1. Introduction
Nuclear power has remained one of the most significant modes of power generation since its inception in the 1950s [
1]. However, one of the key challenges in its continued use is waste management. Countries generally operate either an open or closed fuel cycle [
2]. In an open fuel cycle, the nuclear fuel is used within the power plant and then is disposed of once it is spent. Conversely, in a closed fuel cycle, once the fuel is spent, it is removed from the power plant and reprocessed, with some or all of the fuel being reused to generate power [
3]. Most countries which reprocess their spent nuclear fuel (SNF) utilise aqueous processes, e.g., PUREX (plutonium and uranium recovery by extraction), where solvent extraction combines the use of acids and organic complexants to dissolve the SNF and separate the actinides from the fission products [
4]. However, there are some disadvantages to aqueous processes, such as their vulnerability to proliferation and radiation damage of the solvents [
5].
Molten-salt-based electrochemical reprocessing (also known as pyroprocessing) solves these issues, as it is inherently proliferation-resistant and the inorganic electrolytes are not vulnerable to radiation damage [
6]. The US developed an electrochemical process for the reprocessing of spent metallic fuel from sodium-cooled fast reactors, such as EBR-II [
7]. Its application is being expanded to process spent fuels from oxide fuel reactors [
6]. South Korea also utilises a molten-salt-based process, with the Korea Atomic Energy Institute (KAERI) beginning development of the process in 1997 [
8]. Generally, the primary stage of pyroprocessing is electrorefining, where a metallic feedstock is dissolved in LiCl-KCl eutectic (LKE; 44:56 wt%) salt, which melts at 355 °C, and the re-useable actinides (mainly U and Pu) are deposited at multiple cathodes [
4]. Due to the requirement of a metallic feedstock for electrorefining, an additional stage, known as electroreduction, is required for processing of oxide fuels [
8]. In electroreduction, the oxide fuels are dissolved into a LiCl molten salt, with the oxygen transported to the anode and released [
8]. In both processes, highly radioactive fission products build up in the salt as the useful actinides are electrochemically extracted, leaving the fission products to accumulate, requiring further treatment. These salts become rich in alkalis (Cs, Rb), alkaline earths (Ba, Sr), and rare earths (La, Ce, Pr…) [
9]. To operate efficient and successful pyroprocessing of spent nuclear fuel, the LiCl and LKE molten salts used in electroreducing and electrorefining need to be periodically treated post-use to remove fission products and other impurities [
4]. There are two primary reasons for this: (1) removal of the fission products means that a significant volume of the salts can be reused, increasing the efficiency of the system; and (2) chloride wastes tend to have low solubility in most common waste forms—reducing the volume of chloride waste through treatment will drastically reduce final waste volumes [
4].
Recent research efforts, primarily out of South Korea and the USA, have worked to establish the applicability of zone refining, and other melt crystallisation techniques, for the treatment of pyroprocessing salt wastes [
3,
4]. The melt crystallisation techniques investigated include zone freezing [
10], layer crystallisation (or coldfinger crystallisation) [
10,
11,
12], the Czochralski method [
13], and zone refining [
6,
13]. In general, this research has focused on the separation of CsCl and SrCl
2 from LiCl, i.e., refinement of electroreducing waste salt where Cs-137 and Sr-90 will be the main fission product contaminants. This is due to the difficulty of separating CsCl and SrCl
2 from LiCl using common techniques such as precipitation [
10]. Cho et al. demonstrated separation efficiencies for Cs and Sr of >90% for both the zone freezing and layer crystallisation methods, provided the process rate (crucible rising velocity for zone freezing, crystal growth rate for layer crystallisation) was suitably slow (1.7 mm/h and <5 g/min, respectively) [
10]. In general, the separation efficiency was similar for Cs and Sr. Cho et al. confirmed the efficacy of the layer crystallisation method, observing >90% separation efficiencies for Cs and Sr up to a crystal growth rate of 5 g/min, although the results suggest a greater efficiency of Cs separation compared to Sr separation [
11]. Versey et al. also observed effective separation of Cs from LiCl using layer crystallisation, and they demonstrated that the efficiency can be maximised through increasing the cooling gas flow rate and crystal growth time [
12]. The Czochralski method was investigated by Lee et al., with effective separation of Cs and Sr from LiCl observed, particularly at lower extraction speeds (2.5 mm/h) [
13]. However, the calculated segregation coefficient (
k) values suggest that Cs was separated more efficiently than Sr using this method. Shim et al. investigated zone refining for separation of Cs and Sr from both LiCl and LKE [
14,
15]. The efficiency of separation was inversely proportional to the freezing rate, and proportional to the number of passes. Cs was separated more effectively than Sr, but both elements were more difficult to separate from the LKE than from the LiCl salt. It was also found that the elements were more difficult to separate in LiCl-KCl-CsCl-SrCl
2 compared to LiCl-KCl-CsCl or LiCl-KCl-SrCl
2.
One key issue for melt crystallisation techniques for processing of pyroprocessing wastes is their relatively slow rates. For a recovery yield of 60% LiCl from an LiCl-CsCl-SrCl
2 waste, zone refining would take around 80 h, zone freezing would take around 102 h, and the Czochralski method around 52 h [
14]. However, the advantage of the zone-refining technique is that multiple heating zones can be implemented, which increases the throughput by reducing the number of passes required to refine the waste salt [
14]. Whilst there have been several investigations into melt crystallisation techniques for electroreducing waste salt [
6,
13], the same cannot be said for its electrorefining counterpart, where various rare-earth radionuclides will be the main impurities. This is likely due to the efficacy of other separation methods, such as reactive precipitation and reactive distillation [
4]. However, the study of zone refinement for electrorefining waste treatment is of value for comparison to other methods. One proposed method of molten salt waste treatment is zone refining [
8]. This technique was developed by W. G. Pfann at Bell Labs in the early 1950s [
16], and can be used on any system with a non-zero segregation coefficient,
k, as calculated by
where
CS is the concentration of the impurity/impurities in the solid salt, and
CL is the concentration of the impurity/impurities in the molten salt. A schematic of this method can be seen in
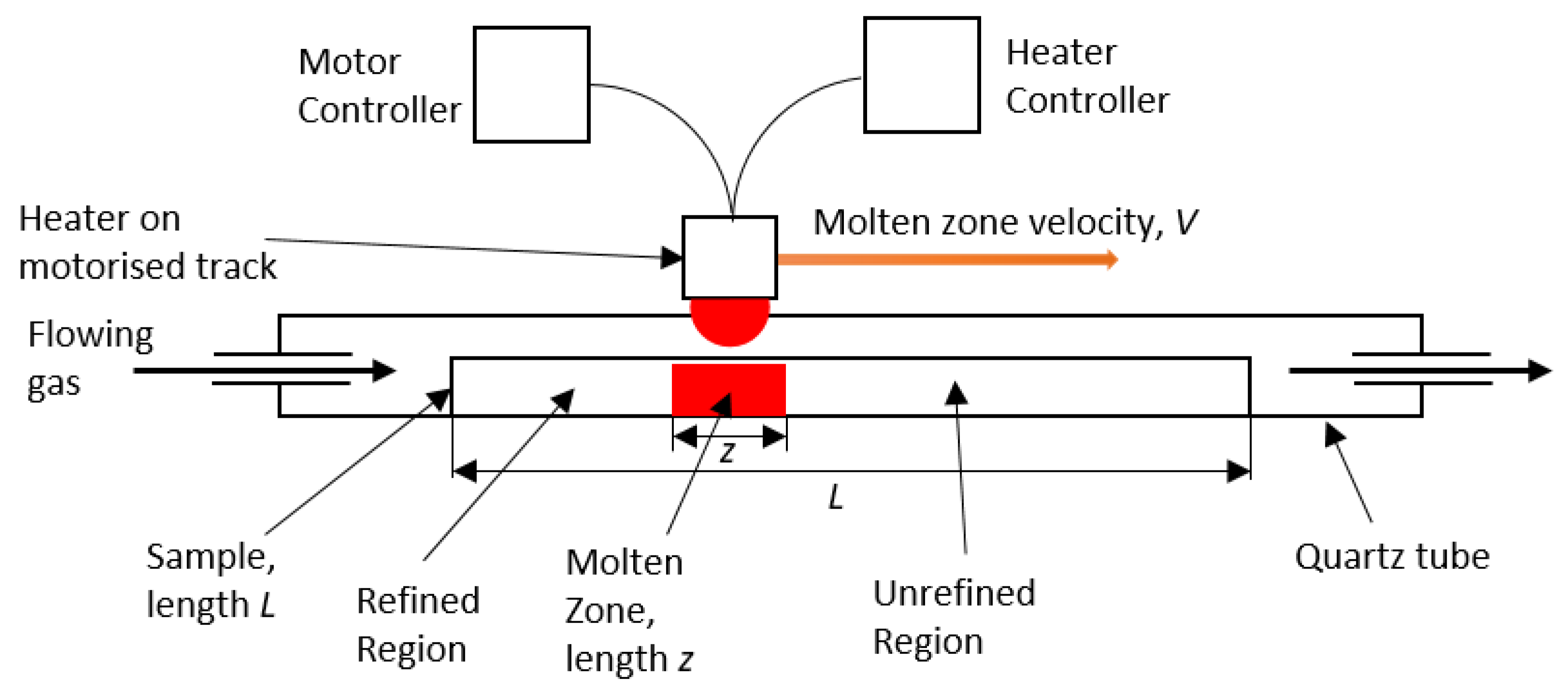
Figure 1. A moving heater generates a moving molten zone, travelling from one end of the sample to the other. When
k < 1, as the salt behind the moving molten zone solidifies, the impurities remain within the molten zone at a higher concentration than in the recrystallised salt. Thus, as the molten zone travels along the sample it ‘sweeps’ the impurities to the end of the sample [
14]. This method has most commonly been used in the preparation of high-purity materials for the production of semi-conductors [
17,
18,
19,
20] and scintillators [
21,
22].
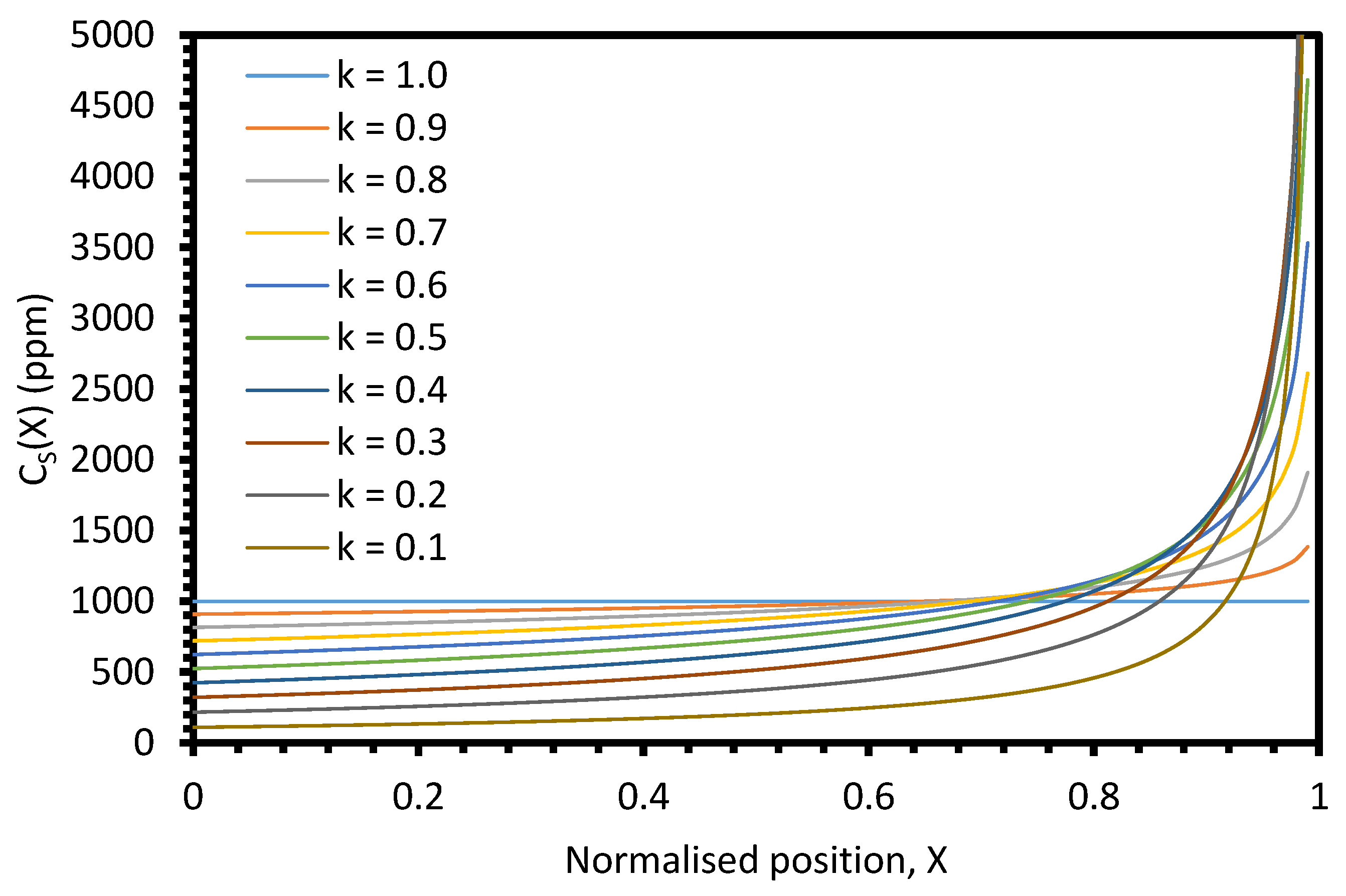
The theoretical distribution of an impurity in a 1D zone-refining system can be calculated through several different equations based on the region within the sample. For a 1D system in equilibrium with sample length,
L; position,
x; normalised position
X = x/L; molten zone length,
z; and normalised molten zone length,
Z = z/L, the sample can be split into three regions: 0 ≤
X ≤ 1 −
Z, 1 −
Z ≤
X < 1, and
X = 1 [
15]. For a single-pass system, the impurity distribution in the 0 ≤
X ≤ 1 −
Z region can be calculated by:
where
CS(
X) is the concentration of the impurity at position
X and
C0 is the initial impurity concentration [
16]. In the 1 −
Z ≤
X < 1 region, no new material is added to the molten zone and the impurity distribution is calculated by [
15]
In the region X = 1, for k < 1, CS(X) approaches infinity; for k = 1, CS(X) = C0; and for k > 1, CS(X) = 0.
However, realistic systems are not in equilibrium, and so the segregation coefficient, k, must be replaced with an effective segregation coefficient,
keff. This can be calculated through the Burton–Prim–Slichter (BPS) theory [
23,
24]:
where
V is the velocity of the molten zone;
δ is the diffusion layer thickness at the solidification interface; and
D is the impurity diffusion coefficient in the liquid. Thus, the separation efficiency of zone refining is positively correlated with increasing impurity diffusion coefficient and decreasing molten zone velocity and diffusion layer thickness.
For a multi-pass, 1D, non-equilibrium system, the sample is broken up into four regions:
X = 0, 0 <
X < 1 –
Z, 1 −
Z ≤
X < 1, and
X = 1. The impurity concentration at
X = 0 after n passes of the heater,
CSn(
X), is given by
where
dx is an individual element of length;
M1 is the control volume, equal in length to
Z; and
q is the index of the dx elements. In the 0 <
X < 1 −
Z region, the impurity distribution can be calculated by [
25]
In this region, the molten zone length is constant and re-solidification of the salt occurs on the left-hand side of the molten zone. In the last zone length section, 1 −
Z ≤
X < 1, the heater begins to travel past the end of the sample and so no further material is added to the molten zone as re-solidification continues to occur on the left-hand side of the molten zone. In this region, the impurity distribution is given by [
26]
where
dX is the displacement between each calculated data point. The impurity concentration where the molten zone fully passes the sample (
X = 1) is calculated by [
26]
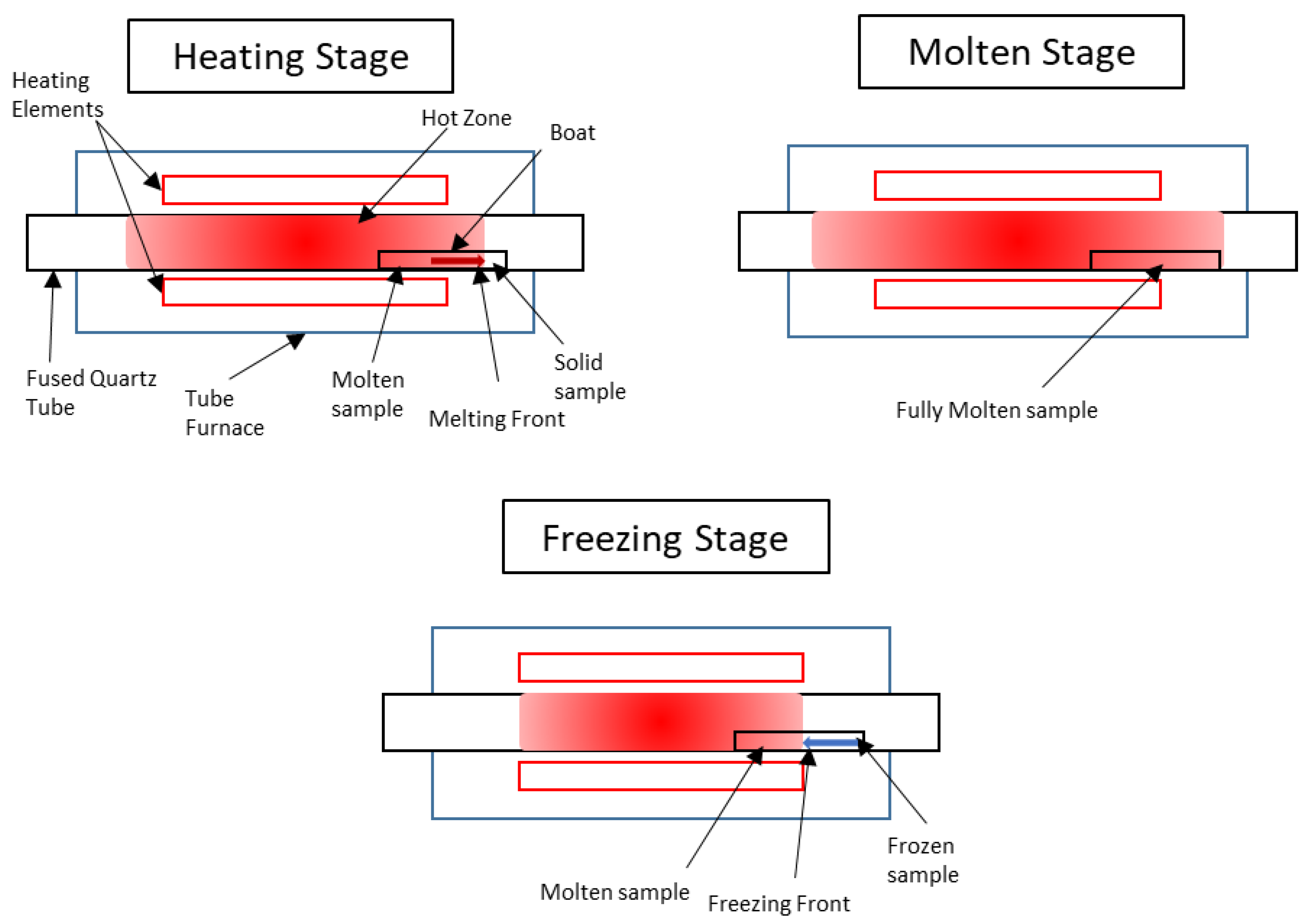
In this work, benchtop apparatus was used to conduct zone-refinement procedures using simulated electrorefiner and electroreducer wastes. These experiments were performed to demonstrate the feasibility of this zone-refining technique for the clean-up of electroreducing and electrorefining wastes. Comparing the experimental data with the theoretical behaviours described through Equations (1)–(8) will illustrate the effective segregation coefficient,
keff, as a measure for the degree of separation in the sample that was achieved. By demonstrating the suitability of laboratory-scale instrumentation, more progress can be made towards resolving the issues of appropriate waste management [
27] for the nuclear sector, enabling more widespread use of nuclear power stations.
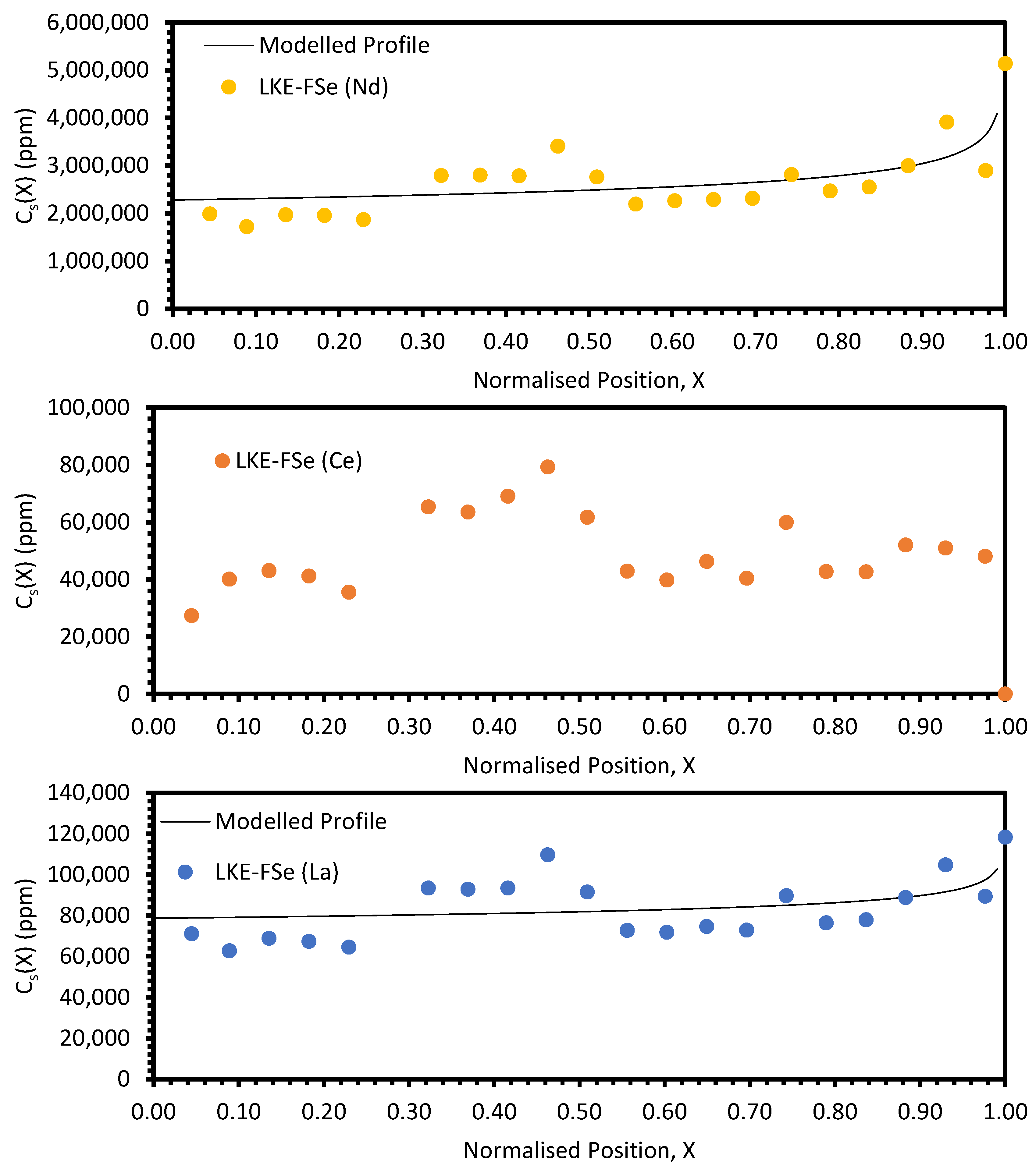
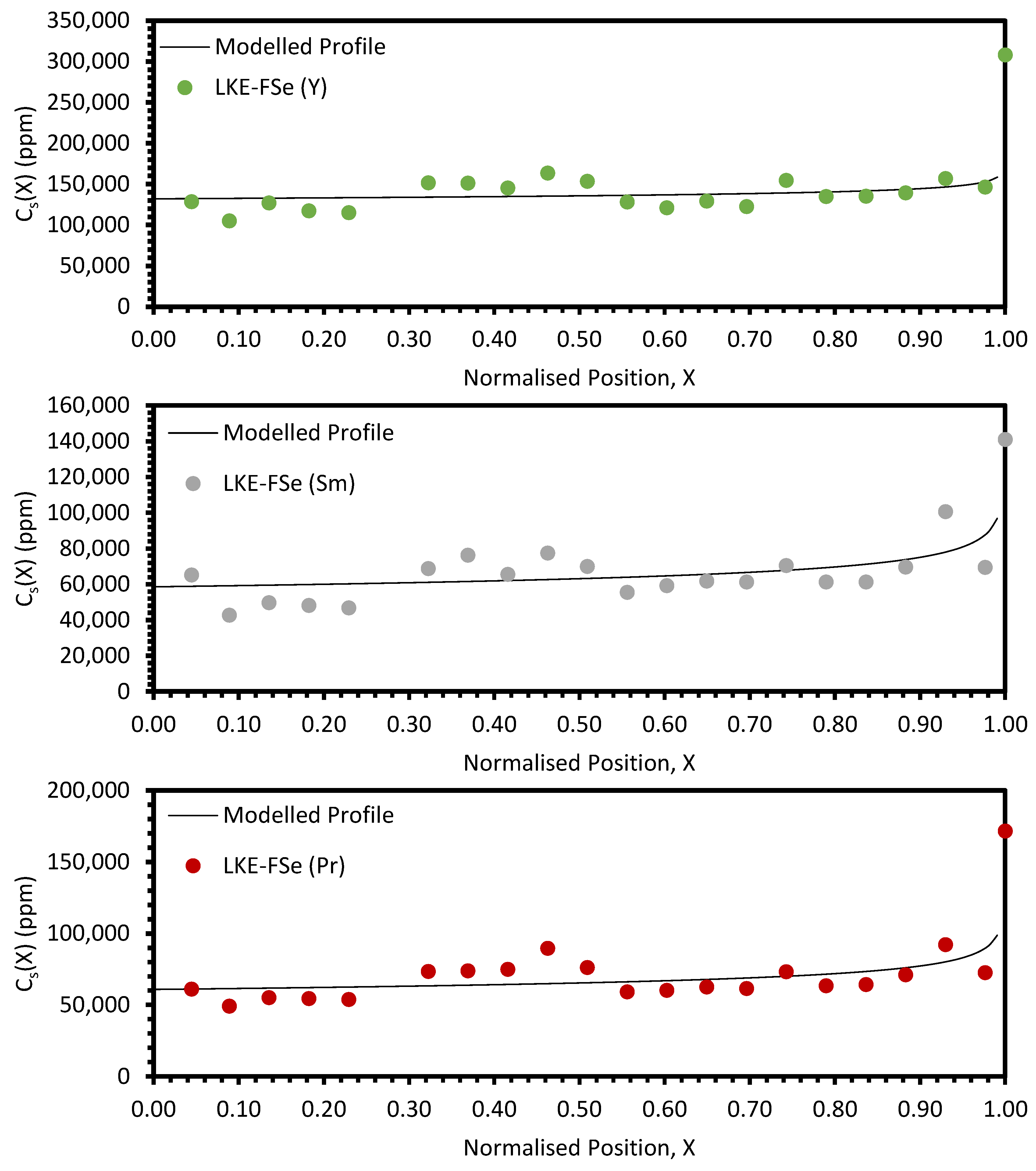
4. Discussion
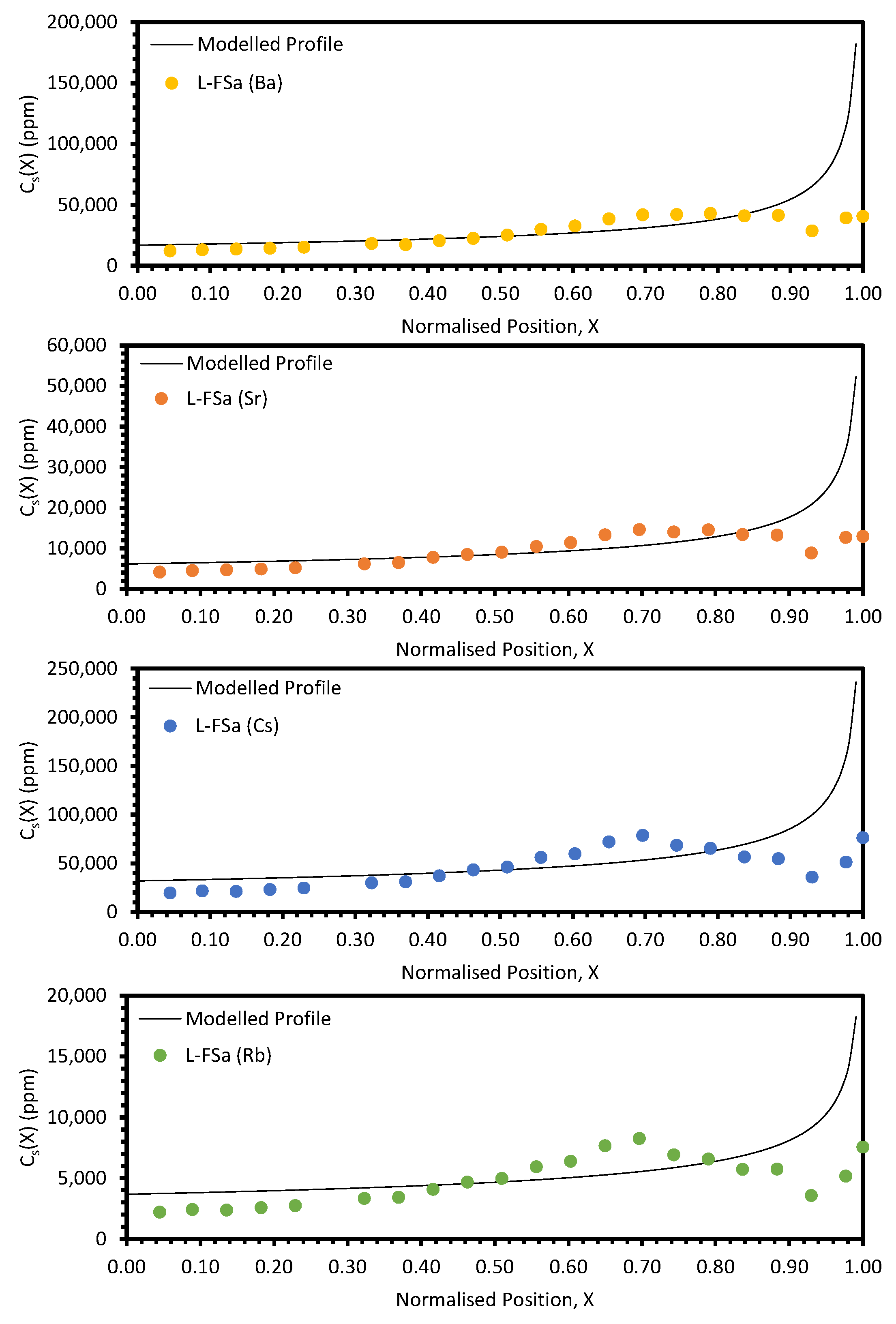
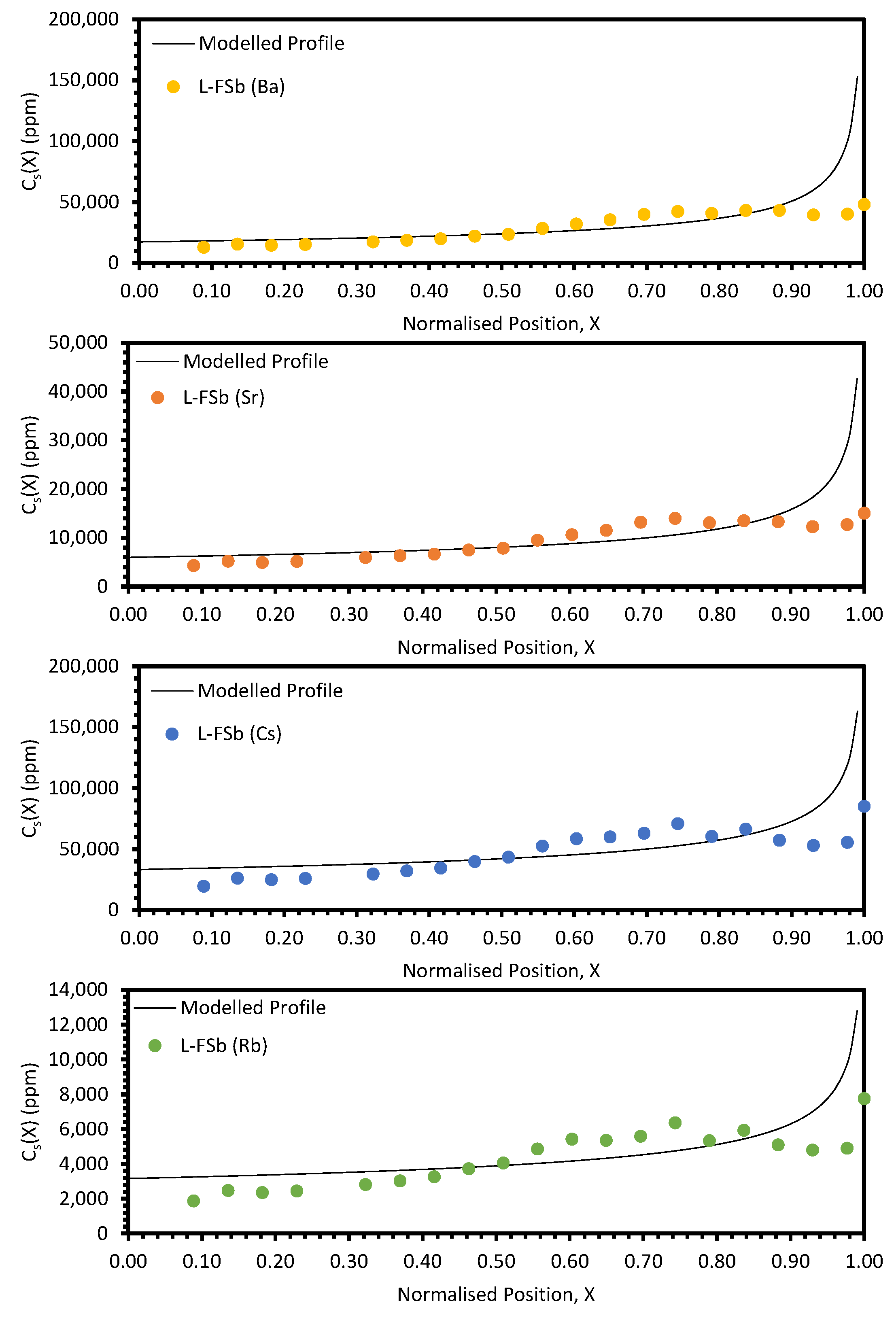
The approach taken in this work regarding the
keff values generated by fitting with Equation (10) was found to be unsuitable when considering the electroreducing wastes, as shown in
Figure 11 and
Figure 12. As such, further discussions will focus on the electrorefining simulant zone-refining efforts.
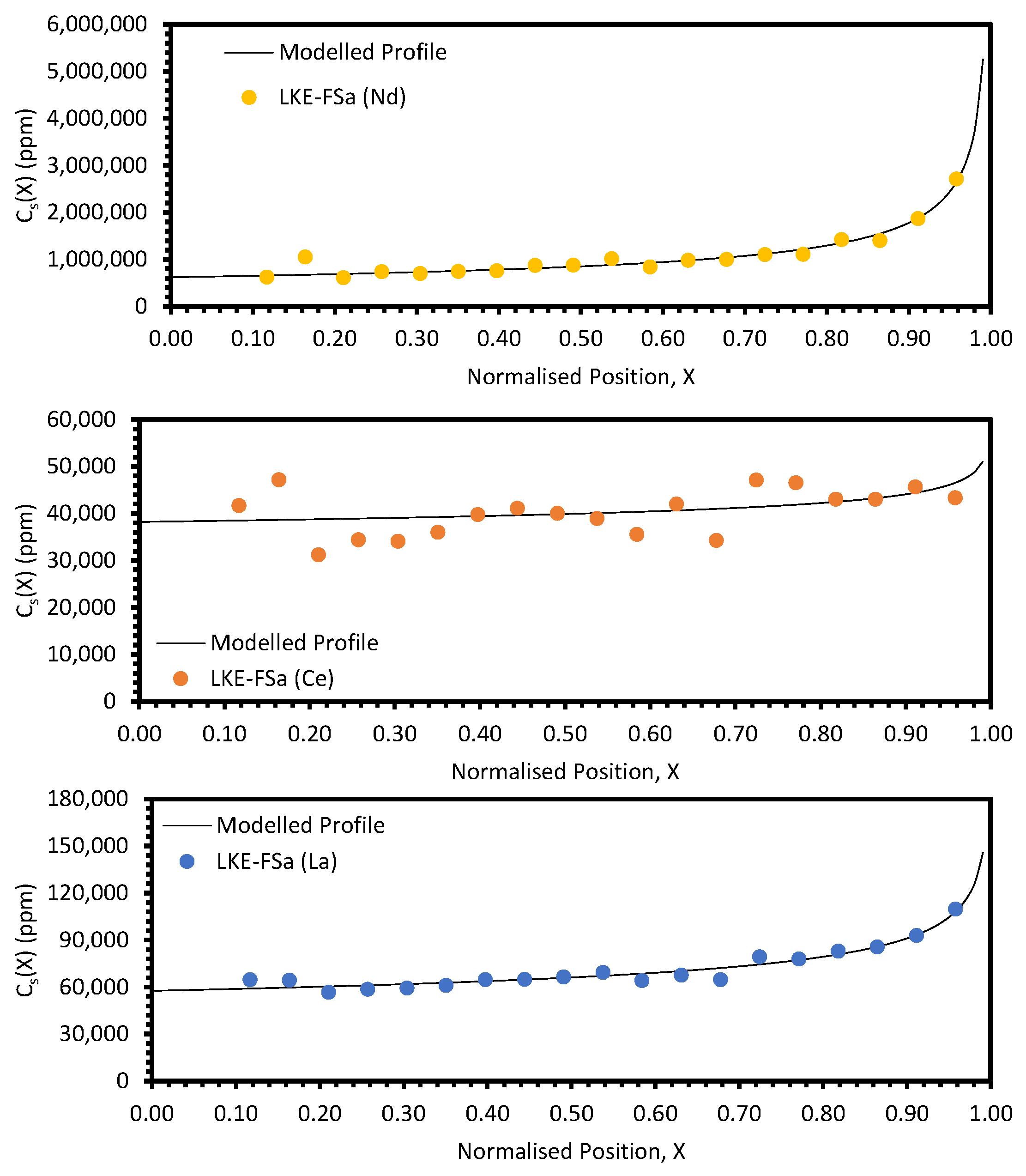
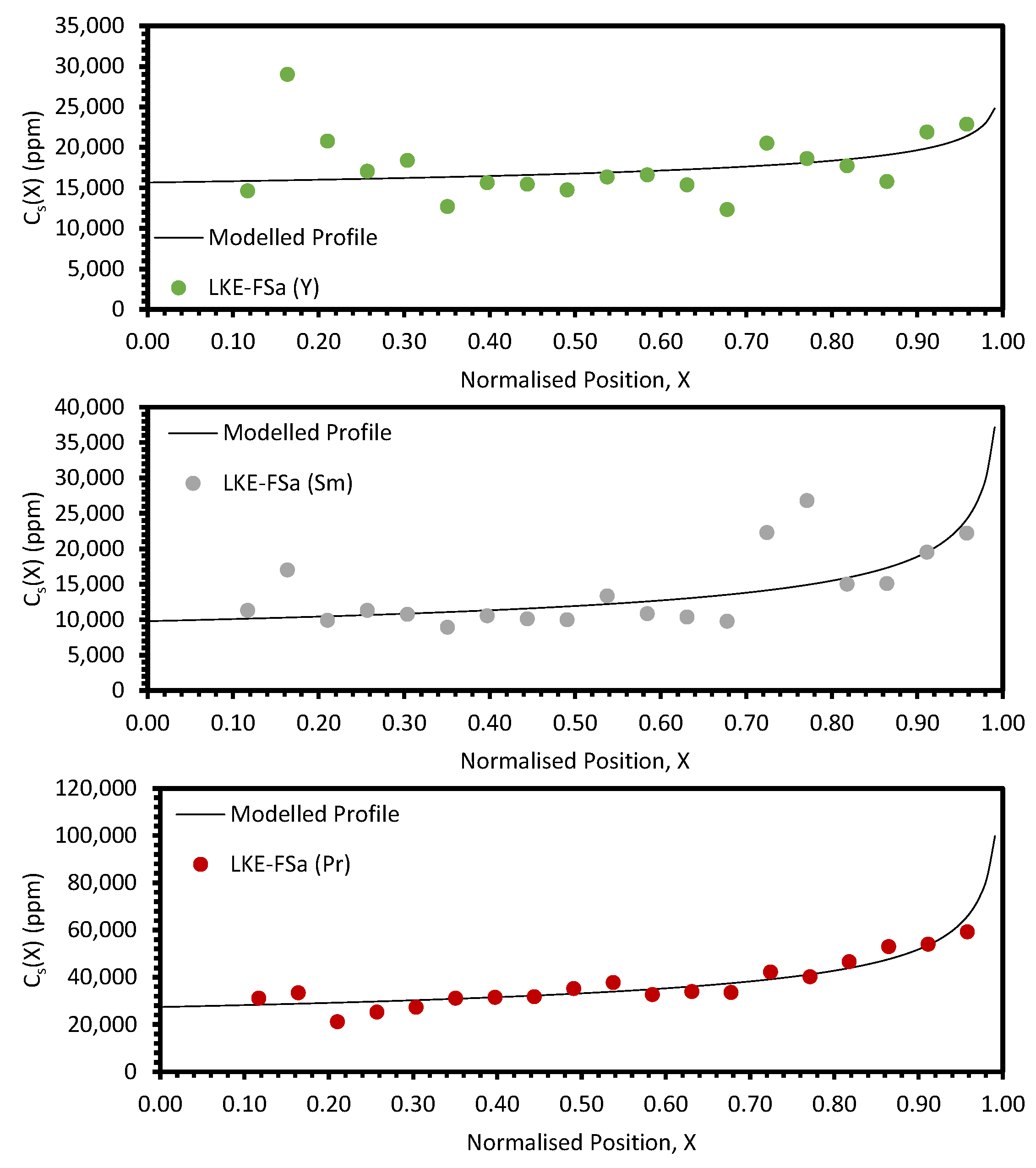
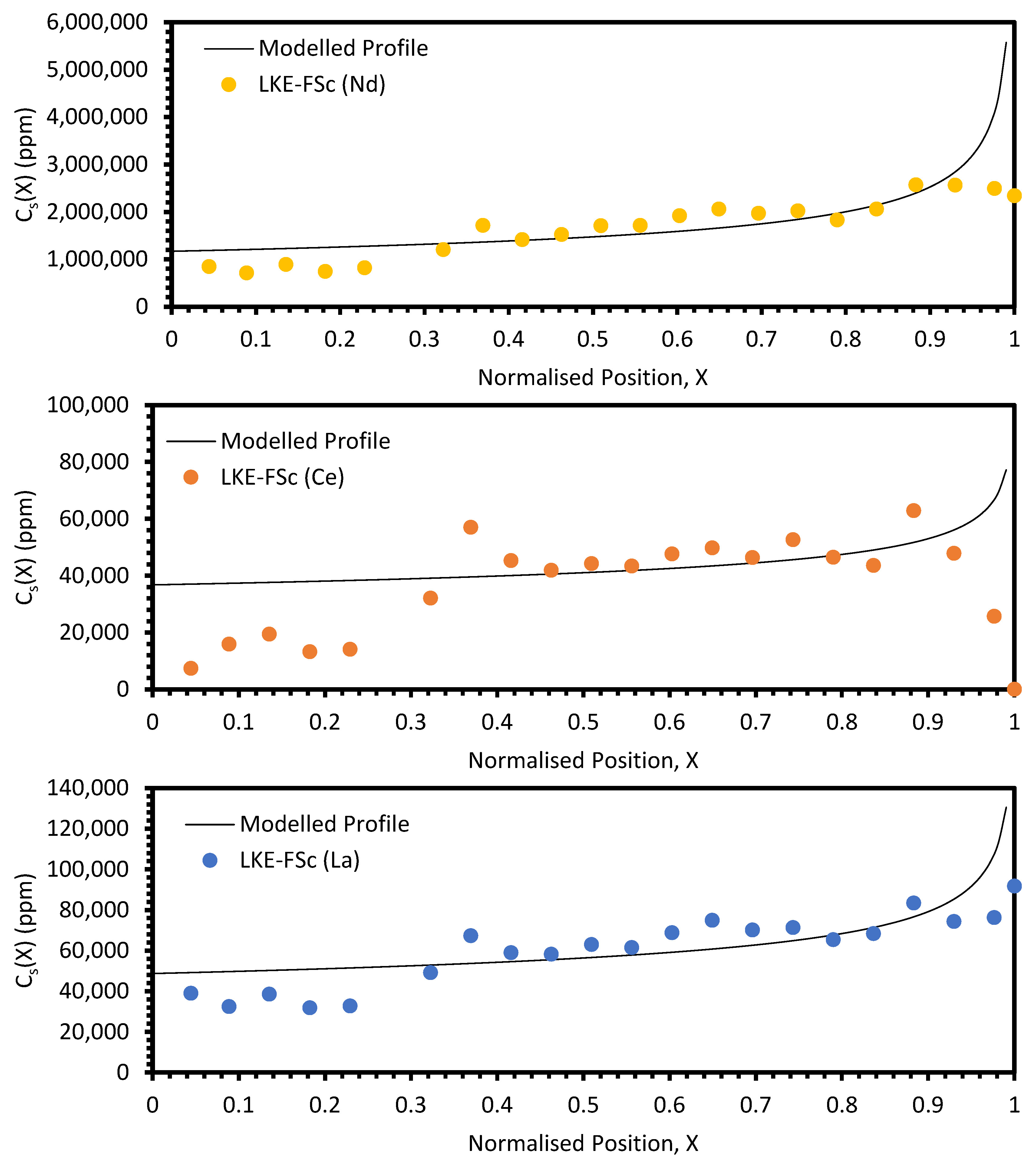
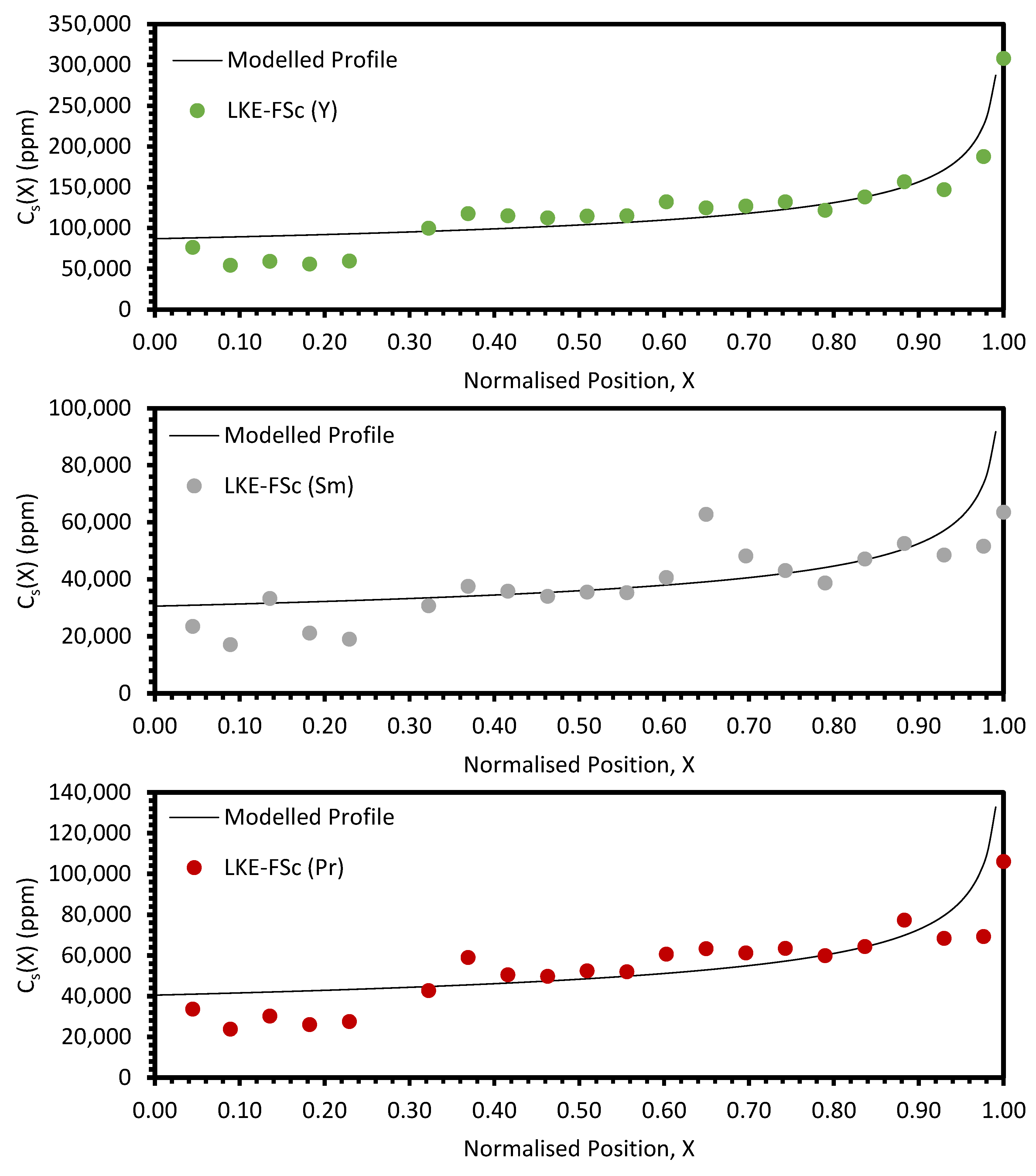
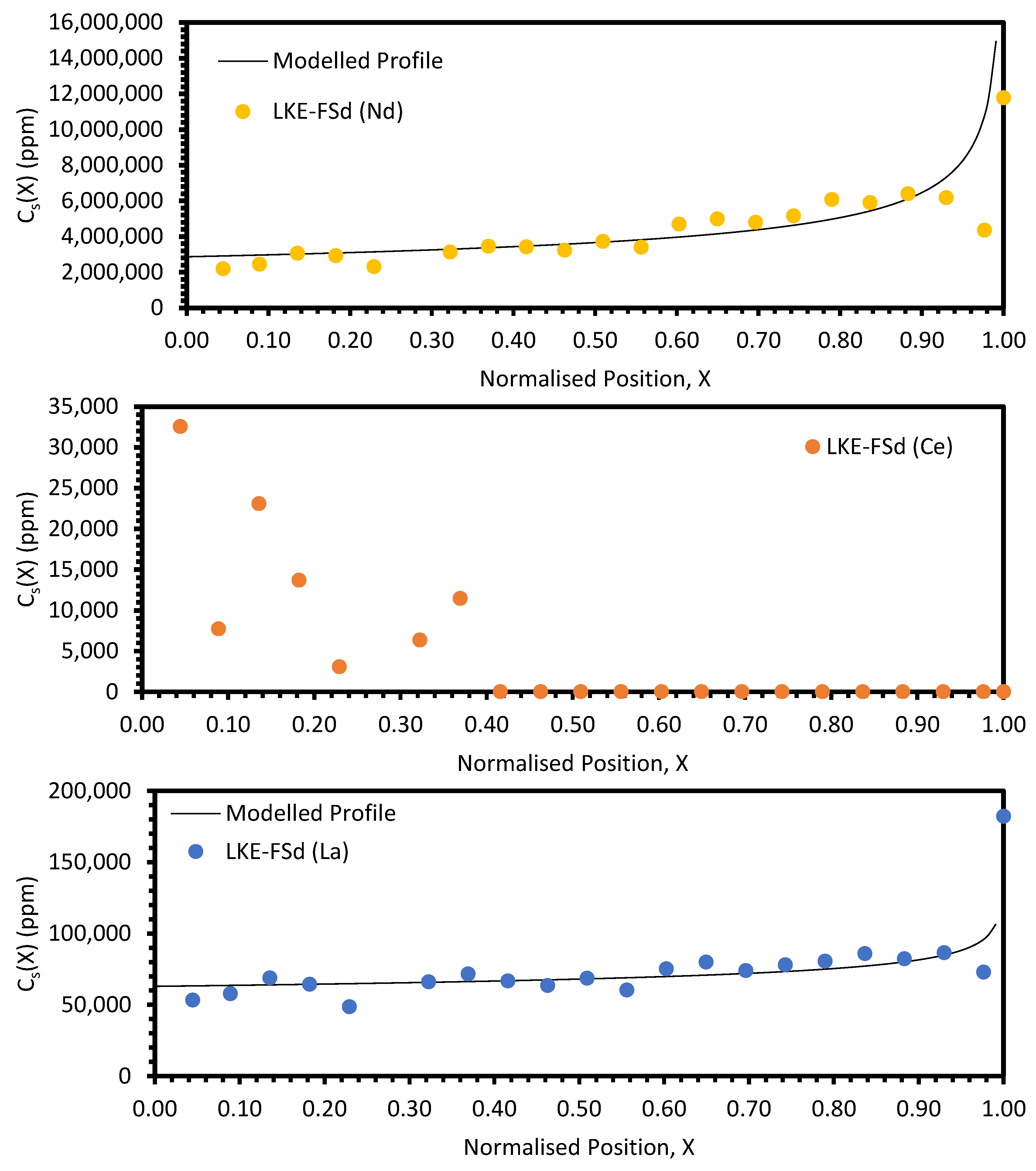
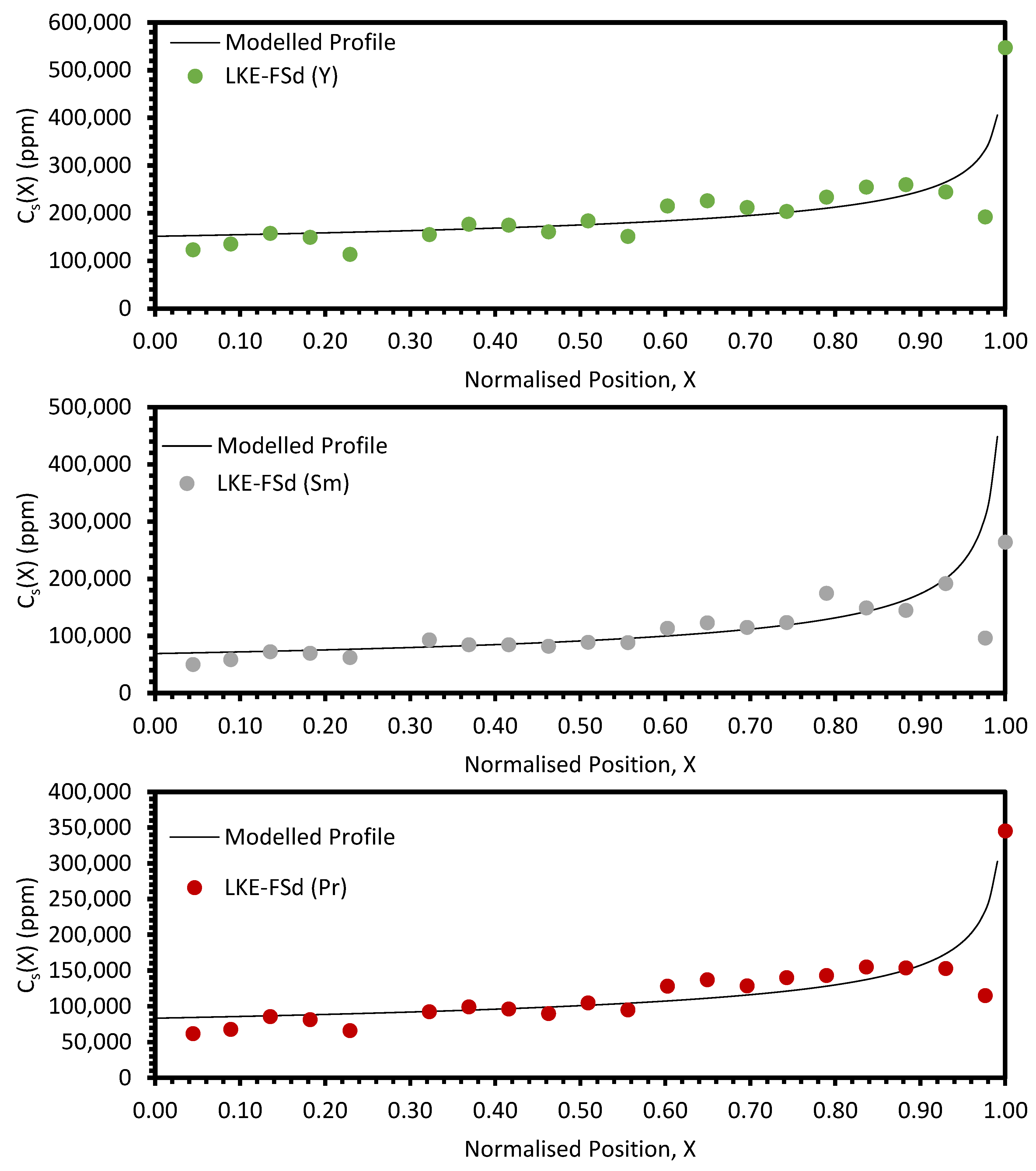
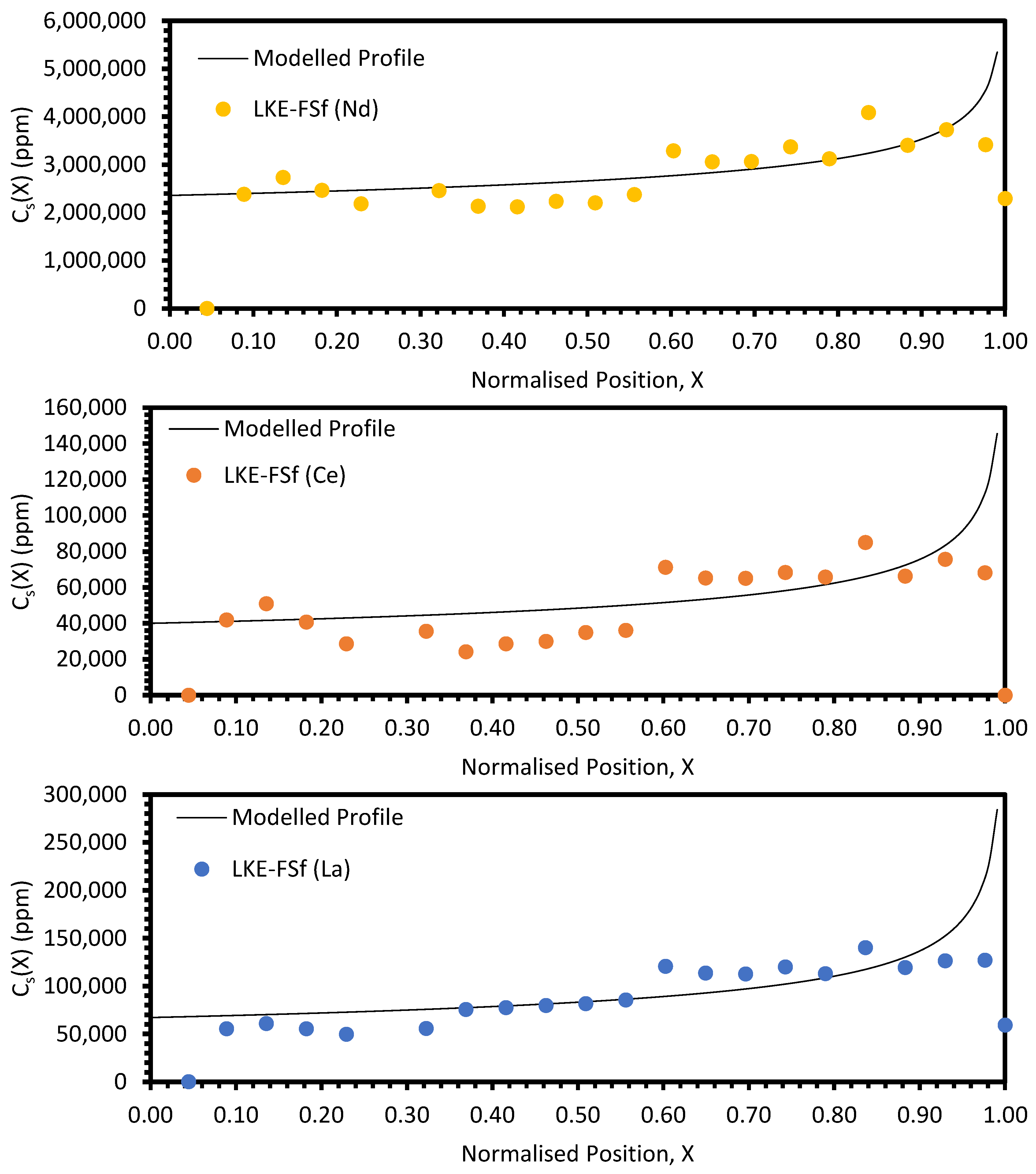
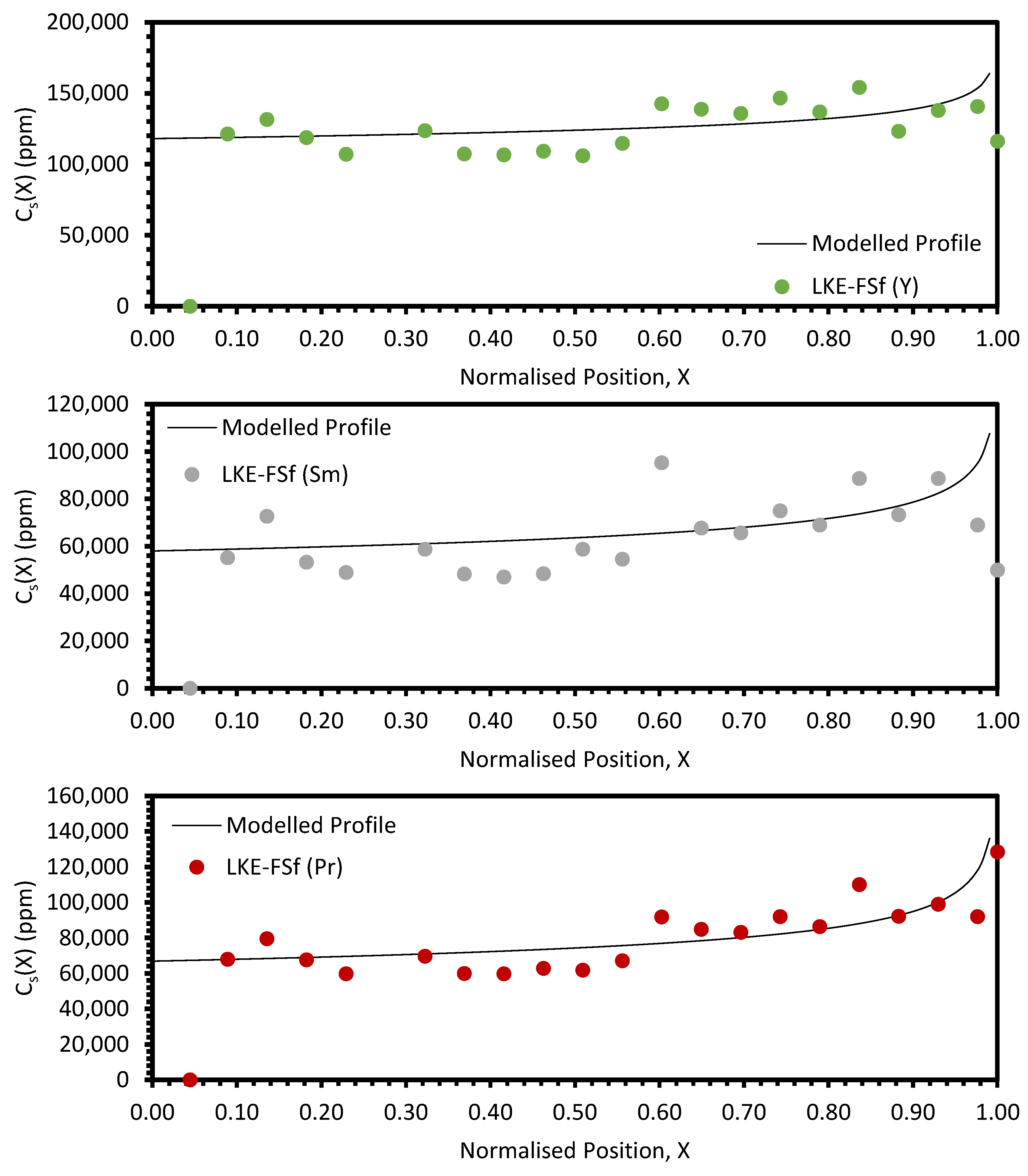
In the electrorefining simulant zone-refining data, the elemental concentration profiles for the RE elements show that some degree of zone refinement was achieved in all five analysed samples (due to volatilisation, ICP-OES analysis of the LKE-FSb sample was not possible). Using the calculated elemental concentration profiles for each element in each sample, decontamination factors and waste compositions can be calculated. The decontamination factor,
DFi, for a given element,
i, can be calculated by
where
XR is a predetermined ‘refinement boundary’. For 0 ≤
X <
XR, the base salt has been refined to remove some of the impurities, and for
XR ≤
X < 1, the impurities have been concentrated to produce a waste material. The average concentrations are calculated by evaluating the definite integral across the two intervals (0 ≤
X <
XR and
XR ≤
X < 1) and dividing by the interval width:
X = 0.99999 is taken to be a reasonable estimate of
X = 1, as there is a singularity in C
S(X) at
X = 1.
DFi values for
XR = 0.8 and 0.9 are shown in
Table 5 and
Table 6, respectively. In general, the minimum enrichment of an RE in the ‘waste’ portion of the sample for
XR = 0.8 is 8% (Y, LKE-FSe) and the maximum is 286% (Nd, LKE-FSa). The equivalent values for
XR = 0.9 are 11% (Y, LKE-FSe) and 360% (Nd, LKE-FSa).
Based on the average elemental concentrations calculated through Equation (13), the composition of the refined salt and the waste salt can be estimated. Li, K, and Cl are assumed to remain in the same proportions throughout each sample. The estimated compositions of the refined and waste salts for
XR = 0.8 and 0.9 are shown in
Table 7,
Table 8,
Table 9 and
Table 10. Due to the variation in
DFi between the REs, their relative proportions vary between the samples, but an estimate of the overall compositional change can be made using the total wt.% of RECl
3. The total RECl
3 content of the waste salt was between 11.4 and 16.2 wt.% for
XR = 0.8, and between 12.1 and 20.3 wt.% for
XR = 0.9. These contaminant-enriched waste salts could be sent for immobilisation, or for further processing, e.g., phosphate precipitation of the RE ions from the chloride salt.
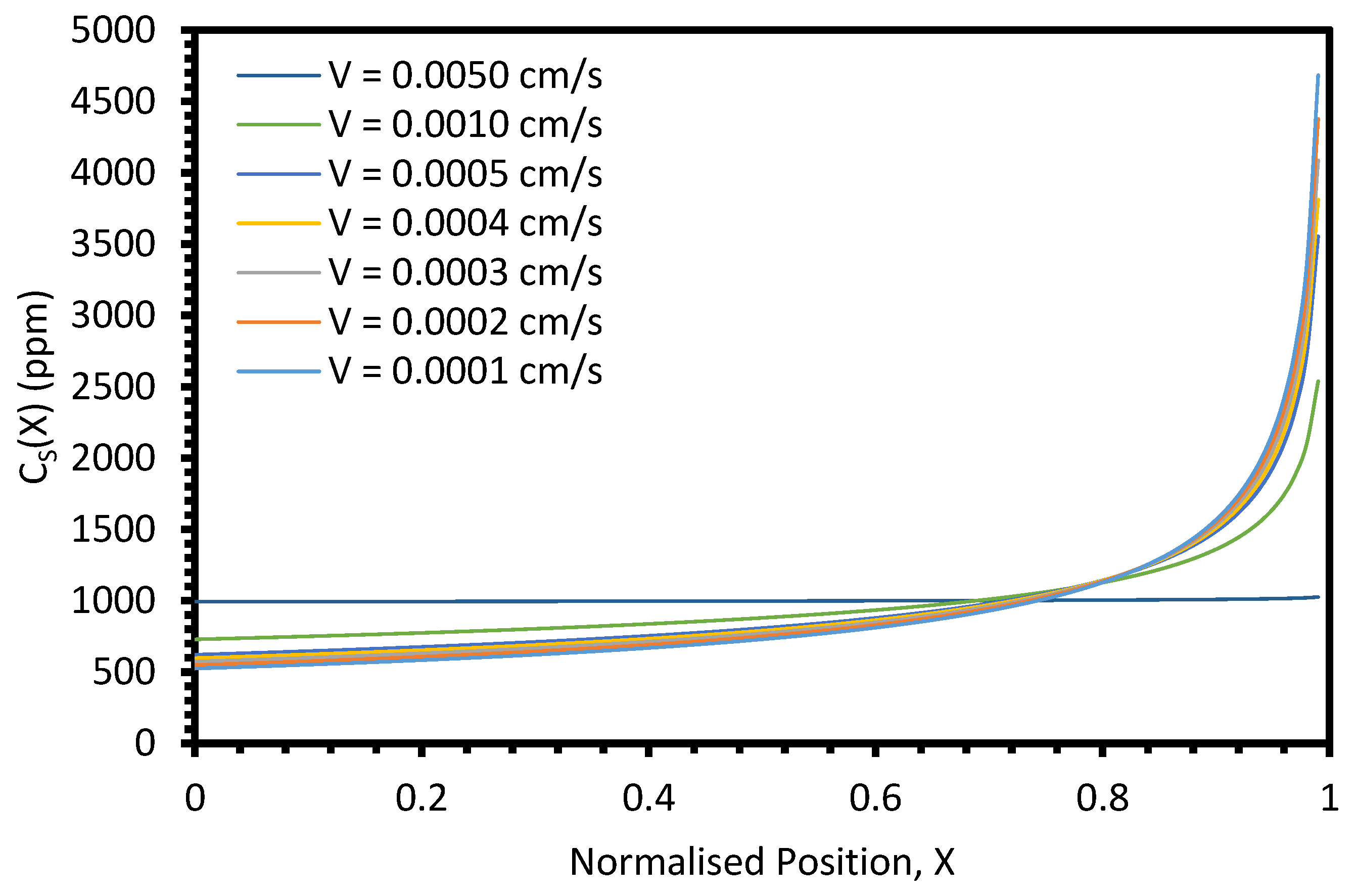
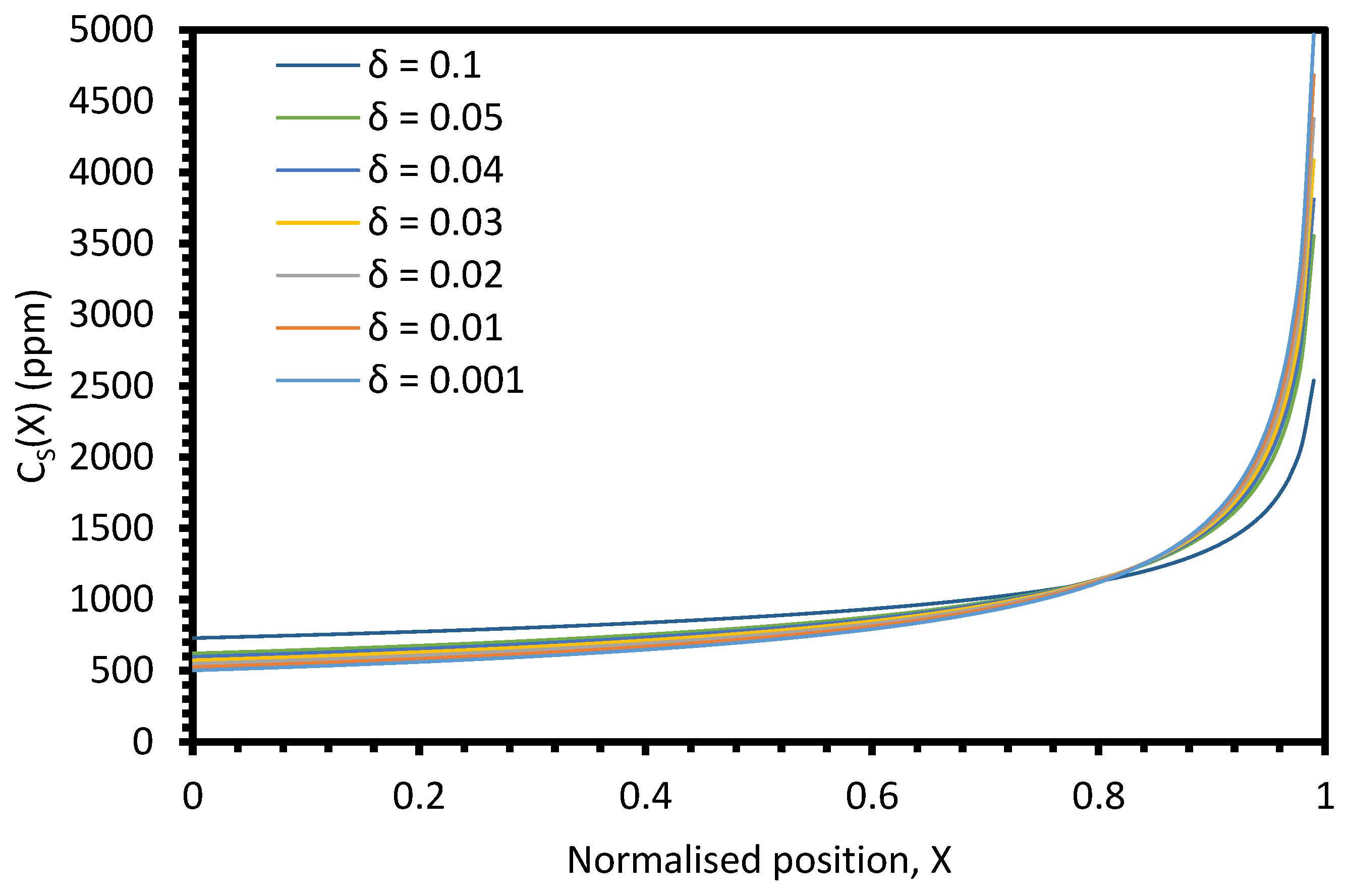
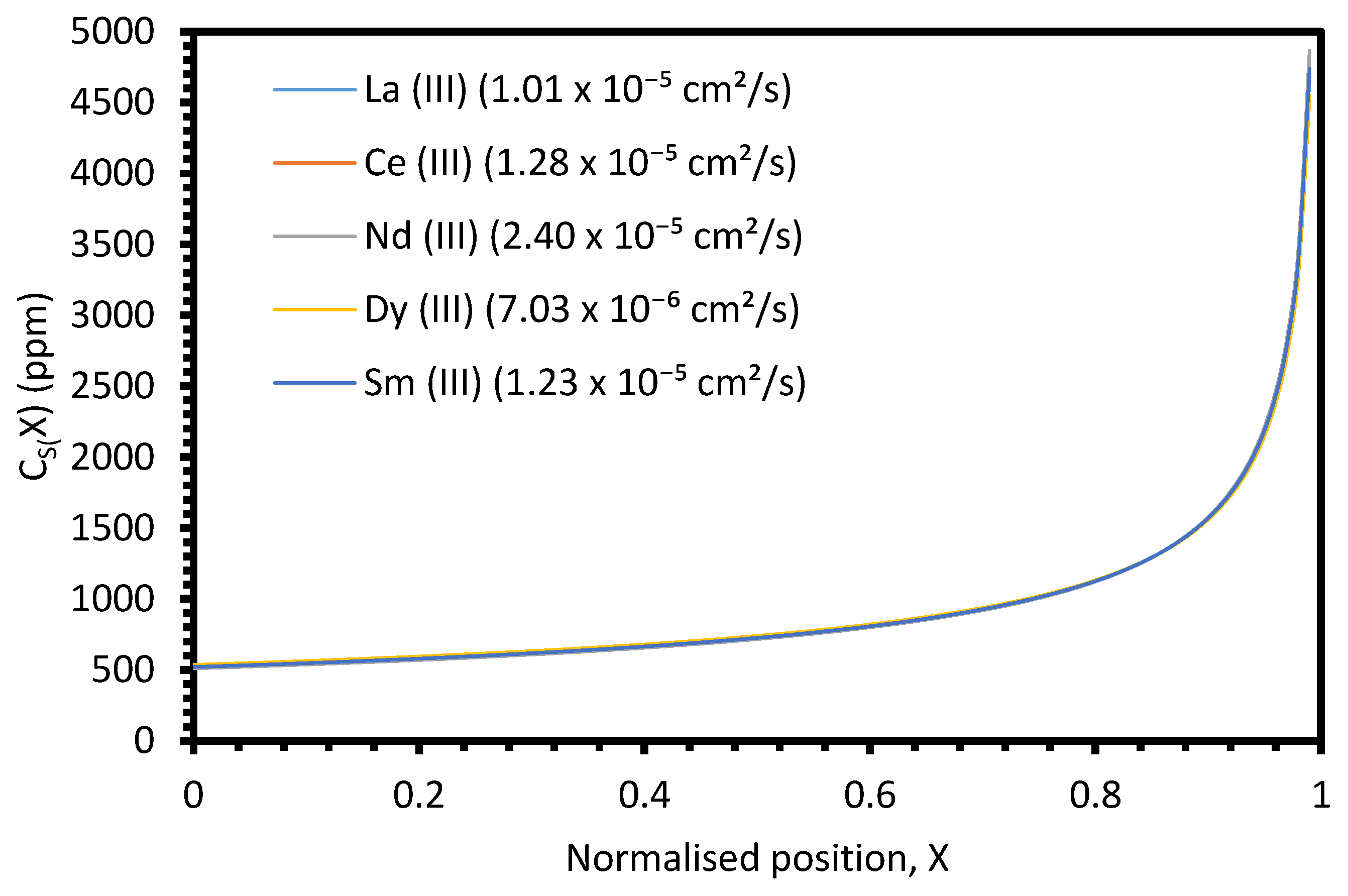
The primary parameter varied between the six electrorefining simulant tests was the cooling rate of the furnace, and hence, the velocity of the freezing front,
V. Based on Equation (4), we would expect
keff to decrease with decreasing
V, assuming
k,
δ, and
D remain constant. For the example system described in
Figure A9 (
k = 0.5,
L = 100 cm,
Z = 1,
C0 = 1000 ppm,
δ = 0.01 cm,
D = 1.01 × 10
−5 cm
2/s), the
keff value would be expected to decrease from 0.800 to 0.688 as
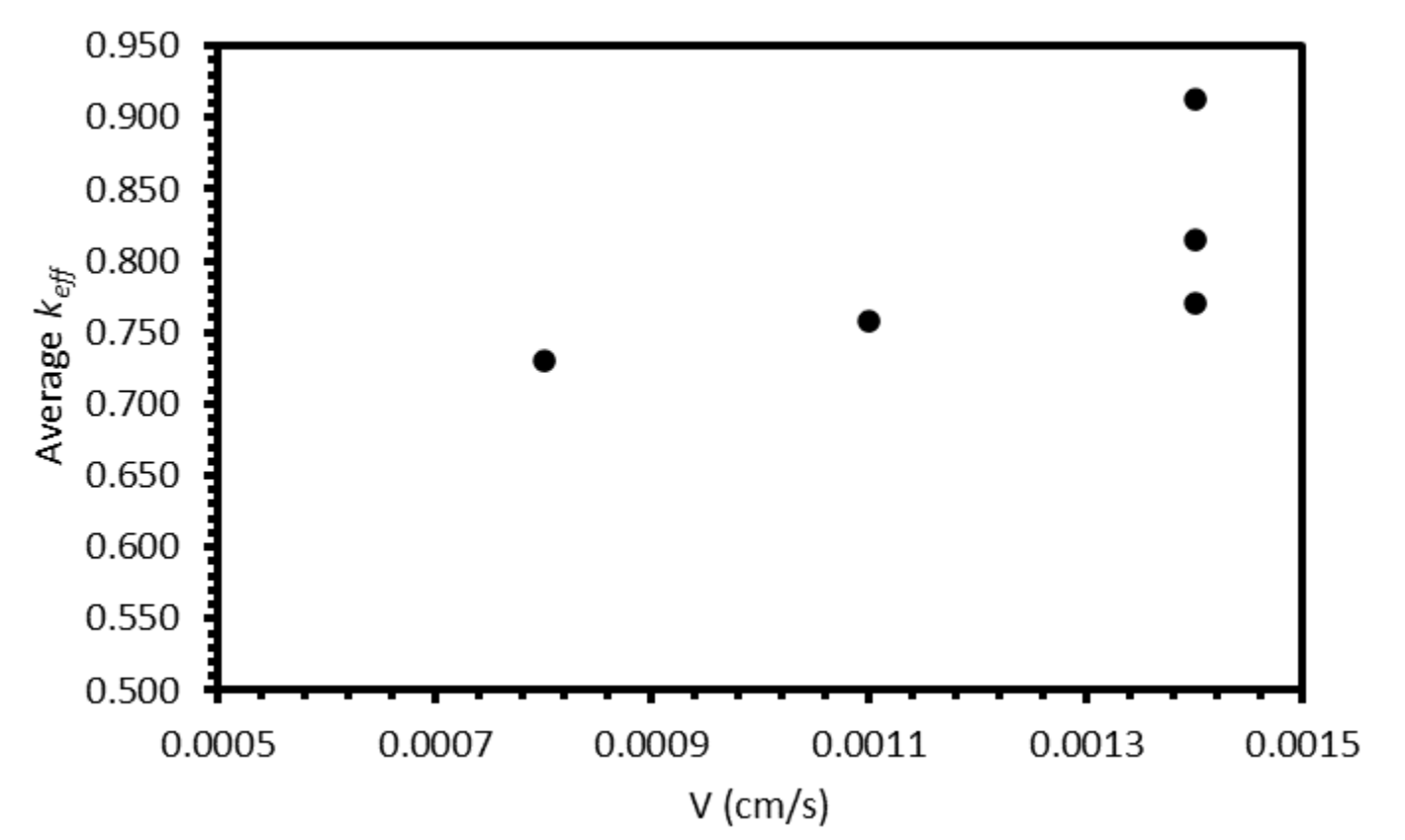
V decreases from 0.0014 to 0.0008 cm/s (the velocity range of the successful experiments). However, when the average
keff values for the six elements for each test are plotted against the estimated
V, the relationship is unclear (
Figure 13). Whilst there may generally be a slight decrease in
keff with
V, the range of the three
keff values at 0.0014 cm/s (0.144) is greater than the difference between
keff at 0.0008 cm/s (0.730) and the average of the three values at 0.0014 cm/s (0.833). Further experiments at a range of
V values, with duplicate or triplicate data, are required to determine whether the relationship is present.
Local maxima in the distribution profiles around the normalised position 0.6 to 0.8, presented in
Figure A7 and
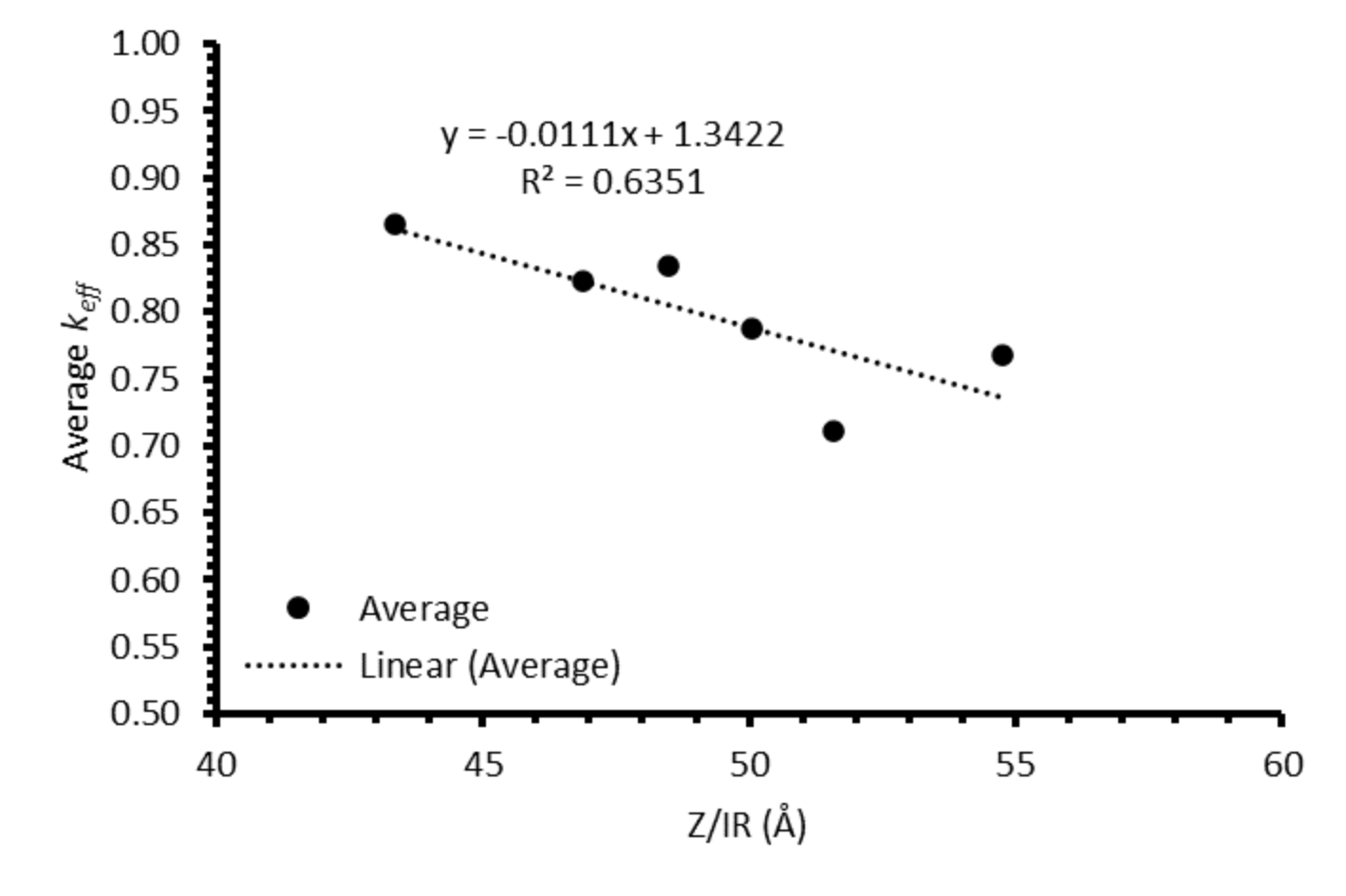
Figure A8, are indicative of incomplete refinement due to the single-pass experimental setup used. Further zone-refinement processing and the use of multi-pass modes could improve the final distribution profiles and result in profiles more consistent with the modelled profiles. However, the variation in the refinement behaviour of the REs is of interest as this will affect the efficacy of zone refining and the composition of the waste generated. The REs are chemically similar, with all six elements present in the salt as 3+ ions. However, there is a slight difference in their coordination within their respective chlorides, with Nd, Ce, La, Sm, and Pr nine-coordinated, whilst Y is six-coordinated. Plotting the average
keff for each ion against their atomic number divided by ionic radius (
Z/
IR) appears to provide a general linear trend of increasing separation with increasing
Z/
IR, although this should be caveated with an
R2 value of 0.64 (
Figure 14). Whether there is a physical explanation for such a relationship remains unclear and would require further work to elucidate.


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

























