
Ab Initio Density Functional Theory Calculation: Americium Hydrolysis Mechanism


Abstract
:1. Introduction
2. Method
3. Results and Discussion
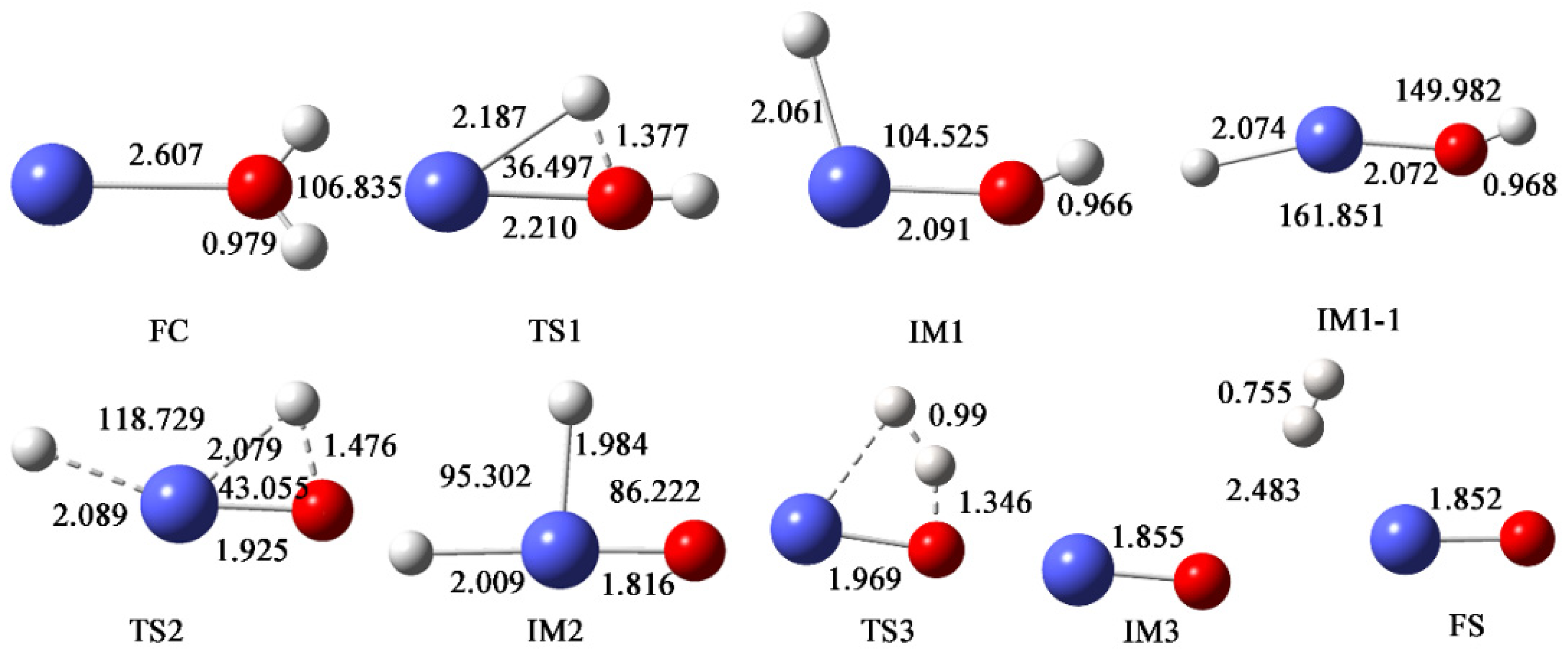
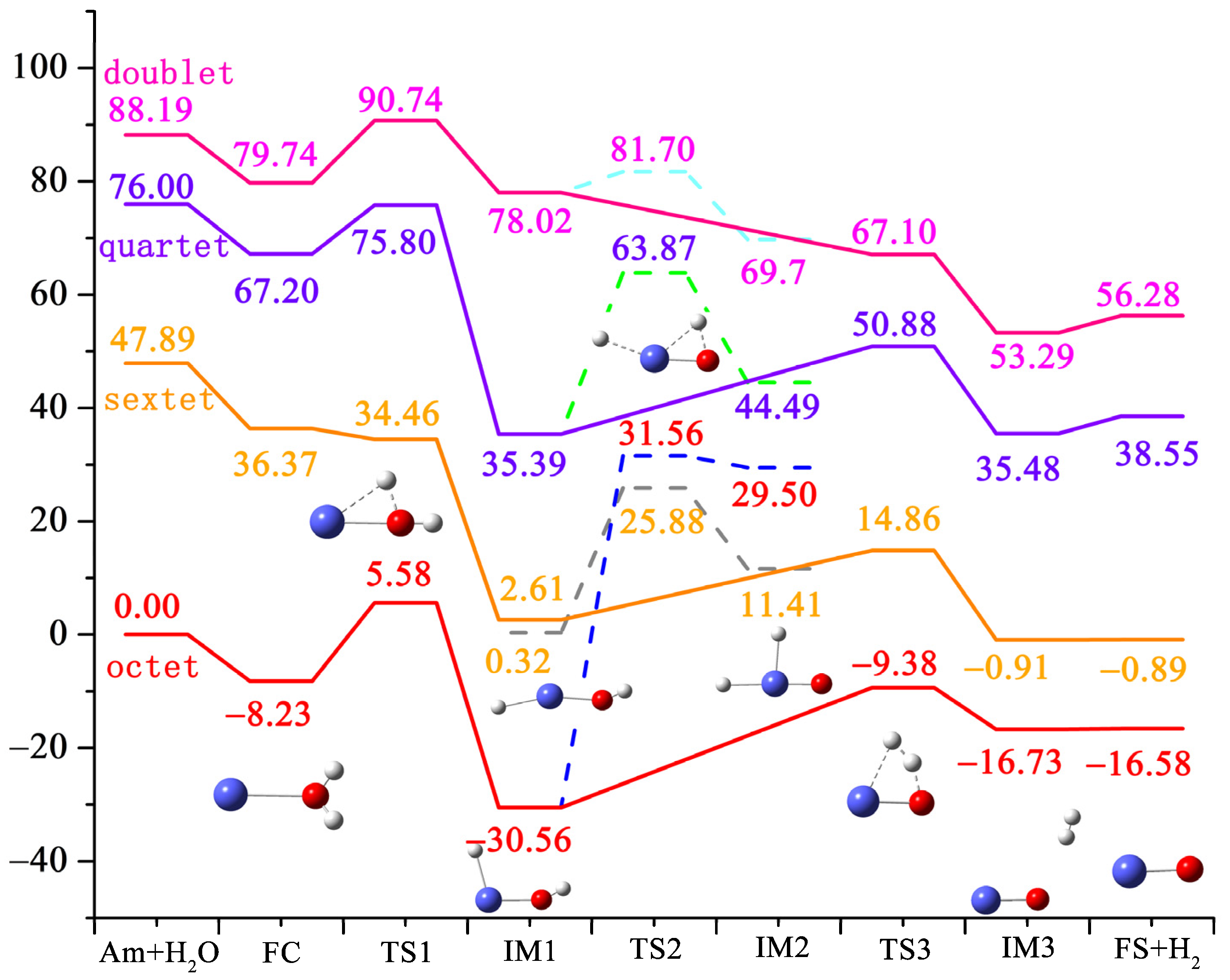
3.1. Ground-State Structures and Energy
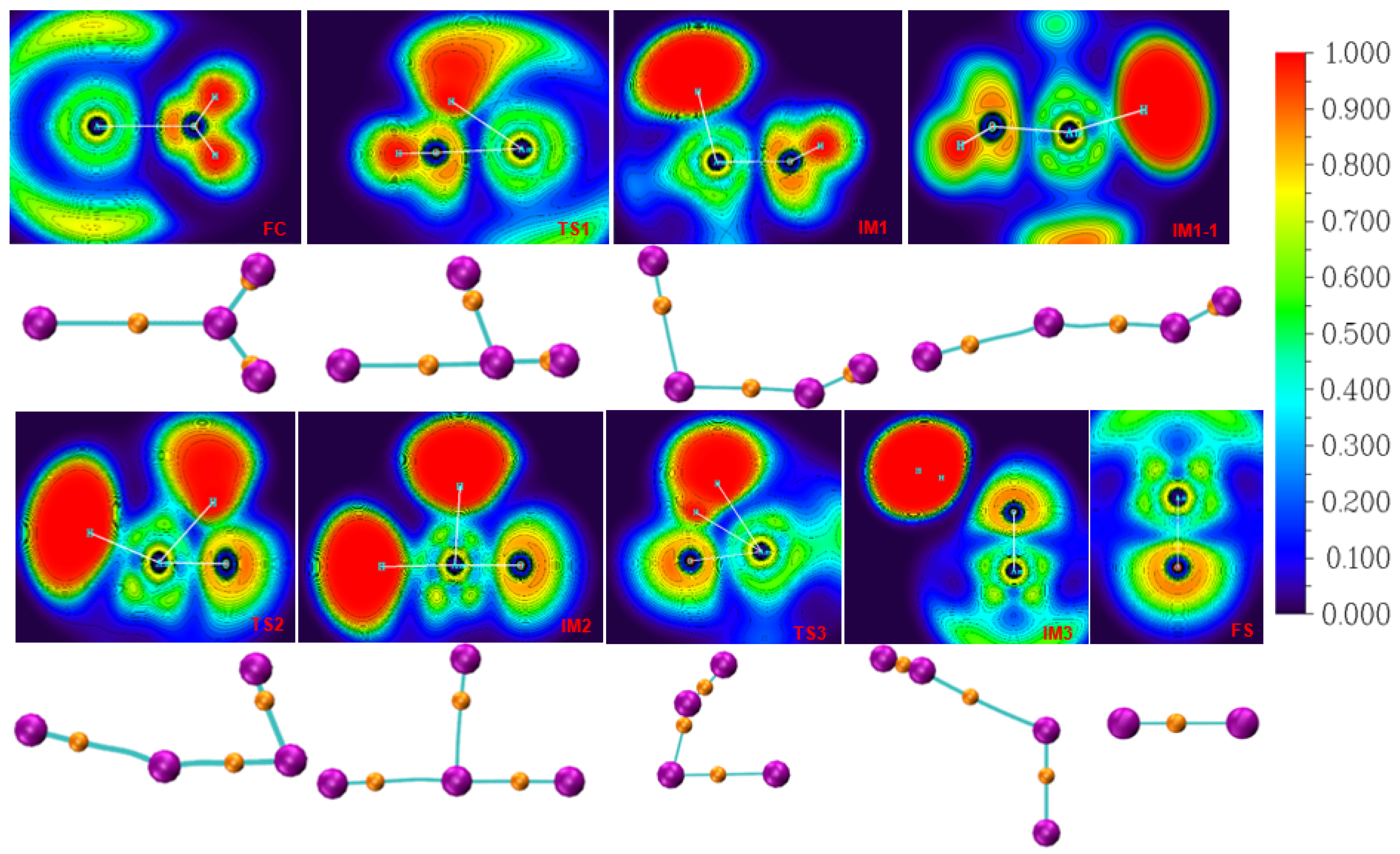
3.2. Bonding Analysis
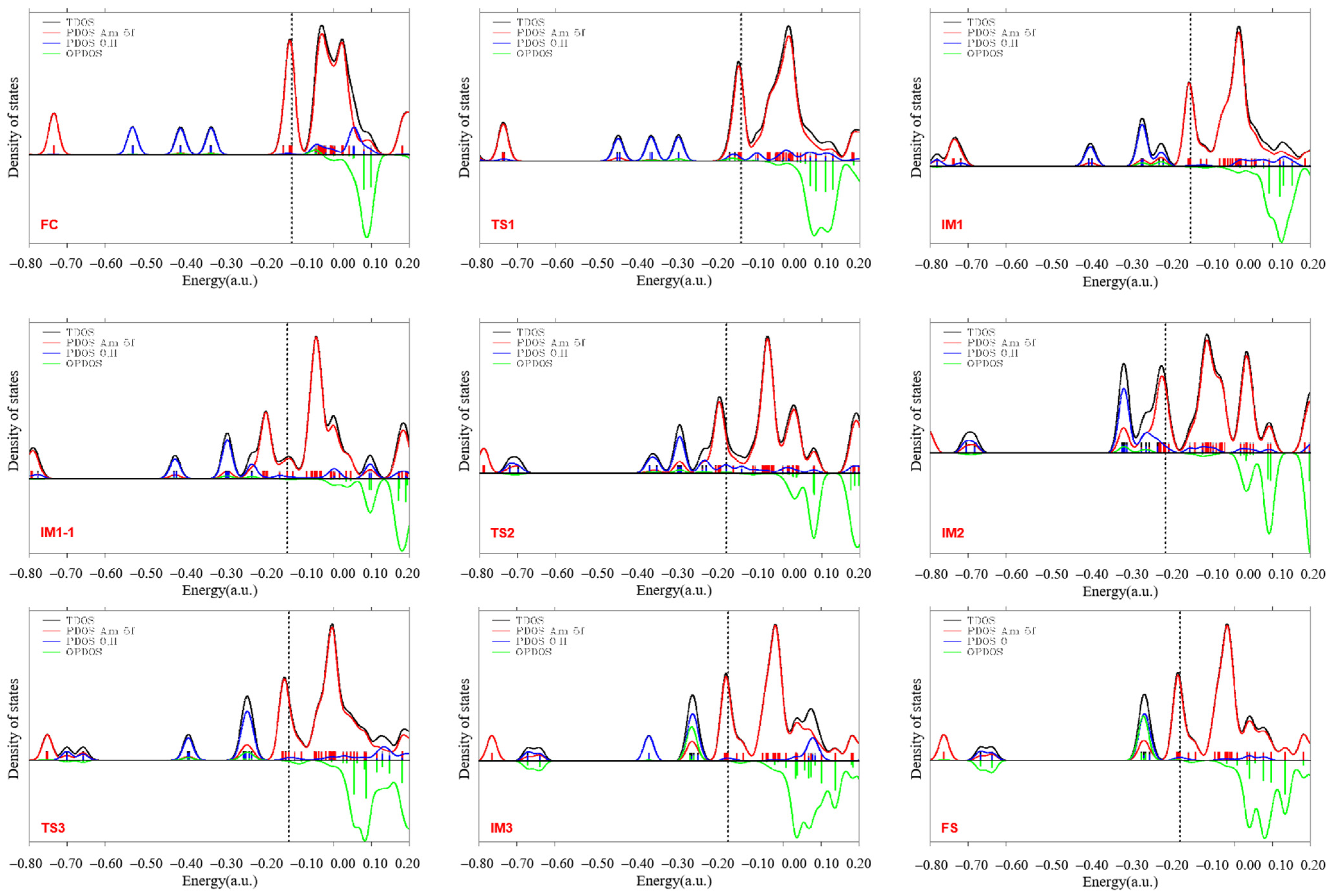
3.3. Density of States (DOS)
3.4. Natural Population Analysis (NPA)
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chien, W.; Anbalagan, V.; Zandler, M.; Van Stipdonk, M.; Hanna, D.; Gresham, G.; Groenewold, G. Intrinsic hydration of monopositive uranyl hydroxide, nitrate, and acetate cations. J. Am. Soc. Mass Spectrom. 2004, 15, 777–783. [Google Scholar] [CrossRef]
- Gresham, G.L.; Gianotto, A.K.; Harrington, P.d.B.; Cao, L.; Scott, J.R.; Olson, J.E.; Appelhans, A.D.; Van Stipdonk, M.J.; Groenewold, G.S. Gas-phase hydration of U(IV), U(V), and U(VI) dioxo monocations. J. Phys. Chem. A 2003, 107, 8530–8538. [Google Scholar] [CrossRef]
- Michelini, M.d.C.; Russo, N.; Sicilia, E. Gas-phase chemistry of actinides ions: New insights into the reaction of UO+ and UO2+ with water. J. Am. Chem. Soc. 2007, 129, 4229–4239. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Yang, Y.; Pande, S.; Qu, B.; Li, D.; Zeng, X.C. Reaction mechanism between small-sized Ce clusters and water molecules: An Ab initio investigation on Cen+H2O. Phys. Chem. Chem. Phys. 2019, 21, 4006–4014. [Google Scholar] [CrossRef] [PubMed]
- Van Stipdonk, M.J.; Chien, W.; Anbalagan, V.; Bulleigh, K.; Hanna, D.; Groenewold, G.S. Gas-phase complexes containing the uranyl ion and acetone. J. Phys. Chem. A 2004, 108, 10448–10457. [Google Scholar] [CrossRef]
- Wang, G.; Batista, E.R.; Yang, P. Excess electrons on reduced AnO2(111) surfaces (An = Th, U, Pu) and their impacts on catalytic water splitting. J. Phys. Chem. C 2019, 123, 30245–30251. [Google Scholar] [CrossRef]
- del Carmen Michelini, M.; Russo, N.; Sicilia, E. How Can Uranium Ions (U+, U2+) Activate the O-H Bond of Water in the Gas Phase? Angew. Chem. Int. Ed. 2006, 45, 1095–1099. [Google Scholar] [CrossRef]
- Vallet, V.; Privalov, T.; Wahlgren, U.; Grenthe, I. The mechanism of water exchange in AmO2(H2O)52+ and in the isoelectronic UO2(H2O) 5+ and NpO2(H2O)52+ complexes as studied by quantum chemical methods. J. Am. Chem. Soc. 2004, 126, 7766–7767. [Google Scholar] [CrossRef]
- Zhou, J.; Schlegel, H.B. Ab initio molecular dynamics study of the reaction between Th+ and H2O. J. Phys. Chem. A 2010, 114, 8613–8617. [Google Scholar] [CrossRef]
- Rios, D.; Michelini, M.C.; Lucena, A.F.; Marçalo, J.; Bray, T.H.; Gibson, J.K. Gas-phase uranyl, neptunyl, and plutonyl: Hydration and oxidation studied by experiment and theory. Inorg. Chem. 2012, 51, 6603–6614. [Google Scholar] [CrossRef]
- Shan, N.; Wang, Q.Q.; Gao, T. Gas-phase reaction process of water activated by bare and Oxo-Ligated protactinium ions. Chem. Phys. Lett. 2023, 825, 140606. [Google Scholar] [CrossRef]
- Morss, L.R.; Edelstein, N.M.; Fuger, J.; Katz, J. The Chemistry of the Actinide and Transactinide Elements; Springer: Dordrecht, The Netherlands, 2006; Volume 1–5. [Google Scholar] [CrossRef]
- Holden, N.E. History of the Origin of the Chemical Elements and Their Discoverers; Brookhaven National Lab (BNL): Upton, NY, USA, 2019. [Google Scholar]
- Cornehl, H.H.; Wesendrup, R.; Diefenbach, M.; Schwarz, H. A comparative study of oxo-ligand effects in the gas-phase chemistry of atomic lanthanide and actinide cations. Chem. Eur. J. 1997, 3, 1083–1090. [Google Scholar] [CrossRef]
- Runde, W.H.; Schulz, W.W. Americium. In The Chemistry of the Actinide and Transactinide Elements; Springer: Dordrecht, The Netherlands, 2006; pp. 1265–1395. [Google Scholar] [CrossRef]
- Street, K., Jr.; Ghiorso, A.; Seaborg, G. The isotopes of americium. Phys. Rev. 1950, 79, 530. [Google Scholar] [CrossRef]
- Pillai, J.S.; Srivastava, A.; Ansari, S.A.; Chaudhury, S. Clean methodology for nuclear laboratory waste remediation: Part-II: Recovery of Americium. J. Clean. Prod. 2021, 280, 124342. [Google Scholar] [CrossRef]
- Gibney, E. How Nuclear Waste Will Help Spacecraft Explore the Moon. Nature 2022, 612, 385–386. [Google Scholar] [CrossRef]
- Mazzone, G.; Michelini, M.d.C.; Russo, N.; Sicilia, E. Mechanistic Aspects of the Reaction of Th+ and Th2+ with Water in the Gas Phase. Inorg. Chem. 2008, 47, 2083–2088. [Google Scholar] [CrossRef]
- Alikhani, M.E.; Michelini, M.d.C.; Russo, N.; Silvi, B. Topological analysis of the reaction of uranium ions (U+, U2+) with N2O in the gas phase. J. Phys. Chem. A 2008, 112, 12966–12974. [Google Scholar] [CrossRef]
- De Almeida, K.; Duarte, H. Dehydrogenation of methane by gas-phase Th, Th+, and Th2+: Theoretical insights into actinide chemistry. Organometallics 2010, 29, 3735–3745. [Google Scholar] [CrossRef]
- Zhou, R.; Ma, S.; Yang, Y.; Li, D.; Qu, B.; Zeng, X.C. Reaction mechanism between small-sized Ce clusters and water molecules II: An ab initio investigation on Cen (n = 1–3) + mH2O (m = 2–6). Phys. Chem. Chem. Phys. 2019, 21, 8945–8955. [Google Scholar] [CrossRef]
- Frish, M.; Trucks, G.; Schlegel, H.; Scuseria, G.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Mennucci, B.; Paterson, G. Gaussian 09, Revision A. 02; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Gonzalez, C.; Schlegel, H.B. An improved algorithm for reaction path following. J. Chem. Phys. 1989, 90, 2154–2161. [Google Scholar] [CrossRef]
- Gonzalez, C.; Schlegel, H.B. Reaction path following in mass-weighted internal coordinates. J. Phys. Chem. 1990, 94, 5523–5527. [Google Scholar] [CrossRef]
- Perveen, M.; Nazir, S.; Arshad, A.W.; Khan, M.I.; Shamim, M.; Ayub, K.; Khan, M.A.; Iqbal, J. Therapeutic potential of graphitic carbon nitride as a drug delivery system for cisplatin (anticancer drug): A DFT approach. Biophys. Chem. 2020, 267, 106461. [Google Scholar] [CrossRef]
- Shamim, M.; Perveen, M.; Nazir, S.; Hussnain, M.; Mehmood, R.; Khan, M.I.; Iqbal, J. DFT study of therapeutic potential of graphitic carbon nitride (g-C3N4) as a new drug delivery system for carboplatin to treat cancer. J. Mol. Liq. 2021, 331, 115607. [Google Scholar] [CrossRef]
- Munir, I.; Perveen, M.; Nazir, S.; Khera, R.A.; Ayub, A.R.; Ayub, K.; Iqbal, J. Therapeutic potential of graphyne as a new drug-delivery system for daunorubicin to treat cancer: A DFT study. J. Mol. Liq. 2021, 336, 116327. [Google Scholar] [CrossRef]
- Bader, R. A Quantum Theory; Clarendon: Oxford, UK, 1990. [Google Scholar]
- Małecki, J. Synthesis, crystal, molecular and electronic structures of thiocyanate ruthenium complexes with pyridine and its derivatives as ligands. Polyhedron 2010, 29, 1973–1979. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | b3lyp | pbe0 | pw91 | Exptl. a | |
|---|---|---|---|---|---|
| Am | [Rn]5f77s2(6) | 50.70 | 68.61 | 47.89 | 42.93 |
| [Rn]5f77s2(8) | 0.00 | 0.00 | 0.00 | 0.00 | |
| [Rn]5f76d7s(10) | 43.30 | 35.94 | 36.23 | 41.47 |
| Am + H2O | FC8 | TS18 | IM18 | IM1-16 | TS26 | IM26 | TS38 | IM38 | FS8 | |
|---|---|---|---|---|---|---|---|---|---|---|
| E | 0.00 | −8.23 | 5.58 | −30.56 | 0.32 | 25.8 | 11.41 | −9.38 | −16.73 | −16.58 |
| <S2> | - | 15.77 | 15.89 | 15.77 | 9.90 | 9.58 | 8.98 | 15.78 | 15.80 | 15.80 |
| s(s + 1) | - | 15.75 | 15.75 | 15.75 | 8.75 | 8.75 | 8.75 | 15.75 | 15.75 | 15.75 |
| Bond | ρ | σ2ρ | G(r) | V(r) | H(r) | η | |
|---|---|---|---|---|---|---|---|
| 8FC | Am-O | 0.034 | 0.147 | 0.035 | −0.034 | 0.001 | 0.163 |
| O-H3 | 0.344 | −2.280 | 0.071 | −0.711 | −0.641 | 1.696 | |
| O-H4 | 0.344 | −2.281 | 0.071 | −0.711 | −0.641 | 1.696 | |
| 8TS1 | Am-O | 0.081 | 0.394 | 0.108 | −0.117 | −0.009 | 0.170 |
| O-H3 | 0.124 | 0.012 | 0.064 | −0.126 | −0.062 | 0.493 | |
| O-H4 | 0.347 | −2.298 | 0.073 | −0.720 | −0.647 | 1.673 | |
| 8IM1 | Am-O | 0.112 | 0.499 | 0.151 | −0.178 | −0.027 | 0.180 |
| Am-H3 | 0.074 | 0.064 | 0.040 | −0.064 | −0.024 | 0.373 | |
| O-H4 | 0.354 | −2.316 | 0.078 | −0.734 | −0.657 | 1.666 | |
| 6IM1-1 | Am-O | 0.118 | 0.488 | 0.152 | −0.182 | −0.030 | 0.194 |
| Am-H3 | 0.076 | 0.033 | 0.033 | −0.058 | −0.025 | 0.433 | |
| O-H4 | 0.351 | −2.316 | 0.075 | −0.729 | −0.654 | 1.674 | |
| 6ts2 | Am-O | 0.194 | 0.483 | 0.222 | −0.323 | −0.101 | 0.294 |
| Am-H3 | 0.073 | 0.044 | 0.033 | −0.056 | −0.023 | 0.424 | |
| O-H4 | 0.103 | 0.072 | 0.057 | −0.096 | −0.039 | 0.438 | |
| 6im2 | Am-O | 0.254 | 0.476 | 0.295 | −0.471 | −0.176 | 0.333 |
| Am-H3 | 0.088 | 0.016 | 0.037 | −0.069 | −0.033 | 0.506 | |
| Am-H4 | 0.090 | 0.031 | 0.043 | −0.078 | −0.035 | 0.448 | |
| 8TS3 | Am-O | 0.171 | 0.533 | 0.212 | −0.291 | −0.079 | 0.252 |
| O-H4 | 0.130 | 0.026 | 0.075 | −0.143 | −0.068 | 0.494 | |
| H3-H4 | 0.143 | −0.311 | 0.025 | −0.128 | −0.103 | 0.943 | |
| 8IM3 | Am-O | 0.228 | 0.537 | 0.277 | −0.420 | −0.143 | 0.290 |
| O-H4 | 0.009 | 0.026 | 0.006 | −0.005 | 0.001 | 0.195 | |
| H3-H4 | 0.254 | −1.006 | 0.001 | −0.253 | −0.252 | 1.197 | |
| 8FS | Am-O | 0.231 | 0.535 | 0.280 | −0.426 | −0.146 | 0.292 |
| NPA | Am | O | H3 | H4 | |
|---|---|---|---|---|---|
| 8FC | Charge | 0.04879 | −1.01363 | 0.48238 | 0.48246 |
| Natural Electron Configuration | 7S1.915f6.996d0.047p0.01 | 2S1.782p5.223S0.013p0.01 | 1S0.512S0.01 | 1S0.512S0.01 | |
| 8TS1 | Charge | 0.65194 | −1.15263 | 0.00025 | 0.50044 |
| Natural Electron Configuration | 7S1.205f6.946d0.207p0.02 | 2S1.842p5.303p0.01 | 1S0.992S0.01 | 1S0.49 | |
| 8IM1 | Charge | 1.27564 | −1.20985 | −0.54767 | 0.48188 |
| Natural Electron Configuration | 7S0.515f6.816d0.417p0.03 | 2S1.802p5.40 | 1S1.55 | 1S0.51 | |
| 6IM1-1 | Charge | 1.07897 | −1.14738 | −0.42137 | 0.48979 |
| Natural Electron Configuration | 7S0.965f6.506d0.437p0.048S0.01 | 2S1.812p5.33 | 1S1.42 | 1S0.51 | |
| 6TS2 | Charge | 1.35518 | −0.8941 | −0.43545 | −0.02563 |
| Natural Electron Configuration | 7S0.445f6.616d0.587p0.06 | 2S1.912p4.973p0.01 | 1S1.43 | 1S1.012S0.01 | |
| 6IM2 | Charge | 1.46476 | −0.75821 | −0.34861 | −0.35794 |
| Natural Electron Configuration | 7S0.325f6.396d0.847p0.08 | 2S1.902p4.85 | 1S1.34 | 1S1.36 | |
| 8TS3 | Charge | 1.13815 | −1.05612 | −0.28414 | 0.20212 |
| Natural Electron Configuration | 7S0.475f6.786d0.577p0.06 | 2S1.902p5.15 | 1S1.28 | 1S0.792S0.01 | |
| 8IM3 | Charge | 1.04945 | −1.03958 | −0.03848 | 0.02862 |
| Natural Electron Configuration | 7S0.705f6.616d0.627p0.08 | 2S1.932p5.10 | 1S1.04 | 1S0.97 | |
| 8FS | Charge | 1.0308 | −1.0308 | ||
| Natural Electron Configuration | 7S0.715f6.606d0.637p0.09 | 2S1.932p5.09 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shan, N.; Gao, T. Ab Initio Density Functional Theory Calculation: Americium Hydrolysis Mechanism. Materials 2024, 17, 572. https://doi.org/10.3390/ma17030572
Shan N, Gao T. Ab Initio Density Functional Theory Calculation: Americium Hydrolysis Mechanism. Materials. 2024; 17(3):572. https://doi.org/10.3390/ma17030572
Chicago/Turabian StyleShan, Na, and Tao Gao. 2024. "Ab Initio Density Functional Theory Calculation: Americium Hydrolysis Mechanism" Materials 17, no. 3: 572. https://doi.org/10.3390/ma17030572
APA StyleShan, N., & Gao, T. (2024). Ab Initio Density Functional Theory Calculation: Americium Hydrolysis Mechanism. Materials, 17(3), 572. https://doi.org/10.3390/ma17030572