Surface Chemistry in Nanoscale Materials

Abstract
:1. Introduction

2. Nanoporous Gold
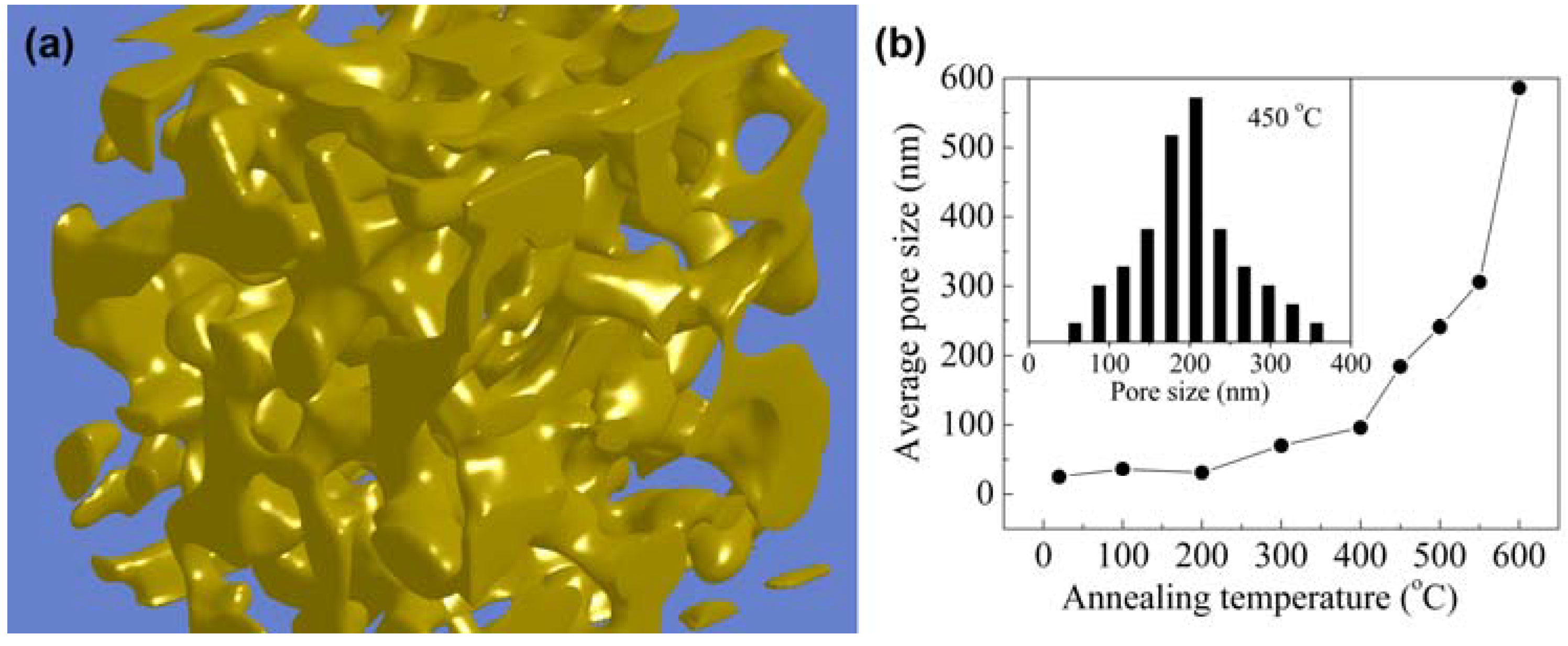
2.1. Synthesis

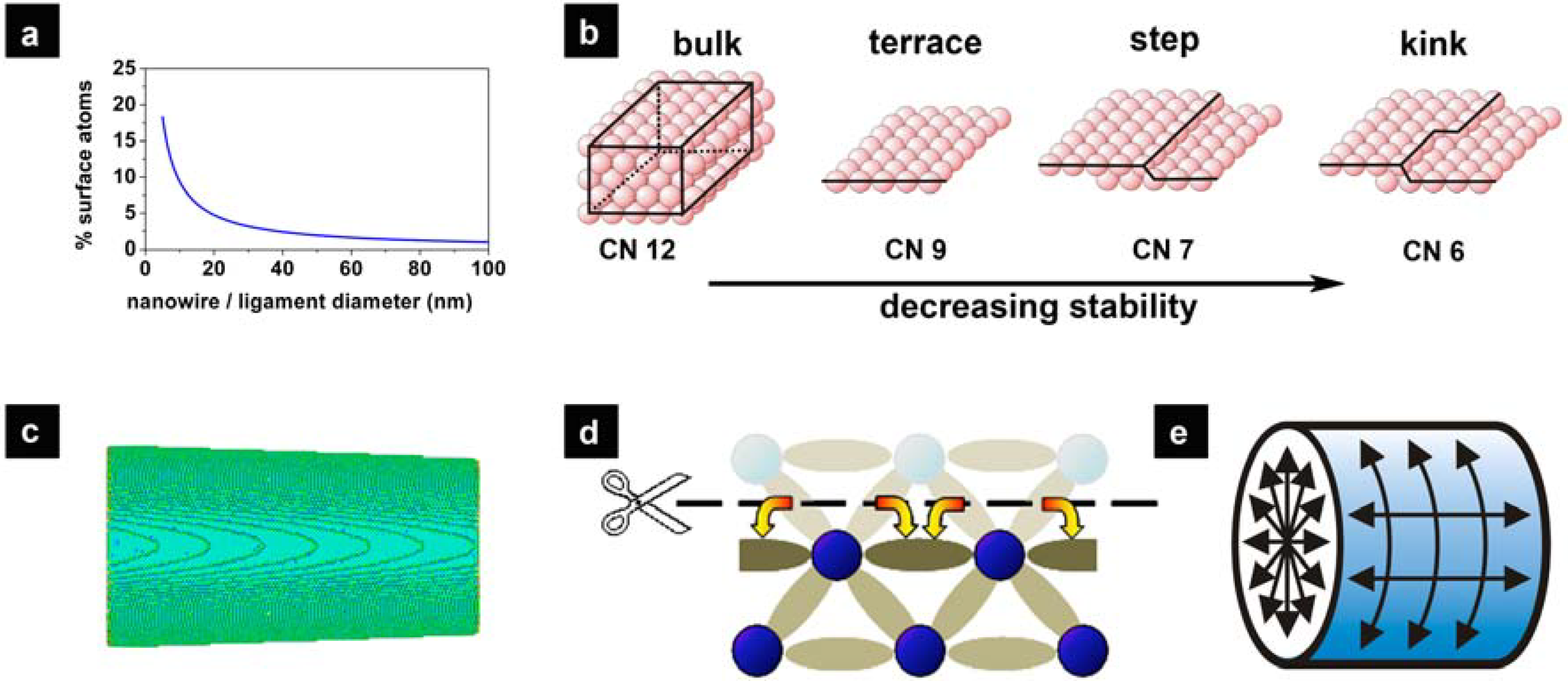
2.2. Surface Chemistry and Stability of np-Au

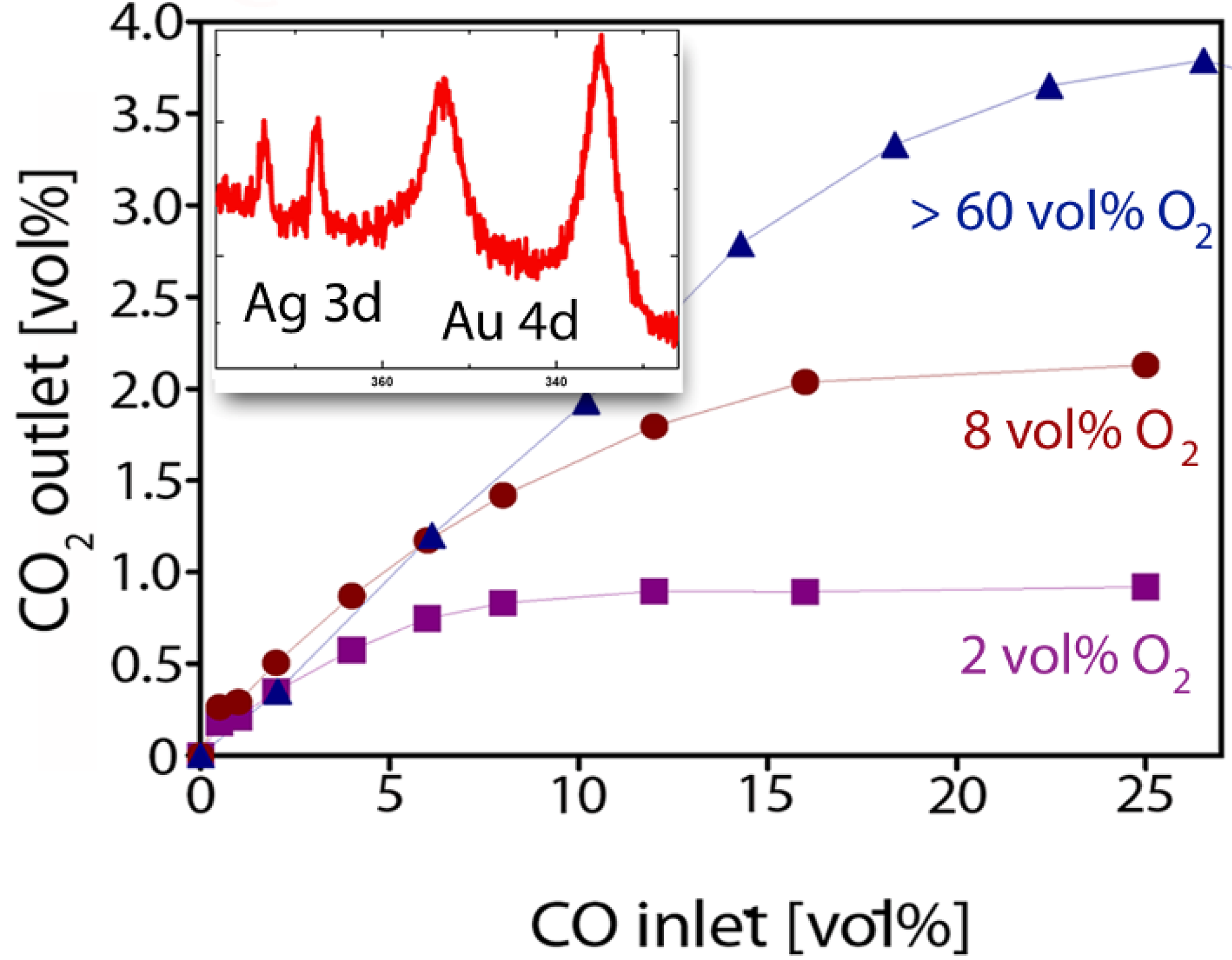
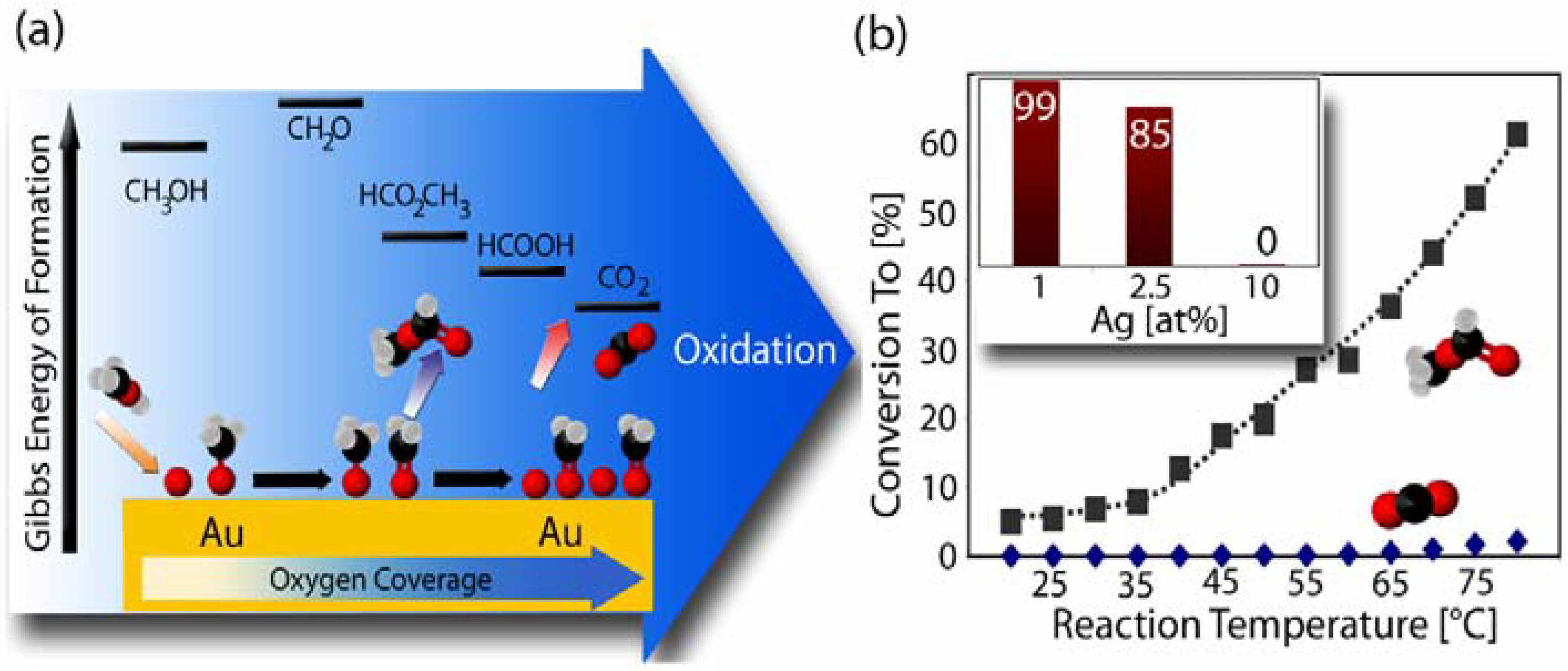
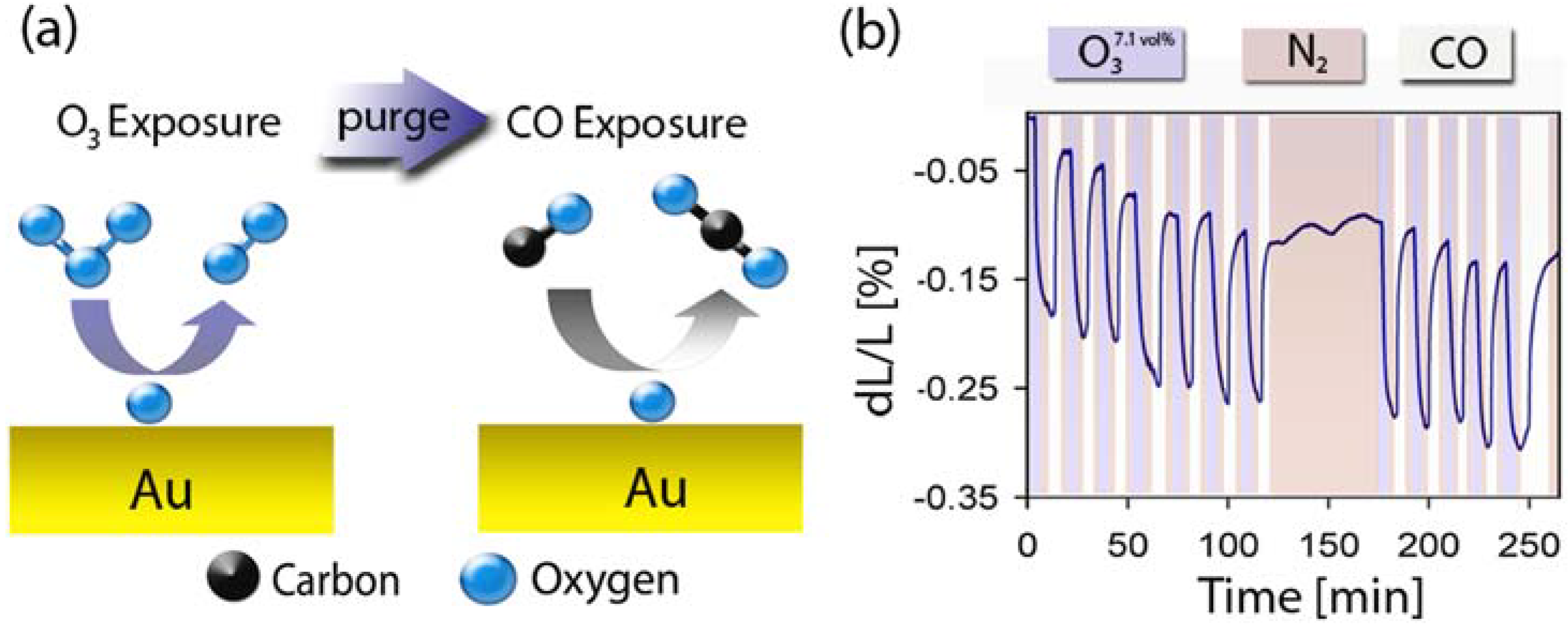
2.3. Catalytic Properties of np-Au


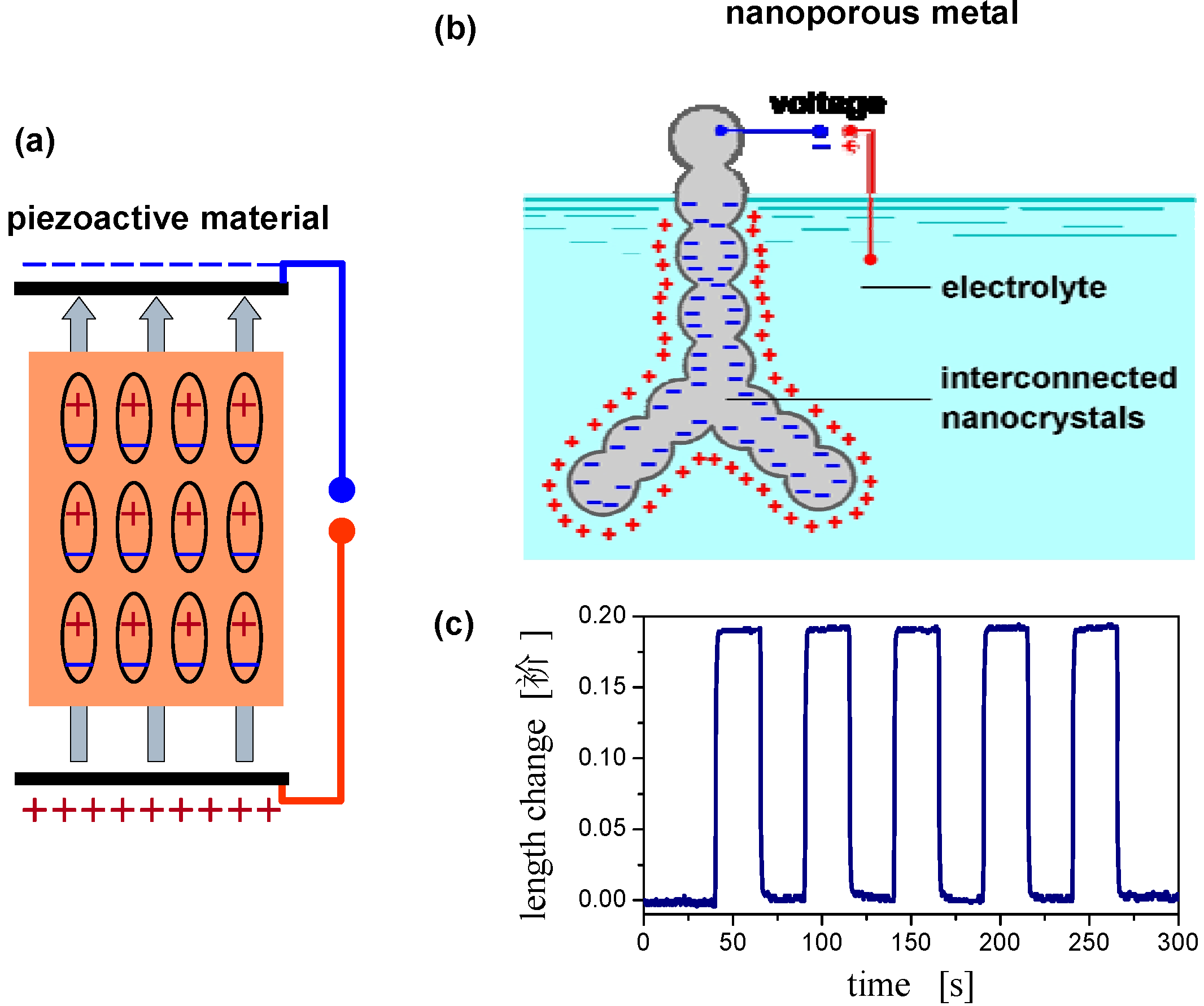
2.4. Charge-Induced Strain and Electrochemical Actuation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fluid | Solid | |
|---|---|---|
| Mechanical balance | ΔP = 2 γ κ | 〈P〉V = ⅔ α 〈f〉A |
| Geometry parameter | curvature, κ | Area per volume, α |
| ± | + | |
| Capillary parameter | surface tension, γ | Surface stress, f = dγ/de |
| + | ± | |
| Response to charging | γ = γ0 – q2 / c | f = f0 + ς q |
| – | ± |
2.5. Surface Chemistry Induced Macroscopic Strain Effects

3. Carbon Aerogels
3.1. Synthesis


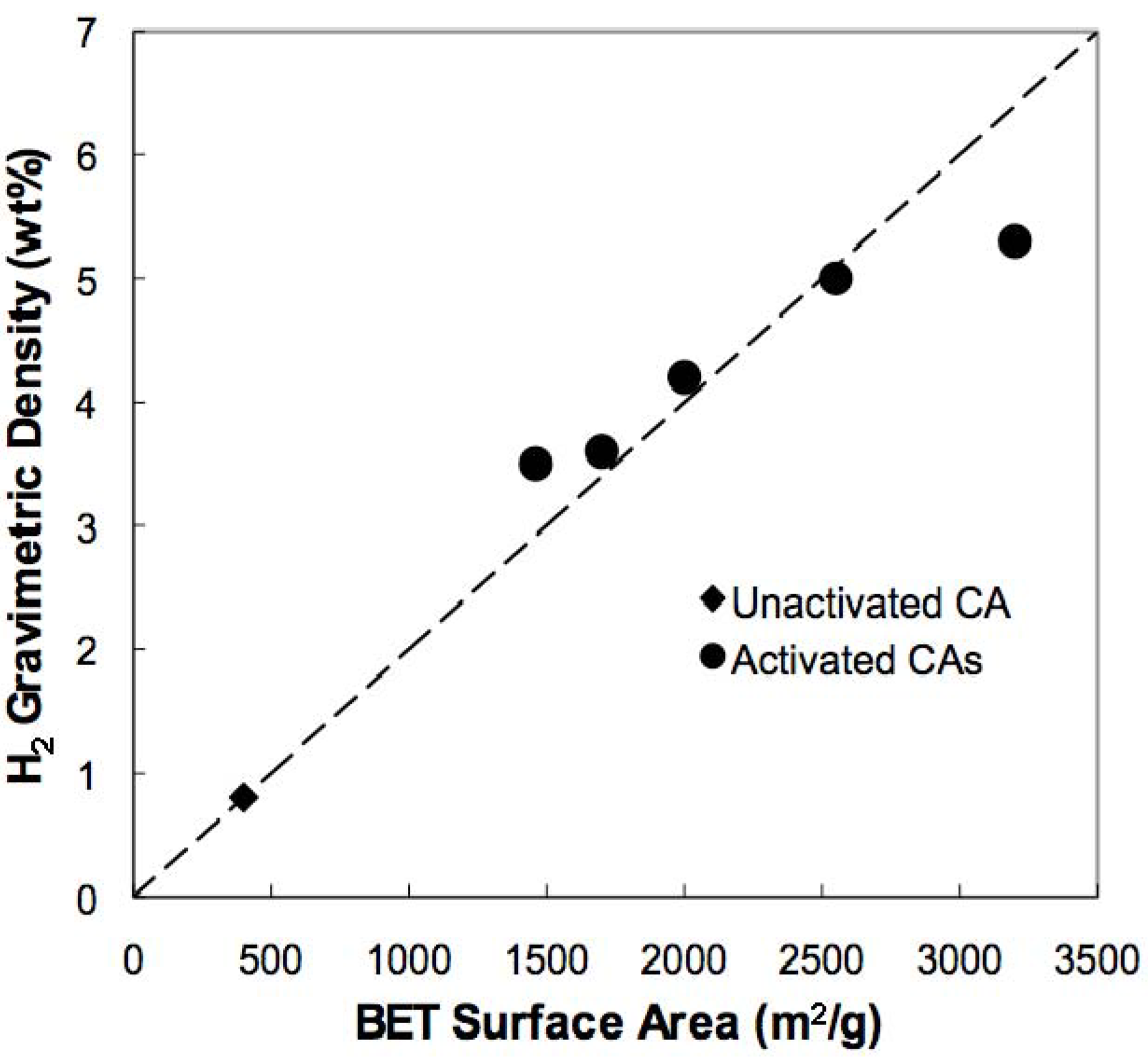
3.2. Hydrogen Storage

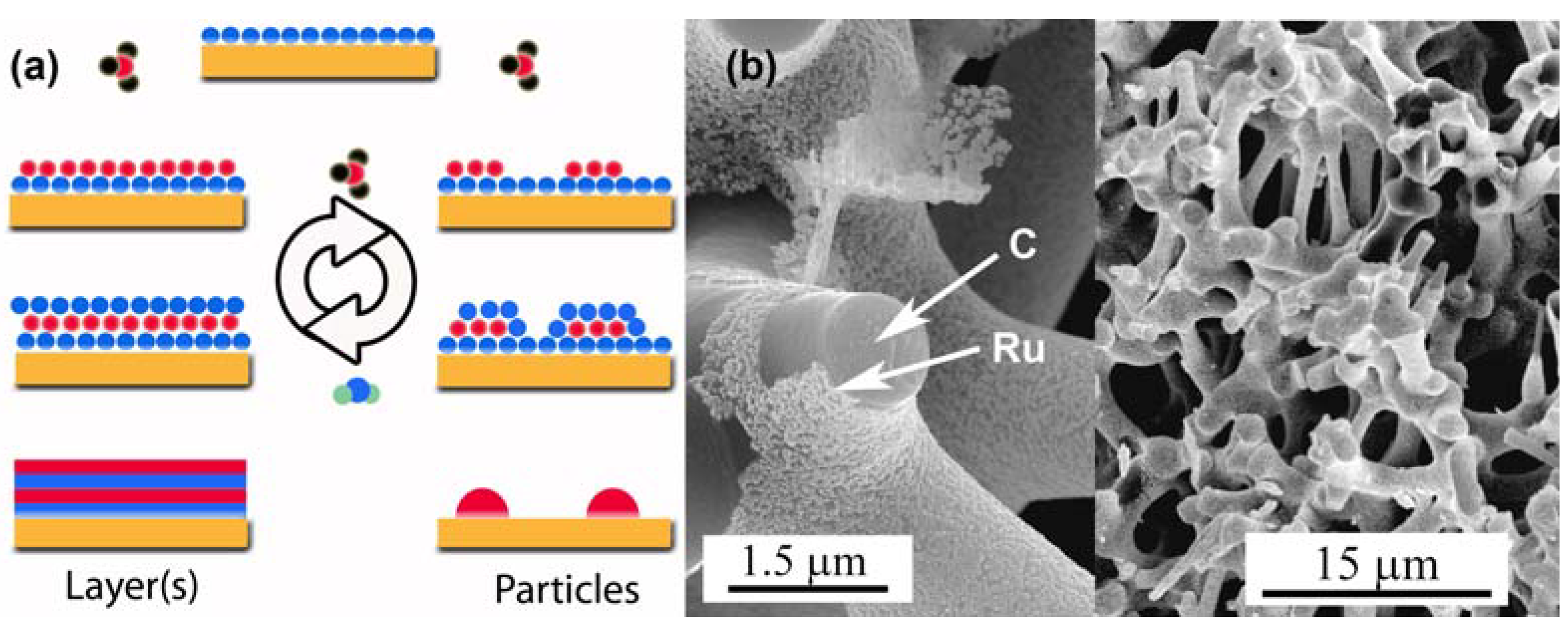
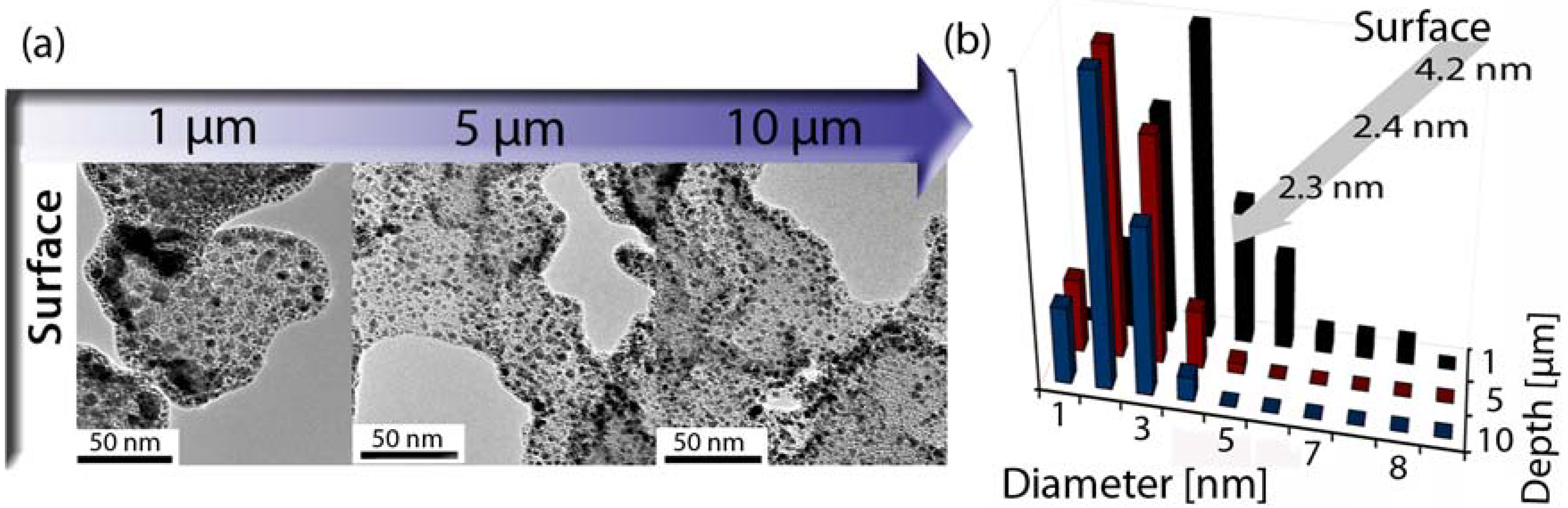
3.3. Surface Modifications Using Atomic Layer Deposition

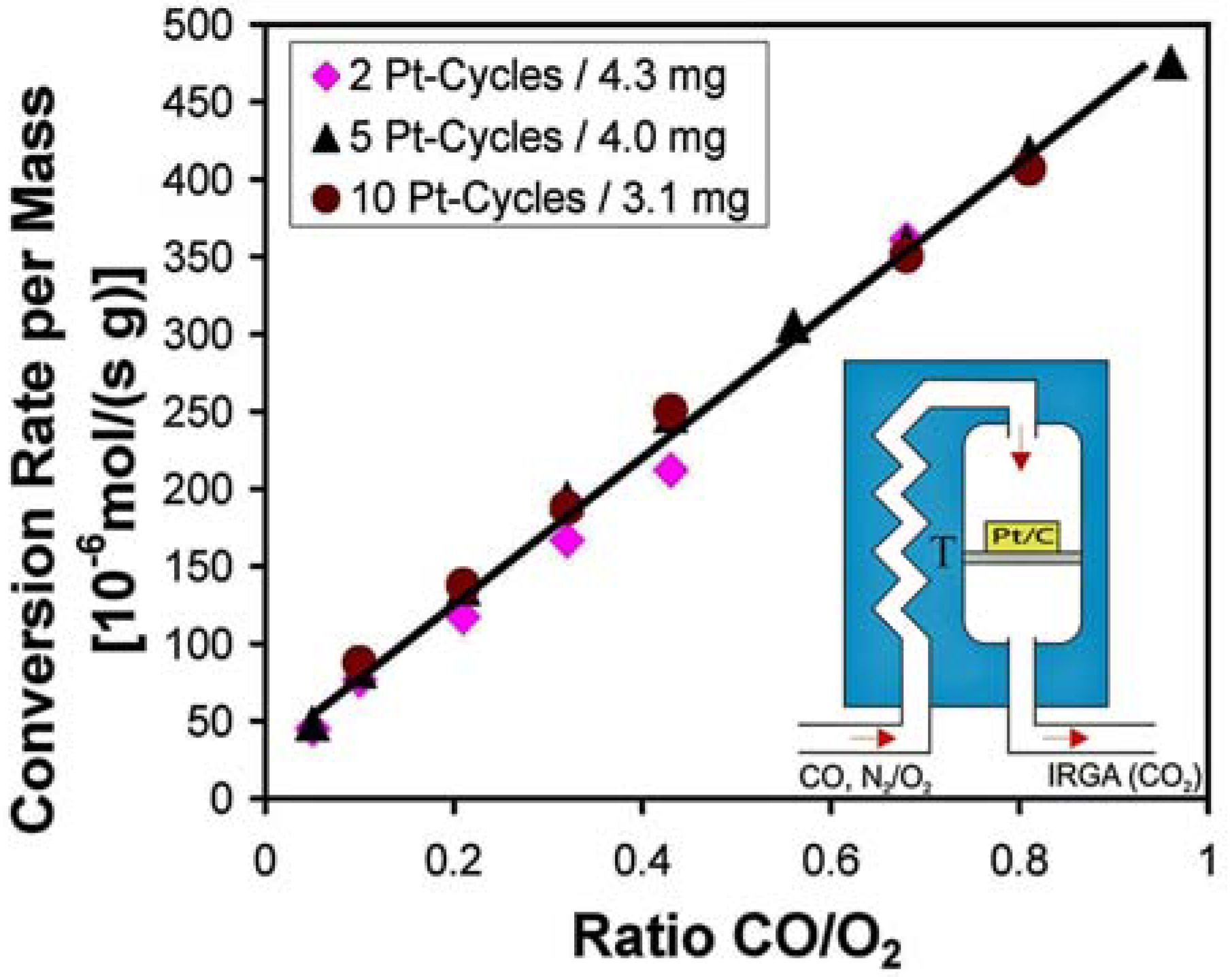
3.4. Catalysis by Pt doped Carbon Aerogels


4. Conclusions
Acknowledgements
References and Notes
- Weigend, F.; Evers, F.; Weissmüller, J. Structural relaxation in charged metal surfaces and cluster ions. Small 2006, 2, 1497–1503. [Google Scholar] [CrossRef] [PubMed]
- Mavrikakis, M.; Stoltze, P.; Norskov, J.K. Making gold less noble. Catal. Lett. 2000, 64, 101–106. [Google Scholar] [CrossRef]
- Xu, Y.; Mavrikakis, M. Adsorption and dissociation of O2 on gold surfaces: Effect of steps and strain. J. Phys. Chem. B 2003, 107, 9298–9307. [Google Scholar] [CrossRef]
- Ding, Y.; Chen, M.W. Nanoporous metals for catalytic and optical applications. MRS Bull. 2009, 34, 569–576. [Google Scholar] [CrossRef]
- Erlebacher, J.; Seshadri, R. Hard materials with tunable porosity. MRS Bull. 2009, 34, 561–568. [Google Scholar] [CrossRef]
- Weissmueller, J.; Newman, R.C.; Jin, H.J.; Hodge, A.M.; Kysar, J.W. Nanoporous metals by alloy corrosion: formation and mechanical properties. MRS Bull. 2009, 34, 577–586. [Google Scholar] [CrossRef]
- Biener, J.; Hodge, A.M.; Hamza, A.V. Microscopic failure behavior of nanoporous gold. Appl. Phys. Lett. 2005, 87, 121908. [Google Scholar] [CrossRef]
- Biener, J.; Hodge, A.M.; Hamza, A.V.; Hsiung, L.M.; Satcher, J.H. Nanoporous Au: A high yield strength material. J. Appl. Phys. 2005, 97, 024301. [Google Scholar] [CrossRef]
- Biener, J.; Hodge, A.M.; Hayes, J.R.; Volkert, C.A.; Zepeda-Ruiz, L.A.; Hamza, A.V.; Abraham, F.F. Size effects on the mechanical behavior of nanoporous Au. Nano Lett. 2006, 6, 2379–2382. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Sieradzki, K. Ductile-Brittle transition in random porous Au. Phys. Rev. Lett. 1992, 68, 1168–1171. [Google Scholar] [CrossRef] [PubMed]
- Sieradzki, K.; Newman, R.C. Stress-Corrosion cracking. J. Phys. Chem. Solids 1987, 48, 1101–1113. [Google Scholar] [CrossRef]
- Forty, A.J. Corrosion micro-morphology of noble-metal alloys and depletion gilding. Nature 1979, 282, 597–598. [Google Scholar] [CrossRef]
- Forty, A.J.; Durkin, P. A micro-morphological study of the dissolution of silver-gold alloys in nitric-acid. Philos. Mag. A 1980, 42, 295–318. [Google Scholar] [CrossRef]
- Fujita, T.; Qian, L.H.; Inoke, K.; Erlebacher, J.; Chen, M.W. Three-dimensional morphology of nanoporous gold. Appl. Phys. Lett. 2008, 92, 251902. [Google Scholar] [CrossRef]
- Roesner, H.; Parida, S.; Kramer, D.; Volkert, C.A.; Weissmueller, J. Reconstructing a nanoporous metal in three dimensions: An electron tomography study of dealloyed gold leaf. Adv. Eng. Mater. 2007, 9, 535–541. [Google Scholar] [CrossRef]
- Erlebacher, J.; Aziz, M.J.; Karma, A.; Dimitrov, N.; Sieradzki, K. Evolution of nanoporosity in dealloying. Nature 2001, 410, 450–453. [Google Scholar] [CrossRef] [PubMed]
- Van Petegem, S.; Brandstetter, S.; Maass, R.; Hodge, A.M.; El-Dasher, B.S.; Biener, J.; Schmitt, B.; Borca, C.; Van Swygenhoven, H. On the microstructure of nanoporous gold: An x-ray diffraction study. Nano Lett. 2009, 9, 1158–1163. [Google Scholar] [CrossRef] [PubMed]
- Parida, S.; Kramer, D.; Volkert, C.A.; Rosner, H.; Erlebacher, J.; Weissmuller, J. Volume change during the formation of nanoporous gold by dealloying. Phys. Rev. Lett. 2006, 97, 035504. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Kim, Y.J.; Erlebacher, J. Nanoporous gold leaf: "Ancient technology"/advanced material. Adv. Mater. 2004, 16, 1897–1900. [Google Scholar] [CrossRef]
- Thorp, J.C.; Sieradzki, K.; Tang, L.; Crozier, P.A.; Misra, A.; Nastasi, M.; Mitlin, D.; Picraux, S.T. Formation of nanoporous noble metal thin films by electrochemical dealloying of Pt xSi1-x. Appl. Phys. Lett. 2006, 88, 033110. [Google Scholar] [CrossRef]
- Biener, J.; Nyce, G.W.; Hodge, A.M.; Biener, M.M.; Hamza, A.V.; Maier, S.A. Nanoporous plasmonic metamaterials. Adv. Mater. 2008, 20, 1211–1217. [Google Scholar] [CrossRef]
- Kucheyev, S.O.; Hayes, J.R.; Biener, J.; Huser, T.; Talley, C.E.; Hamza, A.V. Surface-enhanced Raman scattering on nanoporous Au. Appl. Phys. Lett. 2006, 89, 053102. [Google Scholar] [CrossRef]
- Hodge, A.M.; Biener, J.; Hayes, J.R.; Bythrow, P.M.; Volkert, C.A.; Hamza, A.V. Scaling equation for yield strength of nanoporous open-cell foams. Acta Mater. 2007, 55, 1343–1349. [Google Scholar] [CrossRef]
- Cattarin, S.; Kramer, D.; Lui, A.; Musiani, M.M. Preparation and characterization of gold nanostructures of controlled dimension by electrochemical techniques. J. Phys. Chem. C. 2007, 111, 12643–12649. [Google Scholar] [CrossRef]
- Newman, R.C.; Corcoran, S.G.; Erlebacher, J.; Aziz, M.J.; Sieradzki, K. Alloy corrosion. MRS Bull. 1999, 24, 24–28. [Google Scholar]
- Biener, J.; Biener, M.M.; Nowitzki, T.; Hamza, A.V.; Friend, C.M.; Zielasek, V.; Bäumer, M. On the role of oxygen in stabilizing low-coordinated au atoms. Chemphyschem 2006, 7, 1906–1908. [Google Scholar] [CrossRef] [PubMed]
- Erlebacher, J.; Sieradzki, K. Pattern formation during dealloying. Scr. Mater. 2003, 49, 991–996. [Google Scholar] [CrossRef]
- Michely, T.; Comsa, G. Generation and nucleation of adatoms during ion-bombardment of Pt(111). Phys. Rev. B 1991, 44, 8411–8414. [Google Scholar] [CrossRef]
- Vishnyakov, V.; Donnelly, S.E.; Carter, G. Scanning-Tunneling-Microscopy observations of the evolution of small-scale topography on gold surfaces following irradiation with low-energy argon ions. Philos. Mag. B 1994, 70, 151–157. [Google Scholar] [CrossRef]
- Xu, C.X.; Su, J.X.; Xu, X.H.; Liu, P.P.; Zhao, H.J.; Tian, F.; Ding, Y. Low temperature CO oxidation over unsupported nanoporous gold. J. Am. Chem. Soc. 2007, 129, 42–43. [Google Scholar] [CrossRef] [PubMed]
- Zielasek, V.; Jurgens, B.; Schulz, C.; Biener, J.; Biener, M.M.; Hamza, A.V.; Bäumer, M. Gold catalysts: Nanoporous gold foams. Angew. Chem. Int. Ed. 2006, 45, 8241–8244. [Google Scholar] [CrossRef]
- Haruta, M.; Kobayashi, T.; Sano, H.; Yamada, N. Novel gold catalysts for the oxidation of carbon-monoxide at a temperature far below 0-degrees-C. Chem. Lett. 1987, 405–408. [Google Scholar] [CrossRef]
- Hashmi, A.S.K.; Hutchings, G.J. Gold catalysis. Angew. Chem. Int. Ed. 2006, 45, 7896–7936. [Google Scholar] [CrossRef]
- Hughes, M.D.; Xu, Y.J.; Jenkins, P.; McMorn, P.; Landon, P.; Enache, D.I.; Carley, A.F.; Attard, G.A.; Hutchings, G.J.; King, F.; Stitt, E.H.; Johnston, P.; Griffin, K.; Kiely, C.J. Tunable gold catalysts for selective hydrocarbon oxidation under mild conditions. Nature 2005, 437, 1132–1135. [Google Scholar] [CrossRef] [PubMed]
- Enache, D.I.; Knight, D.W.; Hutchings, G.J. Solvent-free oxidation of primary alcohols to aldehydes using supported gold catalysts. Catal. Lett. 2005, 103, 43–52. [Google Scholar] [CrossRef]
- Hvolbaek, B.; Janssens, T.V.W.; Clausen, B.S.; Falsig, H.; Christensen, C.H.; Norskov, J.K. Catalytic activity of Au nanoparticles. Nano Today 2007, 2, 14–18. [Google Scholar] [CrossRef]
- Hutchings, G.J. Vapor-phase hydrochlorination of acetylene - correlation of catalytic activity of supported metal chloride catalysTS. J. Catal. 1985, 96, 292–295. [Google Scholar] [CrossRef]
- Wittstock, A.; Neumann, B.; Schaefer, A.; Dumbuya, K.; Kubel, C.; Biener, M.M.; Zielasek, V.; Steinruck, H.P.; Gottfried, J.M.; Biener, J.; Hamza, A.; Bäumer, M. Nanoporous Au: An unsupported pure gold catalyst? J. Phys. Chem. C. 2009, 113, 5593–5600. [Google Scholar] [CrossRef]
- Chen, M.S.; Goodman, D.W. The structure of catalytically active gold on titania. Science 2004, 306, 252–255. [Google Scholar] [CrossRef] [PubMed]
- Valden, M.; Lai, X.; Goodman, D.W. Onset of catalytic activity of gold clusters on titania with the appearance of nonmetallic properties. Science 1998, 281, 1647–1650. [Google Scholar] [CrossRef] [PubMed]
- Bond, G.C.; Thompson, D.T. Gold-catalysed oxidation of carbon monoxide. Gold Bull. 2000, 33, 41–51. [Google Scholar] [CrossRef]
- Falsig, H.; Hvolbaek, B.; Kristensen, I.S.; Jiang, T.; Bligaard, T.; Christensen, C.H.; Norskov, J.K. Trends in the catalytic CO oxidation activity of nanoparticles. Angew. Chem. Int. Ed. 2008, 47, 4835–4839. [Google Scholar] [CrossRef]
- Yim, W.L.; Nowitzki, T.; Necke, M.; Schnars, H.; Nickut, P.; Biener, J.; Biener, M.M.; Zielasek, V.; Al-Shamery, K.; Kluner, T.; Bäumer, M. Universal phenomena of CO adsorption on gold surfaces with low-coordinated sites. J. Phys. Chem. C 2007, 111, 445–451. [Google Scholar] [CrossRef]
- Deng, X.Y.; Min, B.K.; Guloy, A.; Friend, C.M. Enhancement of O-2 dissociation on Au(111) by adsorbed oxygen: Implications for oxidation catalysis. J. Am. Chem. Soc. 2005, 127, 9267–9270. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Cai, Y.; Yan, Z.; Goodman, D.W. On the origin of the unique properties of supported Au nanoparticles. J. Am. Chem. Soc. 2006, 128, 6341–6346. [Google Scholar] [CrossRef] [PubMed]
- Haruta, M. New generation of gold catalysts: Nanoporous foams and tubes-is unsupported gold catalytically active? Chemphyschem 2007, 8, 1911–1913. [Google Scholar] [CrossRef] [PubMed]
- Wittstock, A.; Zielasek, V.; Biener, J.; Friend, C.M.; Bäumer, M. Novel nanoporous gold catalysts for green chemistry: Selective gas-phase oxidative coupling of methanol at low temperature. Science 2010, in press. [Google Scholar]
- Yen, C.W.; Lin, M.L.; Wang, A.Q.; Chen, S.A.; Chen, J.M.; Mou, C.Y. CO Oxidation catalyzed by Au-Ag bimetallic nanoparticles supported in mesoporous silica. J. Phys. Chem. C 2009, 113, 17831–17839. [Google Scholar] [CrossRef]
- Jorgensen, B.; Christiansen, S.E.; Thomsen, M.L.D.; Christensen, C.H. Aerobic oxidation of aqueous ethanol using heterogeneous gold catalysts: Efficient routes to acetic acid and ethyl acetate. J. Catal. 2007, 251, 332–337. [Google Scholar] [CrossRef]
- Abad, A.; Concepcion, P.; Corma, A.; Garcia, H. A collaborative effect between gold and a support induces the selective oxidation of alcohols. Angew. Chem. Int. Ed. 2005, 44, 4066–4069. [Google Scholar] [CrossRef]
- Xu, B.J.; Liu, X.Y.; Haubrich, J.; Madix, R.J.; Friend, C.M. selectivity control in gold-mediated esterification of methanol. Angew. Chem. Int. Ed. 2009, 48, 4206–4209. [Google Scholar] [CrossRef]
- Nyce, G.W.; Hayes, J.R.; Hamza, A.V.; Satcher, J.H. Synthesis and characterization of hierarchical porous gold materials. Chem. Mater. 2007, 19, 344–346. [Google Scholar] [CrossRef]
- Gleiter, H. Materials with ultrafine grain sizes. In Second Risø Int. Symp. on Metallurgy and Materials Science, Røskilde, Denmark, 4–18 September 1981; Hansen, N., Leffers, T., Lilholt, H., Eds.; Risø National Laboratory: Røskilde, Denmark, 1981; pp. 15–21. [Google Scholar]
- Gleiter, H. Nanostructured materials: Basic concepts and microstructure. Acta Mater. 2000, 48, 1–29. [Google Scholar] [CrossRef]
- Weissmueller, J.; Viswanath, R.N.; Kramer, D.; Zimmer, P.; Wuerschum, R.; Gleiter, H. Charge-induced reversible strain in a metal. Science 2003, 300, 312–315. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.J.; Parida, S.; Kramer, D.; Weissmüller, J. Sign-inverted surface stress-charge response in nanoporous gold. Surf. Sci. 2008, 602, 3588–3594. [Google Scholar] [CrossRef]
- Kramer, D.; Viswanath, R.N.; Weissmüller, J. Surface-stress induced macroscopic bending of nanoporous gold cantilevers. Nano Lett. 2004, 4, 793–796. [Google Scholar] [CrossRef]
- Umeno, Y.; Elsasser, C.; Meyer, B.; Gumbsch, P.; Nothacker, M.; Weissmüller, J.; Evers, F. Ab initio study of surface stress response to charging. Europhys. Lett. 2007, 78, 13001. [Google Scholar] [CrossRef]
- Umeno, Y.; Elsasser, C.; Meyer, B.; Gumbsch, P.; Weissmüller, J. Reversible relaxation at charged metal surfaces: An ab initio study. Europhys Lett 2008, 84, 13002. [Google Scholar] [CrossRef]
- Weissmueller, J.; Cahn, J.W. Mean stresses in microstructures due to interface stresses: A generalization of a capillary equation for solids. Acta Mater. 1997, 45, 1899–1906. [Google Scholar] [CrossRef]
- Weissmüller, J.; Duan, H.-L.; Farkas, D. Deformation of solids with nanoscale pores by the action of capillary forces. Acta Mater. 2009. [Google Scholar] [CrossRef]
- Crowson, D.A.; Farkas, D.; Corcoran, S.G. Mechanical stability of nanoporous metals with small ligament sizes. Scripta Mater. 2009, 61, 497–499. [Google Scholar] [CrossRef]
- Young, T. An Essay on the Cohesion of Fluids. Philos. Trans. Roy. Soc. (London) 1805, 95, 65–87. [Google Scholar] [CrossRef]Laplace, P.S. Méchanique Céleste (Gauthier-Villars, Paris, 1805); Reprinted by Chelsea Publishing Comp.; New York, NY, USA, 1966; Volume 4, Supplements au Livre X, IV; p. 685. [Google Scholar]
- Kramer, D.; Weissmüller, J. A note on surface stress and surface tension and their interrelation via Shuttleworth's equation and the Lippmann equation. Surf. Sci. 2007, 601, 3042–3051. [Google Scholar] [CrossRef]
- Lippmann, G. Relations entre les phénomènes électrique et capillaries. Ann. Chim. Phys. 1875, 5, 494–549. [Google Scholar]
- Smetanin, M.; Viswanath, R.N.; Kramer, D.; Beckmann, D.; Koch, T.; Kibler, L.A.; Kolb, D.M.; Weissmüller, J. Surface stress-charge response of a (111)-textured gold electrode under conditions of weak ion adsorption. Langmuir 2008, 24, 8561–8567. [Google Scholar] [CrossRef] [PubMed]
- Tabard-Cossa, V.; Godin, M.; Burgess, I.J.; Monga, T.; Lennox, R.B.; Grutter, P. Microcantilever-based sensors: Effect of morphology, adhesion, and cleanliness of the sensing surface on surface stress. Anal. Chem. 2007, 79, 8136–8143. [Google Scholar] [CrossRef] [PubMed]
- Viswanath, R.N.; Kramer, D.; Weissmüller, J. Adsorbate effects on the surface stress-charge response of platinum electrodes. Electrochim. Acta 2008, 53, 2757–2767. [Google Scholar] [CrossRef]
- Haiss, W. Surface stress of clean and adsorbate-covered solids. Rep. Prog. Phys. 2001, 64, 591–648. [Google Scholar] [CrossRef]
- Viswanath, R.N.; Kramer, D.; Weissmüller, J. Variation of the surface stress-charge coefficient of platinum with electrolyte concentration. Langmuir 2005, 21, 4604–4609. [Google Scholar] [CrossRef] [PubMed]
- Biener, J.; Wittstock, A.; Zepeda-Ruiz, L.A.; Biener, M.M.; Zielasek, V.; Kramer, D.; Viswanath, R.N.; Weissmuller, J.; Bäumer, M.; Hamza, A.V. Surface-chemistry-driven actuation in nanoporous gold. Nat. Mater. 2009, 8, 47–51. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Samano, E.; Koel, B.E. Oxygen adsorption and oxidation reactions on Au(211) surfaces: Exposures using O2 at high pressures and ozone (O3) in UHV. Surf. Sci. 2006, 600, 4622–4632. [Google Scholar] [CrossRef]
- Chai, G.S.; Shin, I.S.; Yu, J.S. Synthesis of ordered, uniform, macroporous carbons with mesoporous walls templated by aggregates of polystyrene spheres and silica particles for use as catalyst supports in direct methanol fuel cells. Adv. Mater. 2004, 16, 2057–2061. [Google Scholar] [CrossRef]
- Chmiola, J.; Yushin, G.; Gogotsi, Y.; Portet, C.; Simon, P.; Taberna, P.L. Anomalous increase in carbon capacitance at pore sizes less than 1 nanometer. Science 2006, 313, 1760–1763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.S.; Adelhelm, P.; Smarsly, B.M.; Hore, S.; Antonietti, M.; Maier, J. Synthesis of hierarchically porous carbon monoliths with highly ordered microstructure and their application in rechargeable lithium batteries with high-rate capability. Adv. Funct. Mater. 2007, 17, 1873–1878. [Google Scholar] [CrossRef]
- Lee, K.T.; Lytle, J.C.; Ergang, N.S.; Oh, S.M.; Stein, A. Synthesis and rate performance of monolithic macroporous carbon electrodes for lithium-ion secondary batteries. Adv. Funct. Mater. 2005, 15, 547–556. [Google Scholar] [CrossRef]
- Wang, Z.Y.; Li, F.; Ergang, N.S.; Stein, A. Effects of hierarchical architecture on electronic and mechanical properties of nanocast monolithic porous carbons and carbon-carbon nanocomposites. Chem. Mater. 2006, 18, 5543–5553. [Google Scholar] [CrossRef]
- Fricke, J.; Petricevic, R. Carbon aerogels. In Handbook of Porous Solids; Schüth, F., Sing, K.S.W., Weitkamp, J., Eds.; Wiley-VCH: Weinheim, Germany, 2002. [Google Scholar]
- Pekala, R.W. Organic aerogels from the polycondensation of resorcinol with formaldehyde. J. Mater. Sci. 1989, 24, 3221–3227. [Google Scholar] [CrossRef]
- Lu, X.P.; Nilsson, O.; Fricke, J.; Pekala, R.W. Thermal and electrical-conductivity of monolithic carbon aerogels. J. Appl. Phys. 1993, 73, 581–584. [Google Scholar] [CrossRef]
- Baumann, T.F.; Worsley, M.A.; Han, T.Y.-J.; Satcher, J.H., Jr. High surface area carbon aerogel monoliths with hierarchical porosity. J. Non-Cryst. Solids 2008, 354, 3513–3515. [Google Scholar] [CrossRef]
- Brandt, R.; Petricevic, R.; Probstle, H.; Fricke, J. Acetic acid catalyzed carbon aerogels. J. Porous Mat. 2003, 10, 171–178. [Google Scholar] [CrossRef]
- Moreno-Castilla, C.; Maldonado-Hodar, F.J.; Perez-Cadenas, A.F. Physicochemical surface properties of Fe, Co, Ni, and Cu-doped monolithic organic aerogels. Langmuir 2003, 19, 5650–5655. [Google Scholar] [CrossRef]
- Hanzawa, Y.; Kaneko, K.; Pekala, R.W.; Dresselhaus, M.S. Activated carbon aerogels. Langmuir 1996, 12, 6167–6169. [Google Scholar] [CrossRef]
- Saliger, R.; Fischer, U.; Herta, C.; Fricke, J. High Surface Area Carbon Aerogels for Supercapacitors. J. Non-Cryst. Solids 1998, 225, 81–85. [Google Scholar] [CrossRef]
- Lin, C.; Ritter, J.A. Carbonization and activation of sol-gel derived carbon xerogels. Carbon 2000, 38, 849–861. [Google Scholar] [CrossRef]
- Feaver, A.; Cao, G.Z. Activated carbon cryogels for low pressure methane storage. Carbon 2006, 44, 590–593. [Google Scholar] [CrossRef]
- Gogotsi, Y.; Dash, R.K.; Yushin, G.; Yildirim, T.; Laudisio, G.; Fischer, J.E. Tailoring of nanoscale porosity in carbide-derived carbons for hydrogen storage. J. Am. Chem. Soc. 2005, 127, 16006–16007. [Google Scholar] [CrossRef] [PubMed]
- Panella, B.; Hirscher, M.; Roth, S. Hydrogen adsorption in different carbon nanostructures. Carbon 2005, 43, 2209–2214. [Google Scholar] [CrossRef]
- Xia, K.S.; Gao, Q.M.; Wu, C.D.; Song, S.Q.; Ruan, M.L. Activation, characterization and hydrogen storage properties of the mesoporous carbon CMK-3. Carbon 2007, 45, 1989–1996. [Google Scholar] [CrossRef]
- Yang, Z.X.; Xia, Y.D.; Mokaya, R. Enhanced hydrogen storage capacity of high surface area zeolite-like carbon materials. J. Am. Chem. Soc. 2007, 129, 1673–1679. [Google Scholar] [CrossRef] [PubMed]
- Schlapbach, L.; Zuttel, A. Hydrogen-storage materials for mobile applications. Nature 2001, 414, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Denbigh, K. The Principles of Chemical Equilibrium; Cambridge University Press: Cambridge, UK, 1971. [Google Scholar]
- Kabbour, H.; Baumann, T.F.; Satcher, J.H.; Saulnier, A.; Ahn, C.C. Toward new candidates for hydrogen storage: High-surface-area carbon aerogels. Chem. Mater. 2006, 18, 6085–6087. [Google Scholar] [CrossRef]
- Murata, K.; Kaneko, K.; Kanoh, H.; Kasuya, D.; Takahashi, K.; Kokai, F.; Yudasaka, M.; Iijima, S. Adsorption mechanism of supercritical hydrogen in internal and interstitial nanospaces of single-wall carbon nanohorn assembly. J. Phys. Chem. B 2002, 106, 11132–11138. [Google Scholar] [CrossRef]
- Patchkovskii, S.; Tse, J.S.; Yurchenko, S.N.; Zhechkov, L.; Heine, T.; Seifert, G. Graphene nanostructures as tunable storage media for molecular hydrogen. Proc. Natl. Acad. Sci. USA 2005, 102, 10439–10444. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, S.K.; Myers, A.L. Optimum conditions for adsorptive storage. Langmuir 2006, 22, 1688–1700. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.W.; Yang, R.T. Hydrogen storage on platinum nanoparticles doped on superactivated carbon. J. Phys. Chem. C 2007, 111, 11086–11094. [Google Scholar] [CrossRef]
- Robell, A.J.; Ballou, E.V.; Boudart, M. Surface diffusion of hydrogen on carbon. J. Phys. Chem. 1964, 68, 2748–2753. [Google Scholar] [CrossRef]
- Yoo, E.; Gao, L.; Komatsu, T.; Yagai, N.; Arai, K.; Yamazaki, T.; Matsuishi, K.; Matsumoto, T.; Nakamura, J. Atomic hydrogen storage in carbon nanotubes promoted by metal catalysts. J. Phys. Chem. B 2004, 108, 18903–18907. [Google Scholar] [CrossRef]
- Kim, Y.H.; Zhao, Y.F.; Williamson, A.; Heben, M.J.; Zhang, S.B. Nondissociative adsorption of H2 molecules in light-element-doped fullerenes. Phys. Rev. Lett. 2006, 96, 016102. [Google Scholar] [CrossRef] [PubMed]
- Ritala, M.; Leskelä, M. Handbook of Thin Film Materials; Nalwa, S., Ed.; Academic Press: San Diego, CA, USA, 2002; Volume 1, p. 103. [Google Scholar]
- Leskelä, M.; Ritala, M. Atomic layer deposition chemistry: Recent developments and future challenges. Angew. Chem. Int. Ed. 2003, 42, 5548–5554. [Google Scholar] [CrossRef]
- Kim, H. Atomic layer deposition of metal and nitride thin films: Current research efforts and applications for semiconductor device processing. J. Vac. Sci. Technol. B 2003, 21, 2231–2261. [Google Scholar] [CrossRef]
- Baumann, T.F.; Biener, J.; Wang, Y.M.M.; Kucheyev, S.O.; Nelson, E.J.; Satcher, J.H.; Elam, J.W.; Pellin, M.J.; Hamza, A.V. Atomic layer deposition of uniform metal coatings on highly porous aerogel substrates. Chem. Mater. 2006, 18, 6106–6108. [Google Scholar] [CrossRef]
- Biener, J.; Baumann, T.F.; Wang, Y.M.; Nelson, E.J.; Kucheyev, S.O.; Hamza, A.V.; Kemell, M.; Ritala, M.; Leskela, M. Ruthenium/aerogel nanocomposites via atomic layer deposition. Nanotechnology 2007, 18, 055303. [Google Scholar] [CrossRef]
- King, J.S.; Wittstock, A.; Biener, J.; Kucheyev, S.O.; Wang, Y.M.; Baumann, T.F.; Giri, S.K.; Hamza, A.V.; Bäumer, M.; Bent, S.F. Ultralow loading Pt nanocatalysts prepared by atomic layer deposition on carbon aerogels. Nano Lett. 2008, 8, 2405–2409. [Google Scholar] [CrossRef] [PubMed]
- Kucheyev, S.O.; Biener, J.; Baumann, T.F.; Wang, Y.M.; Hamza, A.V.; Li, Z.; Lee, D.K.; Gordon, R.G. Mechanisms of atomic layer deposition on substrates with ultrahigh aspect ratios. Langmuir 2008, 24, 943–948. [Google Scholar] [CrossRef] [PubMed]
- Ghosal, S.; Baumann, T.F.; King, J.S.; Kucheyev, S.O.; Wang, Y.M.; Worsley, M.A.; Biener, J.; Bent, S.F.; Hamza, A.V. Controlling atomic layer deposition of TiO2 in aerogels through surface functionalization. Chem. Mater. 2009, 21, 1989–1992. [Google Scholar] [CrossRef]
- Kucheyev, S.O.; Biener, J.; Wang, Y.M.; Baumann, T.F.; Wu, K.J.; van Buuren, T.; Hamza, A.V.; Satcher, J.H.; Elam, J.W.; Pellin, M.J. Atomic layer deposition of ZnO on ultralow-density nanoporous silica aerogel monoliths. Appl. Phys. Lett. 2005, 86, 083108. [Google Scholar] [CrossRef]
- Wang, Y.X.; Balbuena, P.B. Ab initio molecular dynamics simulations of the oxygen reduction reaction on a Pt(111) surface in the presence of hydrated hydronium (H3O)+(H2O)2: Direct or series pathway? J. Phys. Chem. B 2005, 109, 14896–14907. [Google Scholar] [CrossRef] [PubMed]
- Du, H.D.; Gan, L.; Li, B.H.; Wu, P.; Qiu, Y.L.; Kang, F.Y.; Zeng, Y.Q. Influences of mesopore size on oxygen reduction reaction catalysis of Pt/carbon aerogels. J. Phys. Chem. C 2007, 111, 2040–2043. [Google Scholar] [CrossRef]
- Liu, H.S.; Song, C.J.; Zhang, L.; Zhang, J.J.; Wang, H.J.; Wilkinson, D.P. A review of anode catalysis in the direct methanol fuel cell. J. Power Sources 2006, 155, 95–110. [Google Scholar] [CrossRef]
- Gao, F.; McClure, S.M.; Cai, Y.; Gath, K.K.; Wang, Y.; Chen, M.S.; Guo, Q.L.; Goodman, D.W. CO oxidation trends on Pt-group metals from ultrahigh vacuum to near atmospheric pressures: A combined in situ PM-IRAS and reaction kinetics study. Surf. Sci. 2009, 603, 65–70. [Google Scholar] [CrossRef]
- Santra, A.K.; Goodman, D.W. Catalytic oxidation of CO by platinum group metals: from ultrahigh vacuum to elevated pressures. Electrochim. Acta 2002, 47, 3595–3609. [Google Scholar] [CrossRef]
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Biener, J.; Wittstock, A.; Baumann, T.F.; Weissmüller, J.; Bäumer, M.; Hamza, A.V. Surface Chemistry in Nanoscale Materials. Materials 2009, 2, 2404-2428. https://doi.org/10.3390/ma2042404
Biener J, Wittstock A, Baumann TF, Weissmüller J, Bäumer M, Hamza AV. Surface Chemistry in Nanoscale Materials. Materials. 2009; 2(4):2404-2428. https://doi.org/10.3390/ma2042404
Chicago/Turabian StyleBiener, Jürgen, Arne Wittstock, Theodore F. Baumann, Jörg Weissmüller, Marcus Bäumer, and Alex V. Hamza. 2009. "Surface Chemistry in Nanoscale Materials" Materials 2, no. 4: 2404-2428. https://doi.org/10.3390/ma2042404
APA StyleBiener, J., Wittstock, A., Baumann, T. F., Weissmüller, J., Bäumer, M., & Hamza, A. V. (2009). Surface Chemistry in Nanoscale Materials. Materials, 2(4), 2404-2428. https://doi.org/10.3390/ma2042404