.; Waghmare, U.V. Emergence of ferroelectricity at a metal-semiconductor transition in a 1T monolayer of MoS2. Phys. Rev. Lett. 2014, 112, 157601. [Google Scholar] [CrossRef]
- Naguib, M.; Mochalin, V.N.; Barsoum, M.W.; Gogotsi, Y. 25th anniversary article: MXenes: A new family of two-dimensional materials. Adv. Mater. 2014, 26, 992–1005. [Google Scholar] [CrossRef]
- Peng, J.; Liu, Y.; Lv, H.; Li, Y.; Lin, Y.; Su, Y.; Wu, J.; Liu, H.; Guo, Y.; Zhuo, Z.; et al. Stoichiometric two-dimensional non-van der Waals AgCrS2 with superionic behaviour at room temperature. Nat. Chem. 2021, 13, 1235–1240. [Google Scholar] [CrossRef]
- Liu, J.; Wang, Y.; Wang, L.-Y.; Yao, Q.; Huang, C.-C.; Huang, H.-Y.; Li, D.-F. Several promising non-vdW multiferroic half-metallic nanosheets ACr2S4 (A = Li, Na, K, Rb): The first-principles researches. Eur. Phys. J. Plus. 2023, 138, 224. [Google Scholar] [CrossRef]
- Huang, J.; Shi, B.; Pan, F.; Wang, J.; Liu, J.; Xu, D.; Zhang, H.; Xia, T.; Cheng, P. Anisotropic magnetic properties and tunable conductivity in two-dimensional layered NaCrX2 (X = Te, Se, S) single crystals. Phys. Rev. Mater. 2022, 6, 094013. [Google Scholar] [CrossRef]
- Hahn, H.; Lorent, C.D. Über ternare chalkogenide des chroms mit einwertigem kupfer und silber. Z. Anorg. Allg. Chem. 1957, 290, 68–81. [Google Scholar] [CrossRef]
- Singh, K.; Maignan, A.; Martin, C.; Simon, C. AgCrS2: A spin driven ferroelectric. Chem. Mater. 2009, 21, 5007–5009. [Google Scholar] [CrossRef]
- Damay, F.; Martin, C.; Hardy, V.; André, G.; Petit, S.; Maignan, A. Magnetoelastic coupling and unconventional magnetic ordering in the multiferroic triangular lattice AgCrS2. Phys. Rev. B 2011, 83, 184413. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Liu, Y.; Dai, X.; Liu, G.; Zhang, X. A ferromagnetic hybrid Weyl semimetal in two dimensions: The monolayer AgCrS2. J. Mater. Sci. 2022, 58, 281–290. [Google Scholar] [CrossRef]
- Zhao, Y.; Liu, Q.; Zhang, F.; Jiang, X.; Gao, W.; Zhao, J. Multiferroicity in a two-dimensional non-van der Waals crystal of AgCr2X4 (X = S or Se). J. Phys. Chem. Lett. 2022, 13, 11346–11353. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comp. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Blochl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1997, 78, 1396. [Google Scholar] [CrossRef] [Green Version]
- Dudarev, S.L.; Botton, G.A.; Savrasov, S.Y.; Humphreys, C.J.; Sutton, A.P. Electron-energy-loss spectra and the structural stability of nickel oxide: An LSDA+U study. Phys. Rev. B 1998, 57, 1505–1509. [Google Scholar] [CrossRef]
- Yu, M.; Trinkle, D.R. Accurate and efficient algorithm for Bader charge integration. J. Chem. Phys. 2011, 134, 064111. [Google Scholar] [CrossRef] [Green Version]
- Binder, K.; Ceperley, D.M.; Hansen, J.-P.; Kalos, M.; Landau, D.; Levesque, D.; Mueller-Krumbhaar, H.; Stauffer, D.; Weis, J.-J. Monte Carlo Methods in Statistical Physics; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2012; Volume 7, pp. 195–233. [Google Scholar]
- Ushakov, A.V.; Kukusta, D.A.; Yaresko, A.N.; Khomskii, D.I. Magnetism of layered chromium sulfides MCrS2 (M = Li, Na, K, Ag, and Au): A first-principles study. Phys. Rev. B 2013, 87, 014418. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Huang, Z.; Jia, T.; Zhang, X.; Zeng, Z. Magnetic ordering and exchange striction stabilized geometric ferroelectricity in multiferroic AgCrS2. J. Phys. Condens. Matter 2016, 28, 236002. [Google Scholar] [CrossRef] [PubMed]
- Streltsov, S.V.; Poteryaev, A.I.; Rubtsov, A.N. Magnetostriction and ferroelectric state in AgCrS2. J. Phys. Condens. Matter 2015, 27, 165601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damay, F.; Petit, S.; Braendlein, M.; Rols, S.; Ollivier, J.; Martin, C.; Maignan, A. Spin dynamics in the unconventional multiferroic AgCrS2. Phys. Rev. B 2013, 87, 134413. [Google Scholar] [CrossRef]
- Goodenough, J.B. Theory of the role of covalence in the perovskite-type manganites [La,M(II)]MnO3. Phys. Rev. 1955, 100, 564–573. [Google Scholar] [CrossRef] [Green Version]
- Kanamori, J. Superexchange interaction and symmetry properties of electron orbitals. J. Phys. Chem. Solids 1959, 10, 87–98. [Google Scholar] [CrossRef]
- Anderson, P.W. Theory of magnetic exchange interactions: Exchange in insulators and semiconductors. Solid State Phys. 1963, 14, 99–214. [Google Scholar]
- Landau, D.; Binder, K. A Guide to Monte Carlo Simulations in Statistical Physics, 5th ed.; Cambridge University Press: Cambridge, UK, 2021; pp. 7–49. [Google Scholar]
- Erkişi, A. Ab Initio study on electronic and elastic properties of AgCr2S4. Acta Phys. Pol. A 2021, 140, 243–251. [Google Scholar] [CrossRef]
- Wang, V.; Xu, N.; Liu, J.-C.; Tang, G.; Geng, W.-T. Vaspkit: A user-friendly interface facilitating high-throughput computing and analysis using vasp code. Comput. Phys. Commun. 2021, 267, 108033. [Google Scholar] [CrossRef]
- Maździarz, M. Comment on ‘the computational 2D materials database: High-throughput modeling and discovery of atomically thin crystals’. 2D Mate. 2019, 6, 048001. [Google Scholar] [CrossRef] [Green Version]
- Born, M.; Huang, K. Dynamical Theory of Crystal Lattices; Oxford University Press: New York, NY, USA, 1954; pp. 120–154. [Google Scholar]
- Li, R.; Shao, Q.; Gao, E.; Liu, Z. Elastic anisotropy measure for two-dimensional crystals. Extreme Mech. Lett. 2020, 34, 100615. [Google Scholar] [CrossRef]
- Falin, A.; Cai, Q.; Santos, E.J.; Scullion, D.; Qian, D.; Zhang, R.; Yang, Z.; Huang, S.; Watanabe, K.; Taniguchi, T. Mechanical properties of atomically thin boron nitride and the role of interlayer interactions. Nat. Commun. 2017, 8, 15815. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| A-AFM | DS1-AFM | Z1-AFM | S1-AFM | FM | DS2-AFM | Z2-AFM | S2-AFM | |
|---|---|---|---|---|---|---|---|---|
| α | 57.25 | 68.40 | 82.62 | 88.25 | 53.65 | 66.44 | 82.82 | 23.39 |
| β | 0 | 14.99 | 11.69 | 24.00 | 2.88 | 13.67 | 13.40 | 22.41 |
| J1 | J2 | Kb | Kc | |
|---|---|---|---|---|
| β phase | −14.86 | −5.76 | 1.11 | 0.80 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, R.; Wang, Y.; Ding, N.; Dong, S.; An, M. Adsorption Tuning of Polarity and Magnetism in AgCr2S4 Monolayer. Materials 2023, 16, 3058. https://doi.org/10.3390/ma16083058
Li R, Wang Y, Ding N, Dong S, An M. Adsorption Tuning of Polarity and Magnetism in AgCr2S4 Monolayer. Materials. 2023; 16(8):3058. https://doi.org/10.3390/ma16083058
Chicago/Turabian StyleLi, Ranran, Yu Wang, Ning Ding, Shuai Dong, and Ming An. 2023. "Adsorption Tuning of Polarity and Magnetism in AgCr2S4 Monolayer" Materials 16, no. 8: 3058. https://doi.org/10.3390/ma16083058
APA StyleLi, R., Wang, Y., Ding, N., Dong, S., & An, M. (2023). Adsorption Tuning of Polarity and Magnetism in AgCr2S4 Monolayer. Materials, 16(8), 3058. https://doi.org/10.3390/ma16083058