Dinitrogen and Related Chemistry of the Lanthanides: A Review of the Reductive Capture of Dinitrogen, As Well As Mono- and Di-aza Containing Ligand Chemistry of Relevance to Known and Postulated Metal Mediated Dinitrogen Derivatives

Abstract
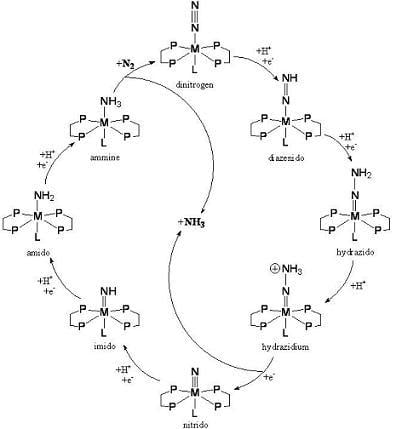
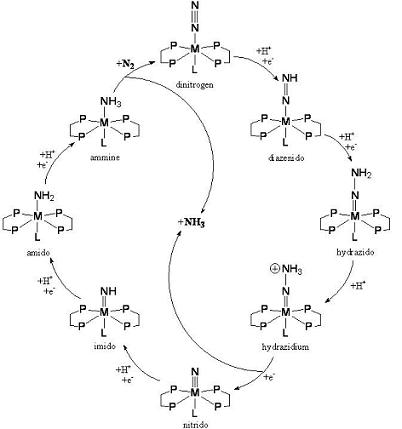
:
1. Introduction to Nitrogen Fixation and Scope of Review

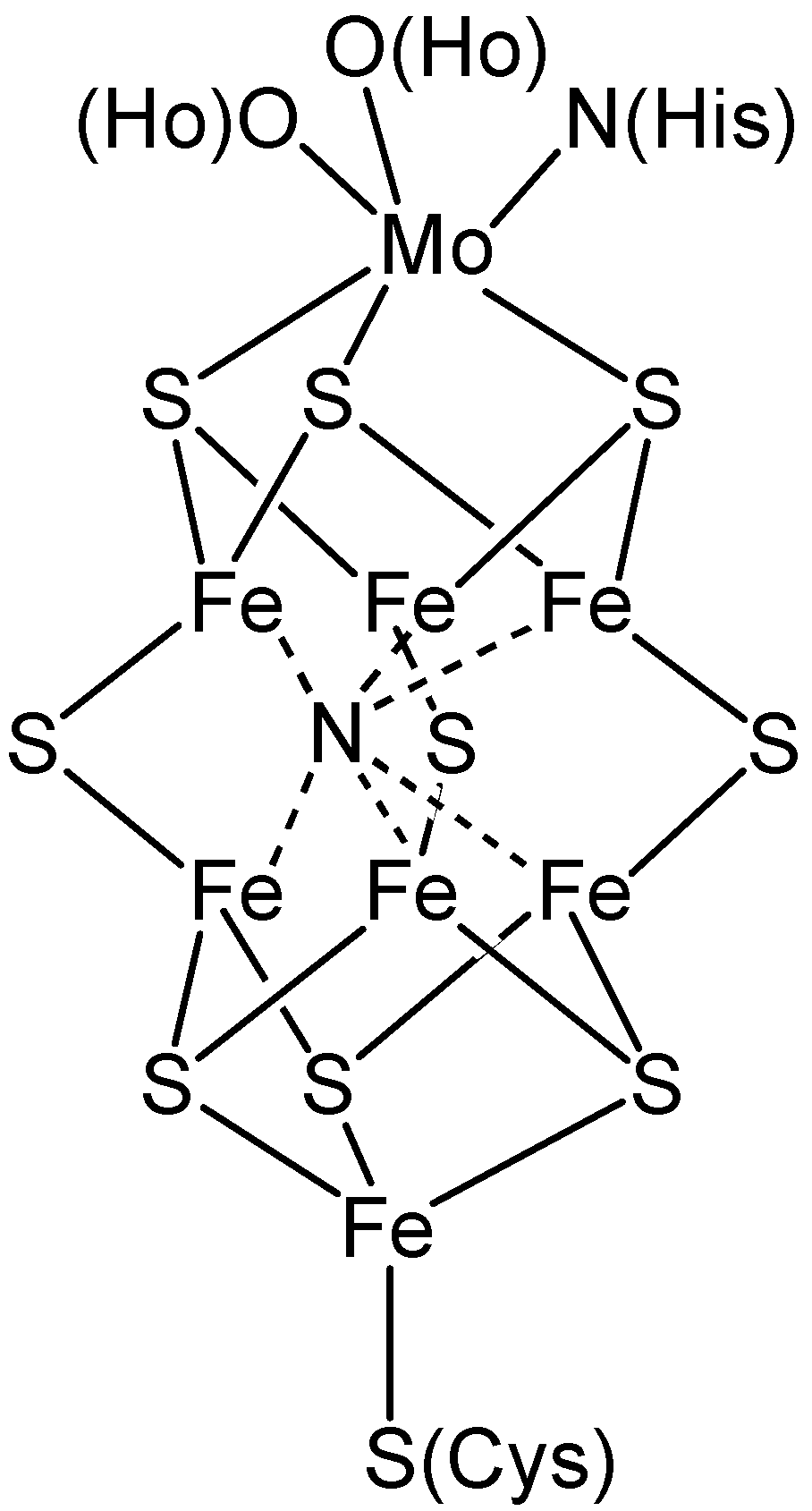
2. Biological Nitrogen Fixation
3. Synthetic Transition Metal Mediated Nitrogen Fixation



4. Lanthanide Mediated Dinitrogen Reduction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
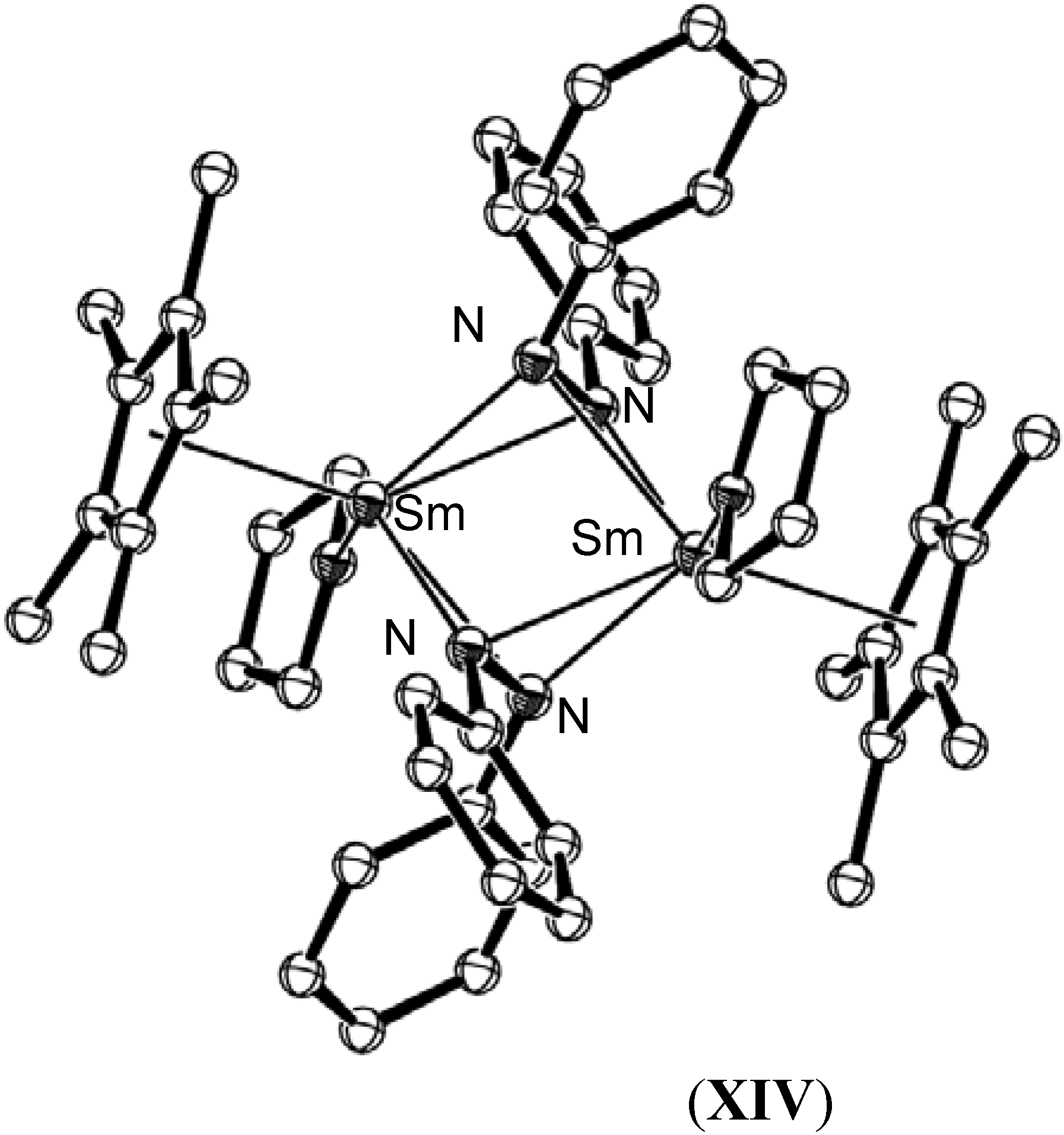
{kind=link}
{kind=link}
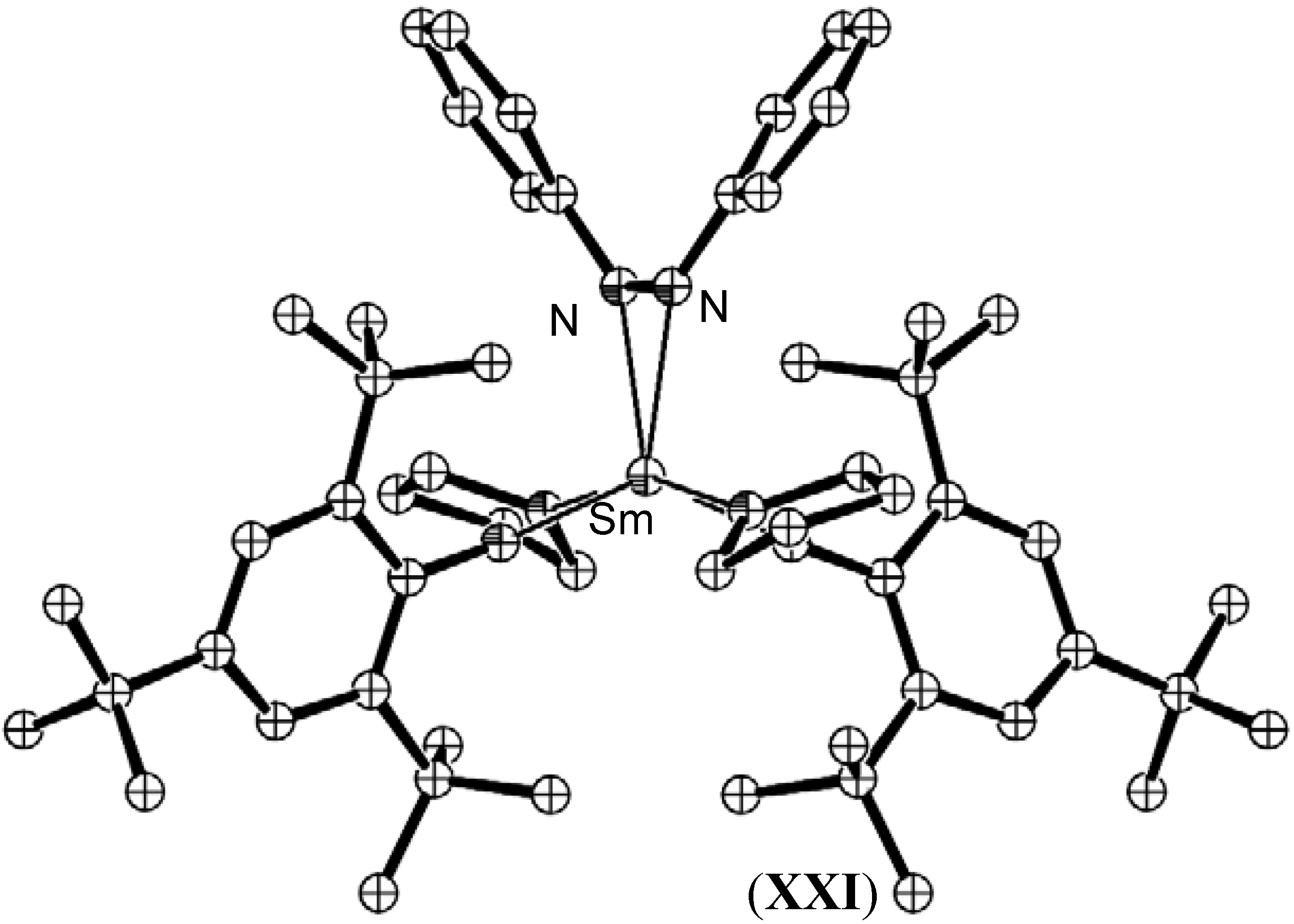{kind=link}
{kind=link}
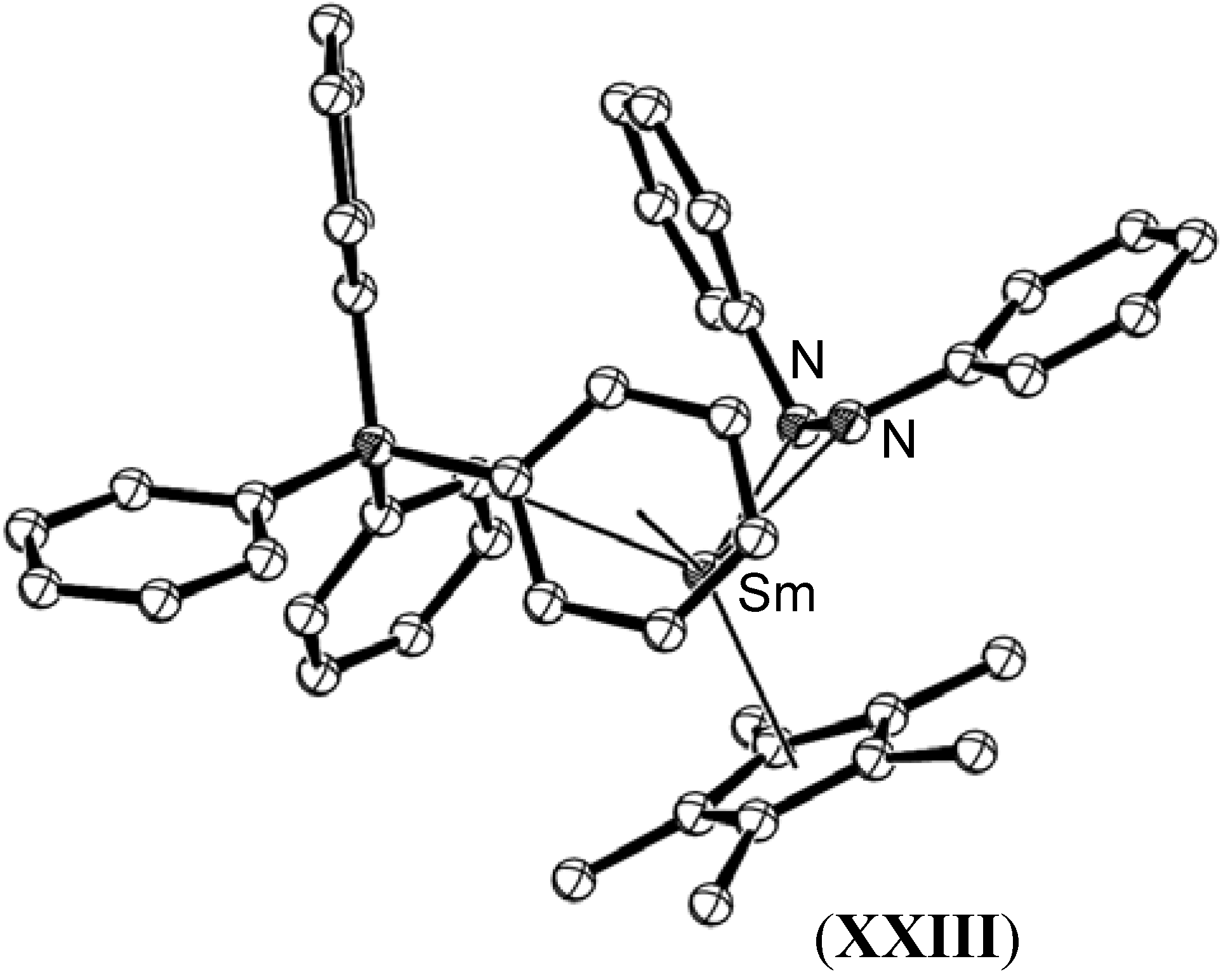
{kind=link}
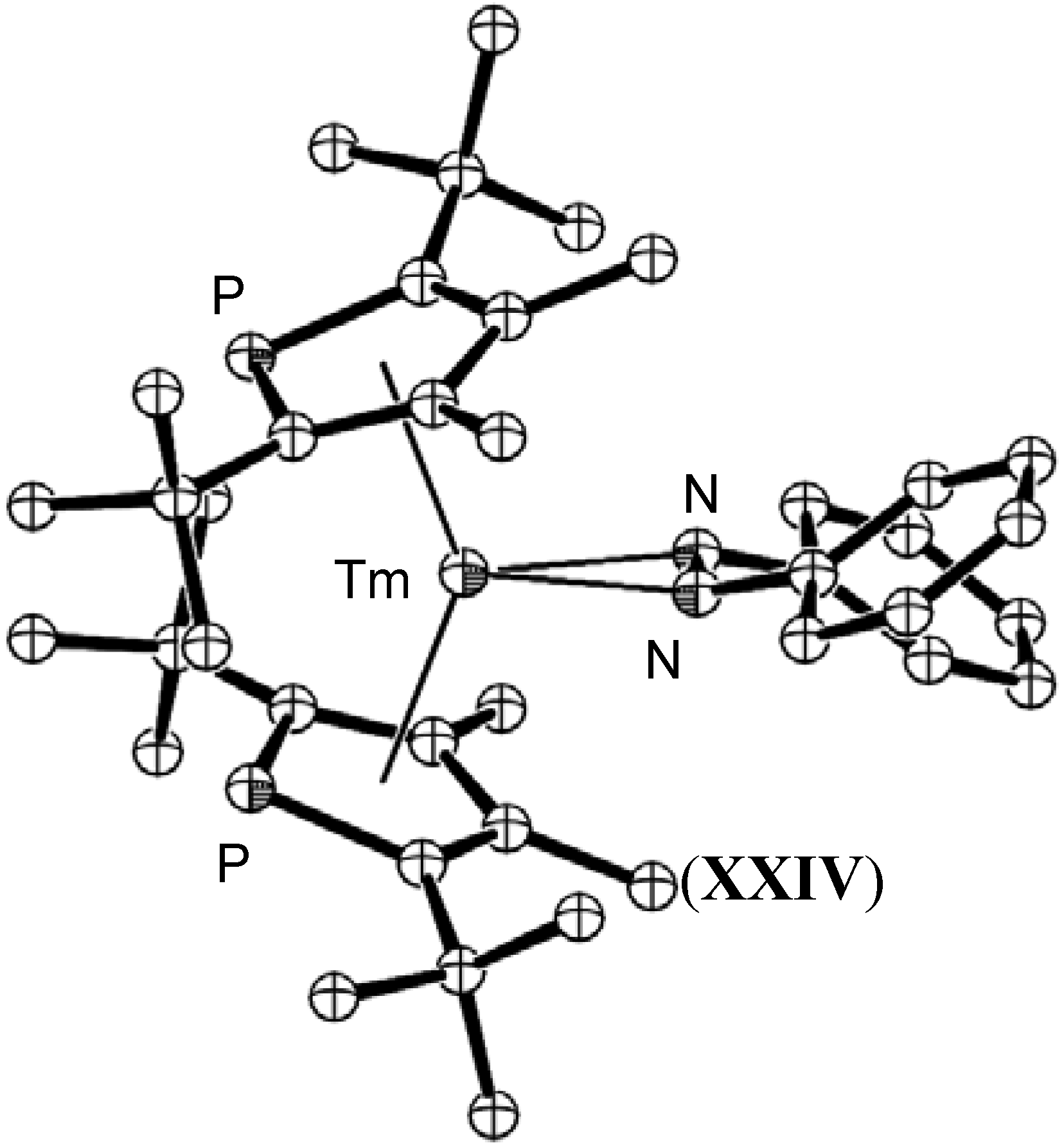
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
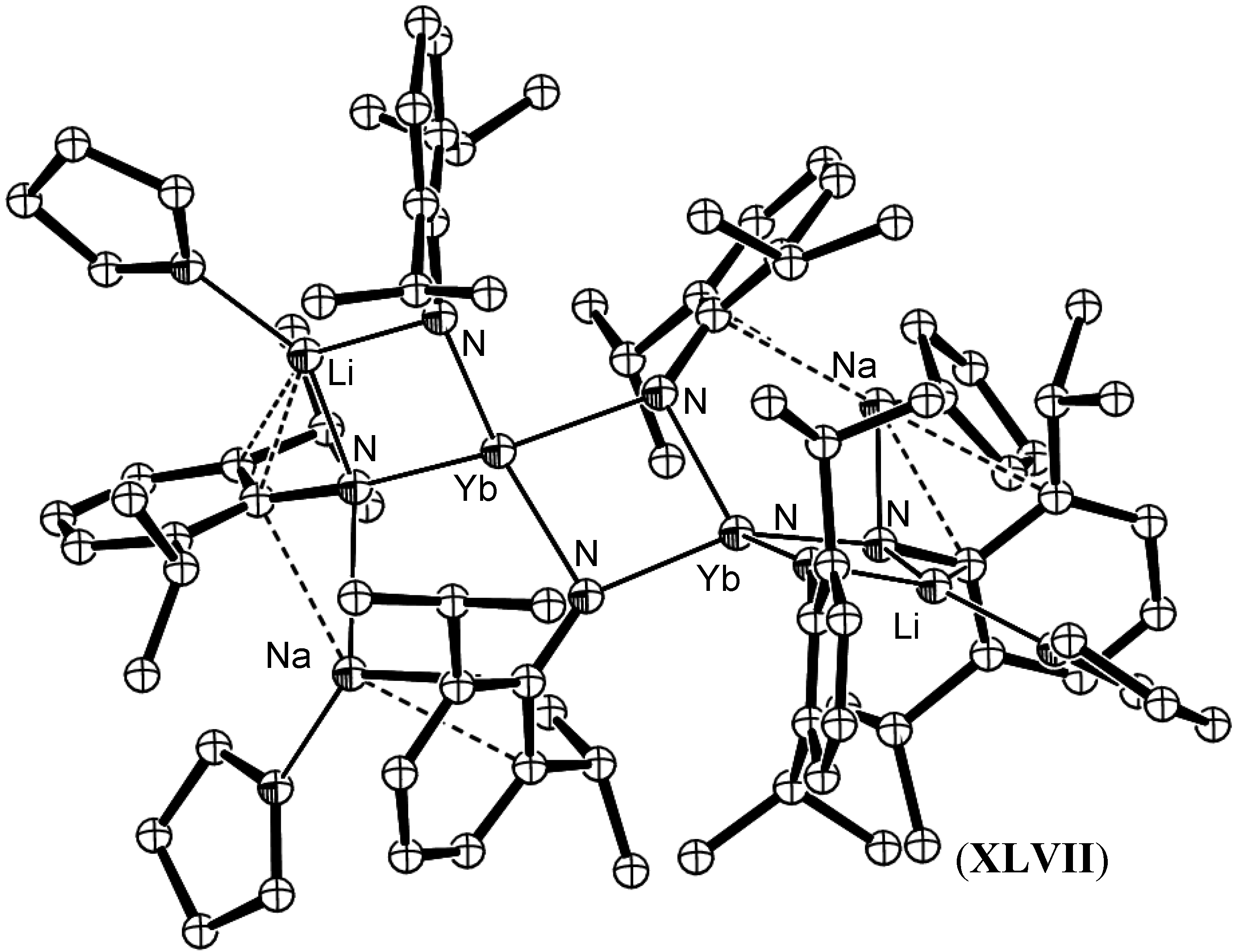{kind=link}
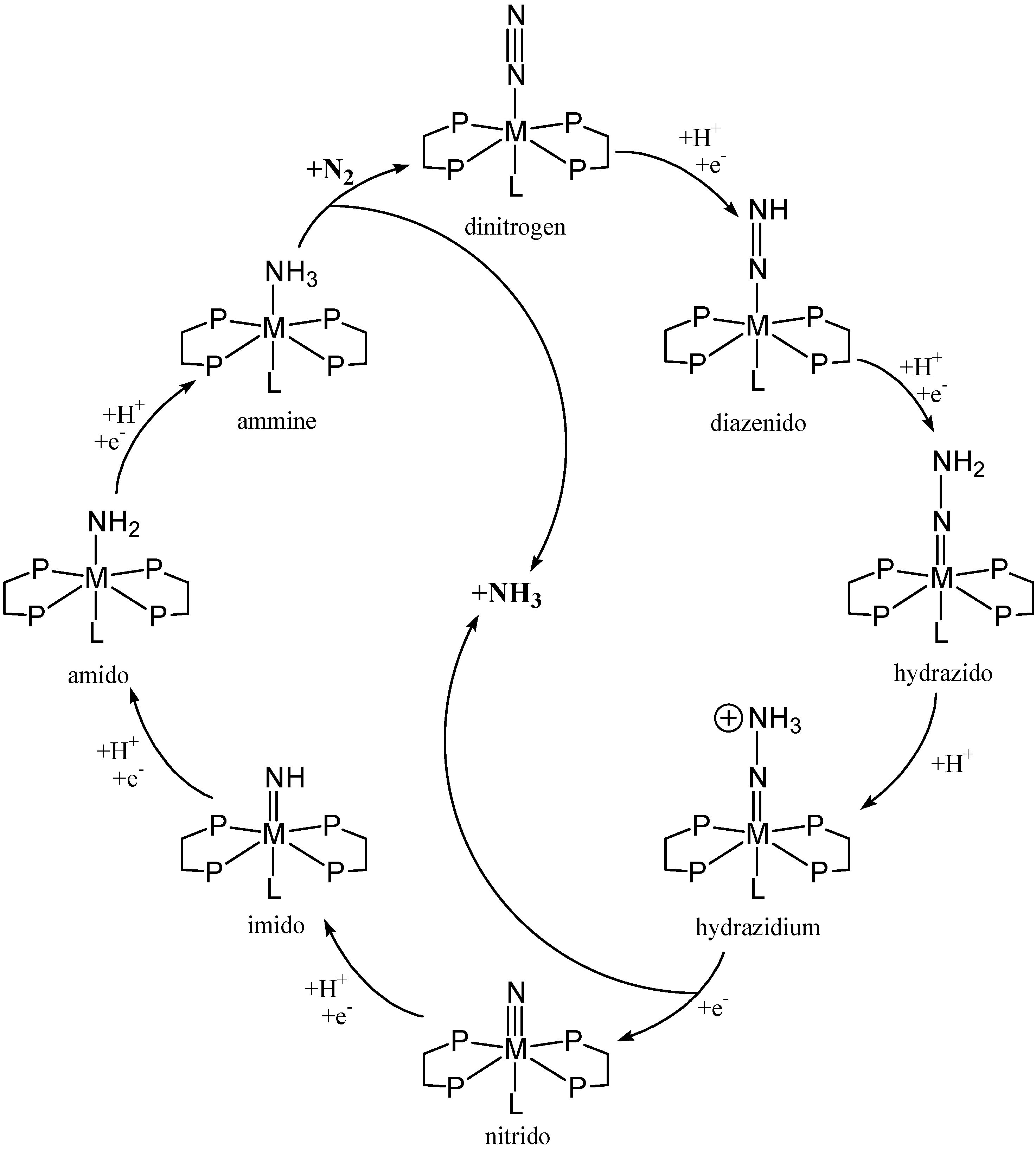{kind=link}
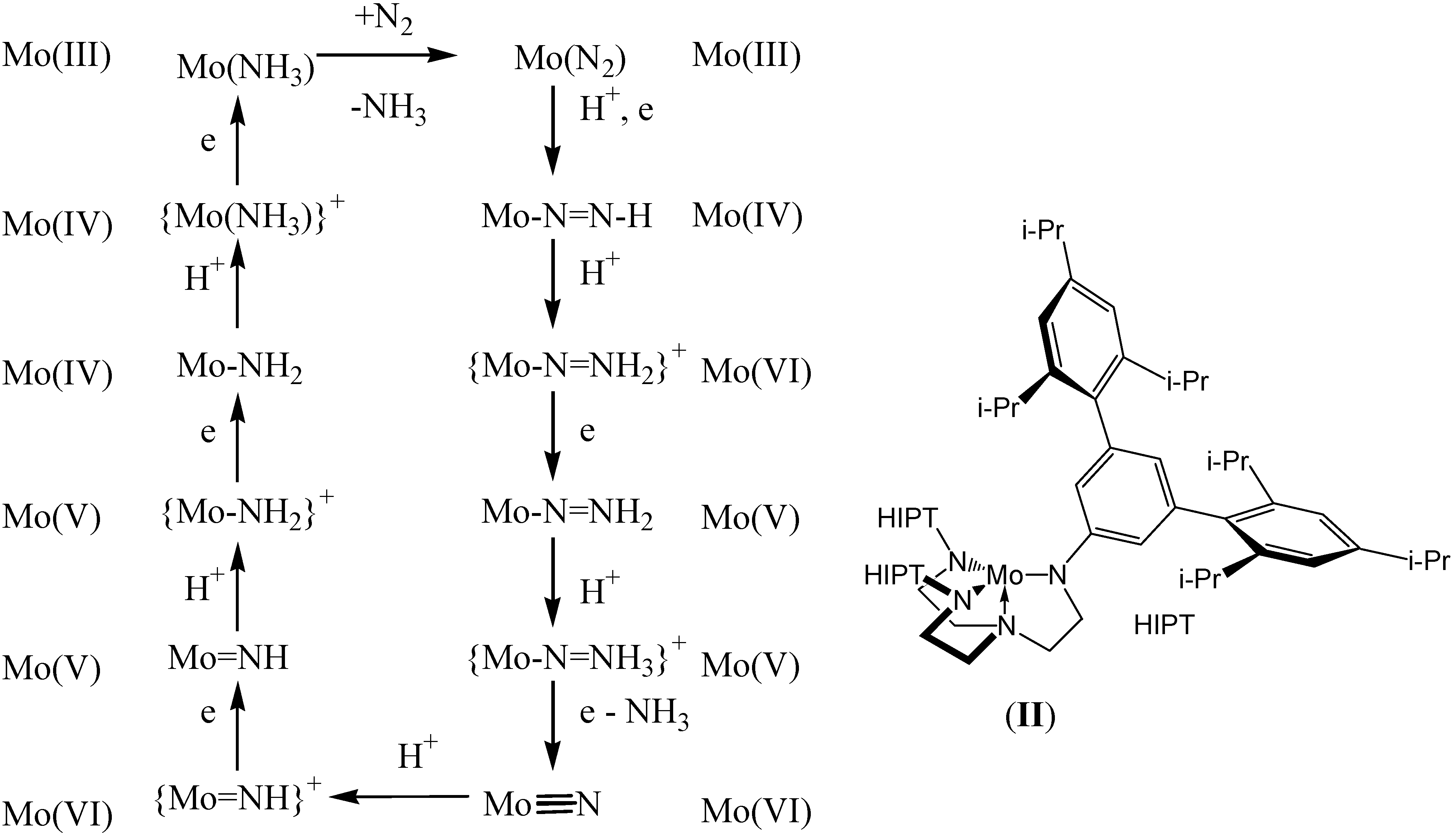{kind=link}
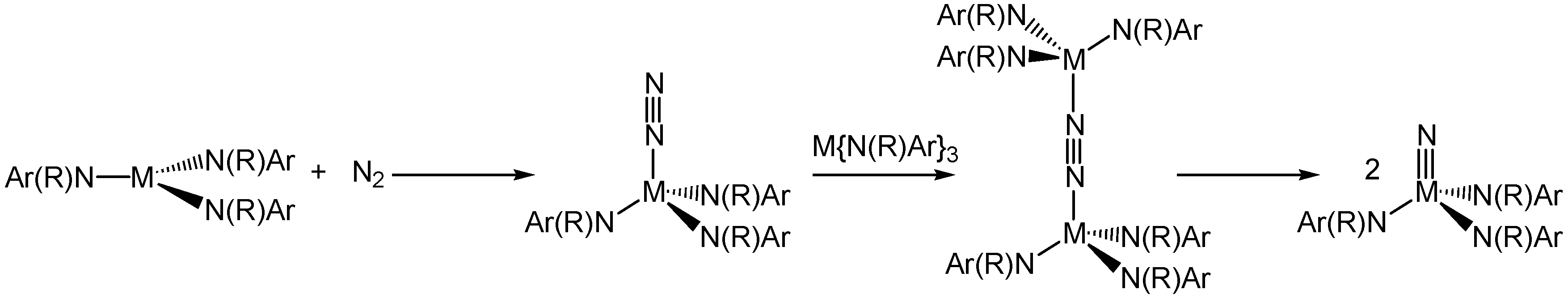{kind=link}
{kind=link}
{kind=link}
| N2 Binding Mode | Weak Activation | Strong Activation |
| End-on mononuclear |  | |
| End-on dinuclear |  |  |
| Side-on dinuclear |  |  |
| Side-on/End-on dinuclear |  |



5. Reductions of RN=NR Species










6. Reactions of R(H)N-N(H)R Species



7. Mono-Nitrogen Containing Lanthanide Complexes



8. Conclusions
References and Notes
- Haber, F.; van Oordt, G. Formation of ammonia from the elements. Z. Anorg. Chem. 1905, 43, 111–115. [Google Scholar] [CrossRef]
- Haber, F. Über die darstellung des ammoniaks aus stickstoff und wasserstoff. Naturwissenschaften 1922, 10, 1041–1049. [Google Scholar] [CrossRef]
- Haber, F. Bemerkung zu vorstehender Notiz. Naturwissenschaften 1923, 11, 339–340. [Google Scholar] [CrossRef]
- Smith, B.E. Nitrogenase reveals its inner secrets. Science 2002, 297, 1654–1655. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Zhang, J.; Han, F.; Zhang, Z.; Weng, L.; Zhou, X. Investigations on organolanthanide derivatives with the hydrazonido (-NHNdCPh2) ligand: Synthesis, crystal structure, and reactivity. Organometallics 2009, 28, 3916–3921. [Google Scholar] [CrossRef]
- Willson, S.P.; Andrews, L. Characterization of the reaction products of laser-ablated early lanthanide metal atoms with dinitrogen. infrared spectra of LnN, LnN2, (LnN)2, and Ln(NN)x molecules. J. Phys. Chem. A 1998, 102, 10238–10249. [Google Scholar] [CrossRef]
- Evans, W.J.; Lee, D.S. Early developments in lanthanide-based dinitrogen reduction chemistry. Can. J. Chem. 2005, 83, 375–384. [Google Scholar] [CrossRef]
- Barney, B.M.; Lee, H.I.; Dos Santos, P.C.; Hoffman, B.M.; Dean, D.R.; Seefeldt, L.C. Breaking the N2 triple bond: insights into the nitrogenase mechanism. Dalton Trans. 2006, 2277–2284. [Google Scholar] [CrossRef]
- Eady, R.R. Structure-function relationships of alternative nitrogenases. Chem. Rev. 1996, 96, 3013–3030. [Google Scholar] [CrossRef] [PubMed]
- Ribbe, M.; Gadkari, D.; Meyer, O. N2 fixation by Streptomyces thermoautotrophicus involves a molybdenum-dinitrogenase and a manganese-superoxide oxidoreductase that couple N2 reduction to the oxidation of superoxide produced from O2 by a molybdenum-CO dehydrogenase. J. Biol. Chem. 1997, 42, 26627–26633. [Google Scholar] [CrossRef]
- Burgess, B.K.; Lowe, D.J. Mechanism of molybdenum nitrogenase. Chem. Rev. 1996, 96, 2983–3012. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.E. Structure, function, and biosynthesis of the metallosulfur clusters in nitrogenases. Adv. Inorg. Chem. 1999, 47, 159–218. [Google Scholar]
- Rehder, D. The coordination chemistry of vanadium as related to its biological functions. Coord. Chem. Rev. 1999, 182, 297–322. [Google Scholar] [CrossRef]
- Allen, A.D.; Senoff, C.V. Nitrogenopentammineruthenium(II) complexes. J. Chem. Soc., Chem. Commun. 1965, 24, 621–622. [Google Scholar]
- Chatt, J.; Dilworth, J.R.; Richards, R.L. Recent advances in the chemistry of nitrogen fixation. Chem. Rev. 1978, 78, 589–625. [Google Scholar] [CrossRef]
- Hidai, M. Chemical nitrogen fixation by molybdenum and tungsten complexes. Coord. Chem. Rev. 1999, 185, 99–108. [Google Scholar] [CrossRef]
- Fryzuk, M.D.; Johnson, S.A. The continuing story of dinitrogen activation. Coord. Chem. Rev. 2000, 200, 379–409. [Google Scholar] [CrossRef]
- Barriere, F. Modeling of the molybdenum center in the nitrogenase FeMo-cofactor. Coord. Chem. Rev. 2003, 236, 71–89. [Google Scholar] [CrossRef]
- Allen, A.D.; Harris, R.O.; Loescher, B.R.; Stevens, J.R.; Whiteley, R.N. Dinitrogen complexes of the transition metals. Chem. Rev. 1973, 73, 11–20. [Google Scholar] [CrossRef]
- Richards, R.L. Reactions of small molecules at transition metal sites: studies relevant to nitrogenase, an organometallic enzyme. Coord. Chem. Rev. 1996, 154, 83–97. [Google Scholar] [CrossRef]
- Chatt, J.; Pearman, A.J.; Richards, R.L. Conversion of dinitrogen in its molybdenum and tungsten complexes into ammonia and possible relevance to the nitrogenase reaction. Dalton Trans. 1977, 1852–1860. [Google Scholar] [CrossRef]
- Schrock, R.R. Catalytic reduction of dinitrogen to ammonia at a single molybdenum center. Acc. Chem. Res. 2005, 38, 955–962. [Google Scholar] [CrossRef] [PubMed]
- MacKay, B.A.; Fryzuk, M.D. Dinitrogen coordination chemistry: On the biomimetic borderlands. Chem. Rev. 2004, 104, 385–402. [Google Scholar] [CrossRef] [PubMed]
- Shilov, A.E. Catalytic reduction of molecular nitrogen in solutions. Russ. Chem. Bull. Int. Ed. 2003, 52, 2555–2562. [Google Scholar] [CrossRef]
- Yandulov, D.V.; Schrock, R.R. Catalytic reduction of dinitrogen to ammonia at a single molybdenum center. Science 2003, 301, 76–78. [Google Scholar] [CrossRef] [PubMed]
- Laplaza, C.E.; Cummins, C.C. Dinitrogen cleavage by a three-coordinate molybdenum (III) complex. Science 1995, 268, 861–863. [Google Scholar] [CrossRef] [PubMed]
- Laplaza, C.E.; Johnson, M.J.A.; Peters, J.C.; Odom, A.L.; Kim, E.; Cummins, C.C.; George, G.N.; Pickering, I.J. Dinitrogen cleavage by three-coordinate molybdenum(III) complexes: Mechanistic and structural data. J. Am. Chem. Soc. 1996, 118, 8623–8638. [Google Scholar] [CrossRef]
- Laplaza, C.E.; Johnson, A.R.; Cummins, C.C. Nitrogen atom transfer coupled with dinitrogen cleavage and Mo−Mo triple bond formation. J. Am. Chem. Soc. 1996, 118, 709–710. [Google Scholar] [CrossRef]
- Christian, G.; Stranger, R.; Yates, B.F.; Cummins, C.C. Rationalizing the different products in the reaction of N2 with three-coordinate MoL3 complexes. Dalton Trans. 2007, 1939–1947. [Google Scholar] [CrossRef]
- Christian, G.; Stranger, R.; Yates, B.F.; Cummins, C.C. Investigating CN– cleavage by three-coordinate M[N(R)Ar]3 complexes. Dalton Trans. 2008, 338–344. [Google Scholar] [CrossRef]
- Ohki, Y.; Fryzuk, M.D. Dinitrogen activation by group 4 metal complexes. Angew. Chem. Int. Ed. 2007, 46, 3180–3183. [Google Scholar] [CrossRef]
- Fryzuk, M.D.; Love, J.B.; Rettig, S.J.; Young, V.G. Transformation of coordinated dinitrogen by reaction with dihydrogen and primary silanes. Science 1997, 275, 1445–1447. [Google Scholar] [CrossRef]
- MacLachlan, E.A.; Fryzuk, M.D. Synthesis and reactivity of side-on-bound dinitrogen metal complexes. Organometallics 2006, 25, 1530–1543. [Google Scholar] [CrossRef]
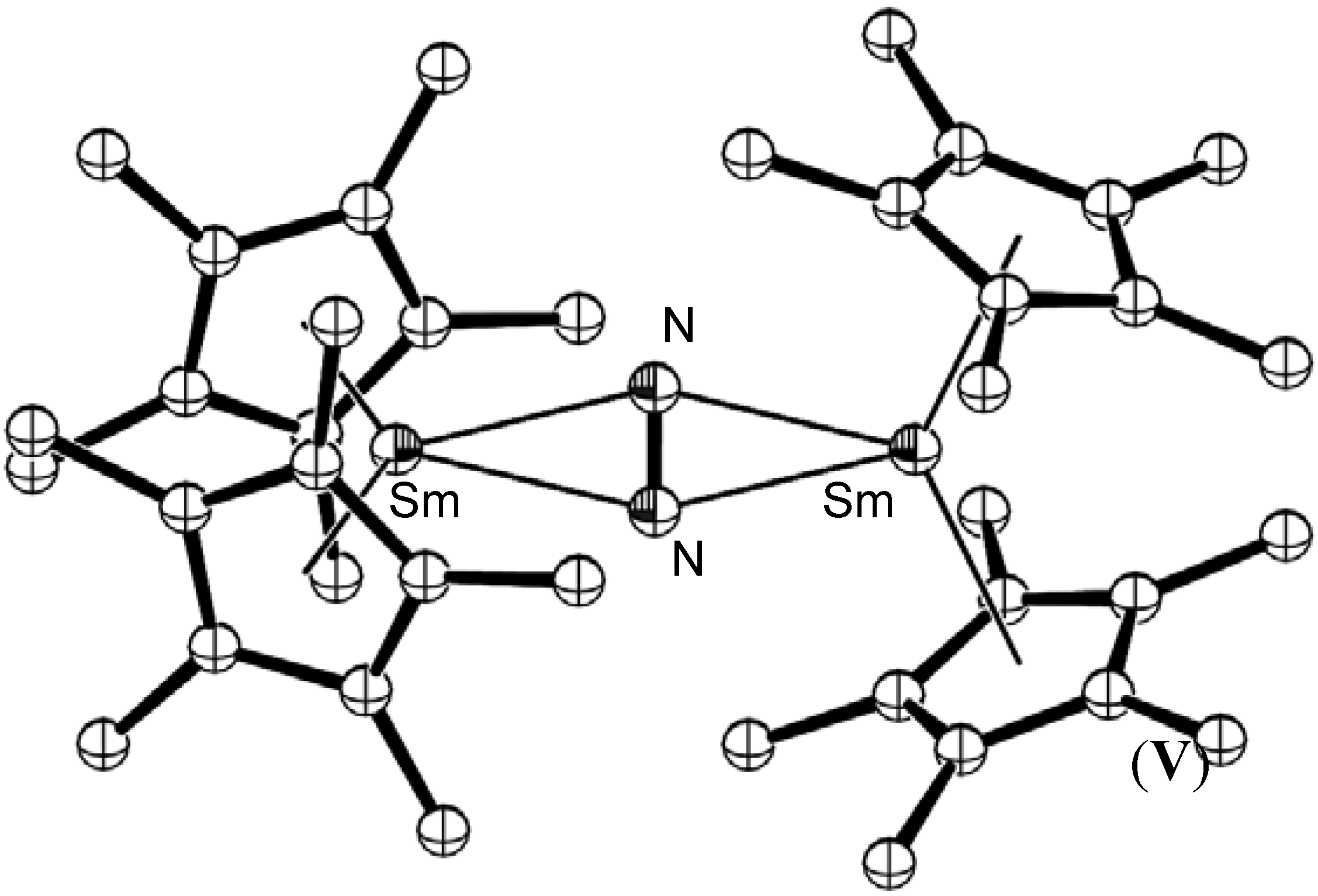
- Evans, W.J.; Ulibarri, T.A.; Ziller, J.W. Isolation and X-ray crystal structure of the first dinitrogen complex of an f-element metal, [(C5Me5)2Sm]2N2. J. Am. Chem. Soc. 1988, 110, 6877–6879. [Google Scholar] [CrossRef]
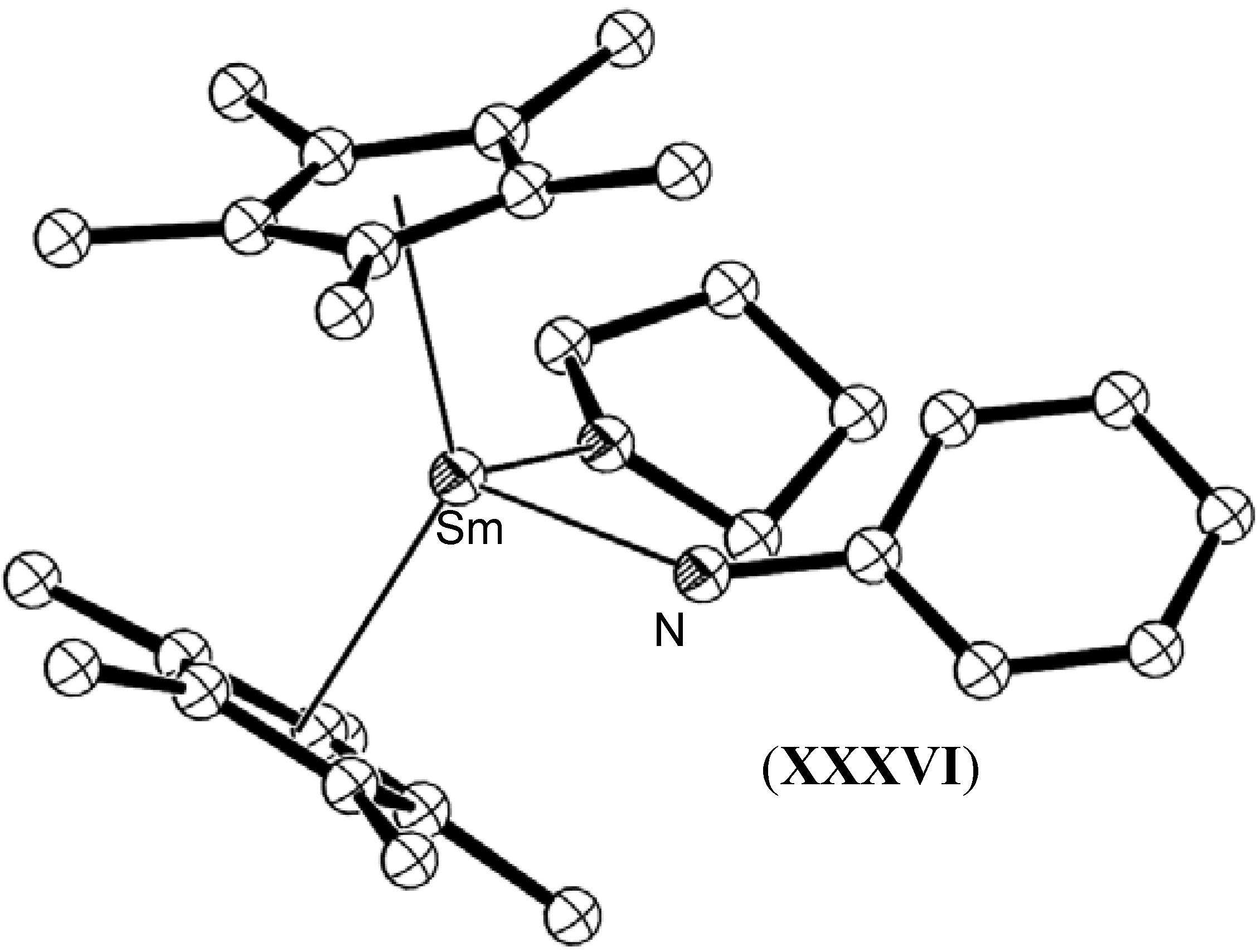
- Evans, W.J.; Kociok-Köhn, G.; Leong, V.S.; Ziller, J.W. Reactivity of hydrazines with organometallic samarium complexes and the X-ray crystal structures of (C5Me5)2Sm(η2-PhNHNPh)(THF), (C5Me5)2Sm(NHPh)(THF), and [(C5Me5)2Sm]2(μ-η2 :η2-HNNH). Inorg. Chem. 1992, 31, 3592–3600. [Google Scholar] [CrossRef]
- Fryzuk, M.D. Side-on end-on bound dinitrogen: An activated bonding mode that facilitates functionalizing molecular nitrogen. Acc. Chem. Res. 2009, 42, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Evans, W.J.; Zucchi, G.; Ziller, J.W. Dinitrogen reduction by TmII, DyII, and NdII with simple amide and aryloxide ligands. J. Am. Chem. Soc. 2003, 125, 10–11. [Google Scholar] [CrossRef] [PubMed]
- Evans, W.J.; Allen, N.T.; Ziller, J.W. Expanding divalent organolanthanide chemistry: The first organothulium(II) complex and the in situ organodysprosium(II) reduction of dinitrogen. Angew. Chem. Int. Ed. 2002, 41, 359–361. [Google Scholar] [CrossRef]
- Evans, W.J.; Fang, M.; Zucchi, G.; Furche, F.; Ziller, J.W.; Hoekstra, R.; Zink, J. Isolation of dysprosium and yttrium complexes of a three-electron reduction product in the activation of dinitrogen, the (N2)3- radical. J. Am. Chem. Soc. 2009, 131, 11195–11202. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.; Dubé, T.; Gambarotta, S.; Yap, G.P.A. Dinitrogen labile coordination versus four-electron reduction, THF cleavage, and fragmentation promoted by a (calix-tetrapyrrole)Sm(II) complex. Organometallics 2000, 19, 4820–4827. [Google Scholar] [CrossRef]
- Jubb, J.; Gambarotta, S. Dinitrogen reduction operated by a samarium macrocyclic complex— encapsulation of dinitrogen into a Sm2Li4 metallic cage. J. Am. Chem. Soc. 1994, 116, 4477–4478. [Google Scholar] [CrossRef]
- Campazzi, E.; Solari, E.; Floriani, C.; Scopelliti, R. The fixation and reduction of dinitrogen using lanthanides: praseodymium and neodymium meso-octaethylporphyrinogen–dinitrogen complexes. Chem. Commun. 1998, 2603–2604. [Google Scholar] [CrossRef]
- Evans, W.J.; Lee, D.S.; Ziller, J.W.; Kaltsoyannis, N. Trivalent [(C5Me5)2(THF)Ln]2(μ-η2:η2-N2) complexes as reducing agents including the reductive homologation of CO to a ketene carboxylate, (μ-η4-O2C-C=C=O)2-. J. Am. Chem. Soc. 2006, 128, 14176–14184. [Google Scholar] [CrossRef] [PubMed]
- Evans, W.J.; Lee, D.S.; Rego, D.B.; Perotti, J.M.; Kozimor, S.A.K.; Moore, E.K.; Ziller, J.W. Expanding dinitrogen reduction chemistry to trivalent lanthanides via the LnZ3/Alkali metal reduction system: Evaluation of the generality of forming Ln2(μ-η2:η2-N2) complexes via LnZ3/K. J. Am. Chem. Soc. 2004, 126, 14574–14582. [Google Scholar] [CrossRef] [PubMed]
- Evans, W.J.; Lee, D.S.; Lie, C.; Ziller, J.W. Expanding the LnZ3/Alkali-metal reduction system to organometallic and heteroleptic precursors: Formation of dinitrogen derivatives of lanthanum. Angew. Chem. Int. Ed. 2004, 43, 5517–5519. [Google Scholar] [CrossRef]
- Evans, W.J.; Lee, D.S.; Johnston, M.A.; Ziller, J.W. The elusive (C5Me4H)3Lu: Its synthesis and LnZ3/K/N2 reactivity. Organometallics 2005, 24, 6393–6397. [Google Scholar] [CrossRef]
- Evans, W.J.; Lorenz, S.E.; Ziller, J.W. Investigating metal size effects in the Ln2(µ-η2:η2-N2) reduction system: Reductive reactivity with complexes of the largest and smallest trivalent lanthanide ions, La3+ and Lu3+. Inorg. Chem. 2009, 48, 2001–2009. [Google Scholar] [CrossRef] [PubMed]
- Evans, W.J.; Drummond, D.K.; Bott, S.G.; Atwood, J.L. Reductive distortion of azobenzene by an organosamarium(II) reagent to form [(C5Me5)2Sm]2(C6H5)2N2: an X-ray crystallographic snapshot of an agostic hydrogen complex on an ortho-metalation reaction coordinate. Organometallics 1986, 5, 2389–2391. [Google Scholar] [CrossRef]
- Evans, W.J.; Drummond, D.K.; Chamberlain, L.R.; Doedens, R.J.; Bott, S.G.; Zhang, H.; Atwood, J.L. Synthetic, structural, and reactivity studies of the reduction and CO derivatization of azobenzene mediated by divalent lanthanide complexes. J. Am. Chem. Soc. 1988, 110, 4983–4994. [Google Scholar] [CrossRef]
- Takats, J.; Zhang, X.W. Synthesis and structure of bis[hydrotris(3,5-dimethylpyrazolyl)borato]samarium(II), Sm[HB(3,5-Me2pz)3]2, and the product of its reaction with azobenzene. Organometallics 1993, 12, 4286–4288. [Google Scholar] [CrossRef]
- Yuan, F.; Liu, X. Materials, nanoscience and catalysis crystallographic report: Crystal structure of bis(2,4,6-tri-tert-butylphenolato-O) bis(tetrahydrofuran-O) samarium (N,N-2-azobenzene) diethyl ether solvate. Appl. Organometal. Chem. 2005, 19, 877–878. [Google Scholar] [CrossRef]
- Evans, W.J.; Champagne, T.M.; Ziller, J.W. Synthesis and reactivity of mono(pentamethylcyclopentadienyl) tetraphenylborate lanthanide complexes of ytterbium and samarium: Tris(ring) precursors to (C5Me5)Ln moieties. Organometallics 2007, 26, 1204–1211. [Google Scholar] [CrossRef]
- Turcitu, D.; Nief, F.; Ricard, L. Structure and reactivity of homoleptic samarium(II) and thulium(II) phospholyl complexes. Chem. Eur. J. 2003, 9, 4916–4923. [Google Scholar] [CrossRef] [PubMed]
- Roitershtein, D.M.; Lyssenko, K.A.; Belyakov, P.A.; Antipin, M.Y.; Petrov, E.S. Reaction of cyclopentadienyllutetium anthracenide with azobenzene. Synthesis and molecular and crystal structure of the binuclear complex C5H5(THF)Lu(μ-η2: η2-PHN-NPH)2(THF)2 and its dynamic behavior in solution. Izv. Akad. Navk SSSR (Russ. Chem. Bull.) 1997, 46, 1590–1594. [Google Scholar]
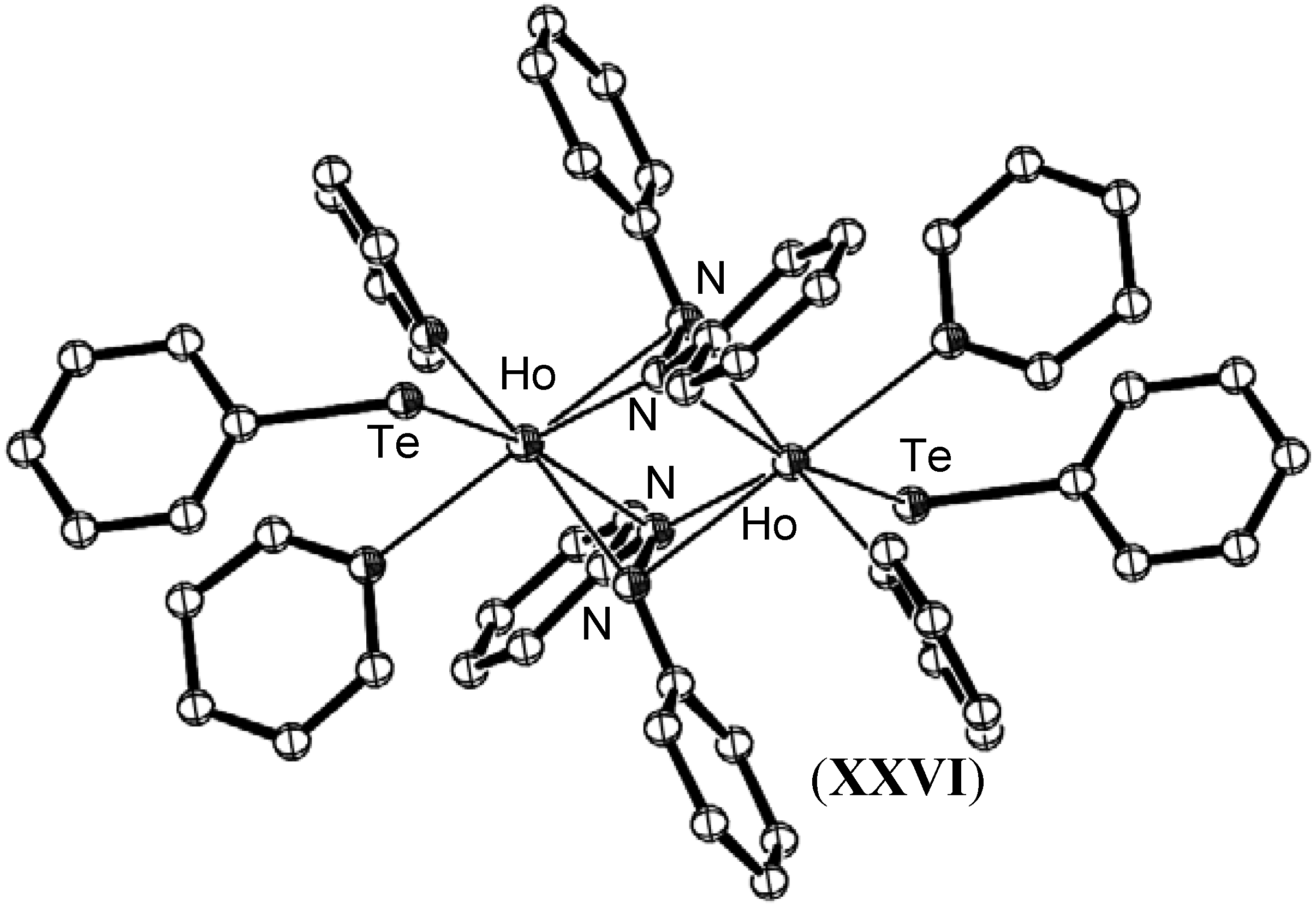
- Kornienko, A.; Freedman, D.; Emge, T.J.; Brennan, J.G. Heteroleptic lanthanide compounds with chalcogenolate ligands: Reduction of PhNNPh/PhEEPh (E = Se or Te) mixtures with Ln (Ln = Ho, Er, Tm, Yb). Thermolysis can give LnN or LnE. Inorg. Chem. 2001, 40, 140–145. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.G.; Stevens, E.D.; Nolan, S.P. Synthesis and structural characterization of a tetranuclear organolanthanide hydrazido complex. Organometallics 1992, 11, 1011–1013. [Google Scholar] [CrossRef]
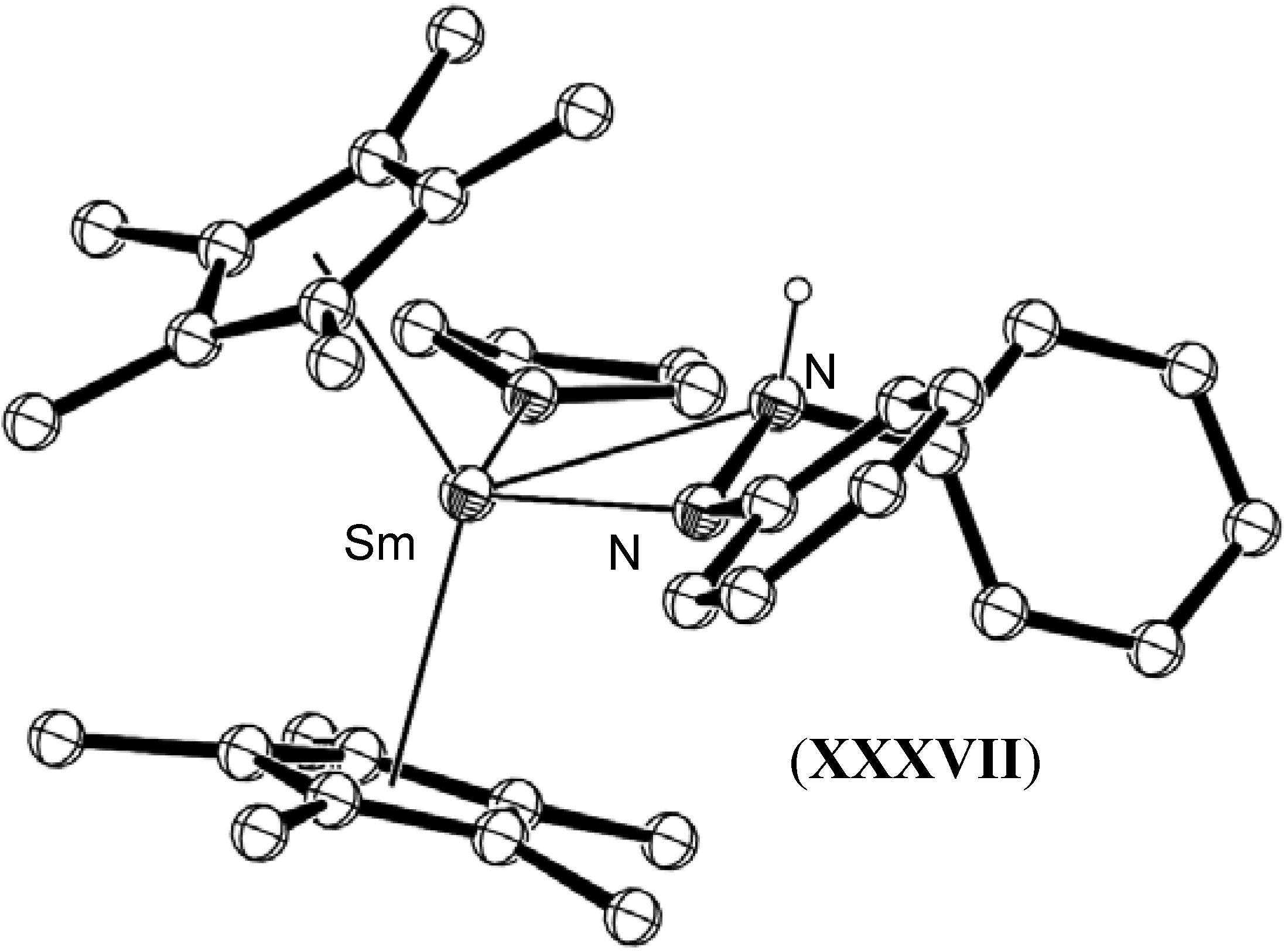
- Evans, W.J.; Kociok-Köhn, G.; Ziller, J.W. Synthesis and structure of a mononuclear η2-hydrazine complex by protonation of an [N2H2]2- complex. Angew. Chem., Int. Ed. Engl. 1992, 31, 1081–1082. [Google Scholar] [CrossRef]
- Emel´Yanova, N.S.; Bochkarev, M.N.; Schumann, H.; Loebel, J.; Esser, L. Synthesis and crystal and molecular structure of a tetranuclear complex of samarium(III) [Sm4(μ-η2:η2-Ph2N2)4(μ3-PhN)2THF6]·2THF. Koord. Khim. 1994, 20, 789–793. [Google Scholar]
- Trifonov, A.A.; Bochkarev, M.N.; Schumann, H; Loebel, J. Reduction of azobenzene by naphthaleneytterbium: A tetranuclear ytterbium(III) complex combining 1,2-diphenylhydrazido(2-) and phenylimido ligands. Angew. Chem., Int. Ed. Engl. 1991, 30, 1149–1151. [Google Scholar] [CrossRef]
- Xie, Z.; Wang, S.; Yang, Q.; Mak, T. Synthesis and structure of the first tetranuclear organolanthanide cluster containing a μ4-imido group. Organometallics 1999, 18, 1578–1579. [Google Scholar] [CrossRef]
- Wang, S.; Yang, Q.; Mak, T.; Xie, Z. Synthesis and structural characterization of novel organolanthanide clusters containing amido and imido groups. Organometallics 1999, 18, 5511–5517. [Google Scholar] [CrossRef]
- Cui, D.; Nishiura, M.; Hou, Z. Lanthanide-imido complexes and their reactions with benzonitrile. Angew. Chem. Int. Ed. 2005, 44, 959–962. [Google Scholar] [CrossRef]
- Cui, D.; Tardif, O.; Hou, Z. Tetranuclear Rare Earth Metal Polyhydrido Complexes Composed of “(C5Me4SiMe3)LnH2” Units. Unique Reactivities toward Unsaturated C-C, C-N, and C-O Bonds. J. Am. Chem. Soc. 2004, 126, 1312–1313. [Google Scholar]
- Pan, C.L.; Chen, W.; Song, S.; Zhang, H.; Li, X. Stabilization of imidosamarium(III) cubane by amidinates. Inorg. Chem. 2009, 48, 6344–6346. [Google Scholar] [CrossRef] [PubMed]
- Gordon, J.C.; Giesbrecht, G.R.; Clark, D.L.; Hay, P.J.; Keogh, D.W.; Poli, R.; Scott, B.L.; Watkin, J.G. The first example of a μ2-imido functionality bound to a lanthanide metal center: X-ray crystal structure and DFT study of [(μ-ArN)Sm(μ-NHAr)(μ-Me)AlMe2]2 (Ar = 2,6-iPr2C6H3). Organometallics 2002, 21, 4726–4734. [Google Scholar] [CrossRef]
- Chan, H.S.; Li, H.W.; Xie, Z. Synthesis and structural characterization of imido-lanthanide complexes with a metal-nitrogen multiple bond. J. Chem. Soc., Chem. Commun. 2002, 652–653. [Google Scholar] [CrossRef]
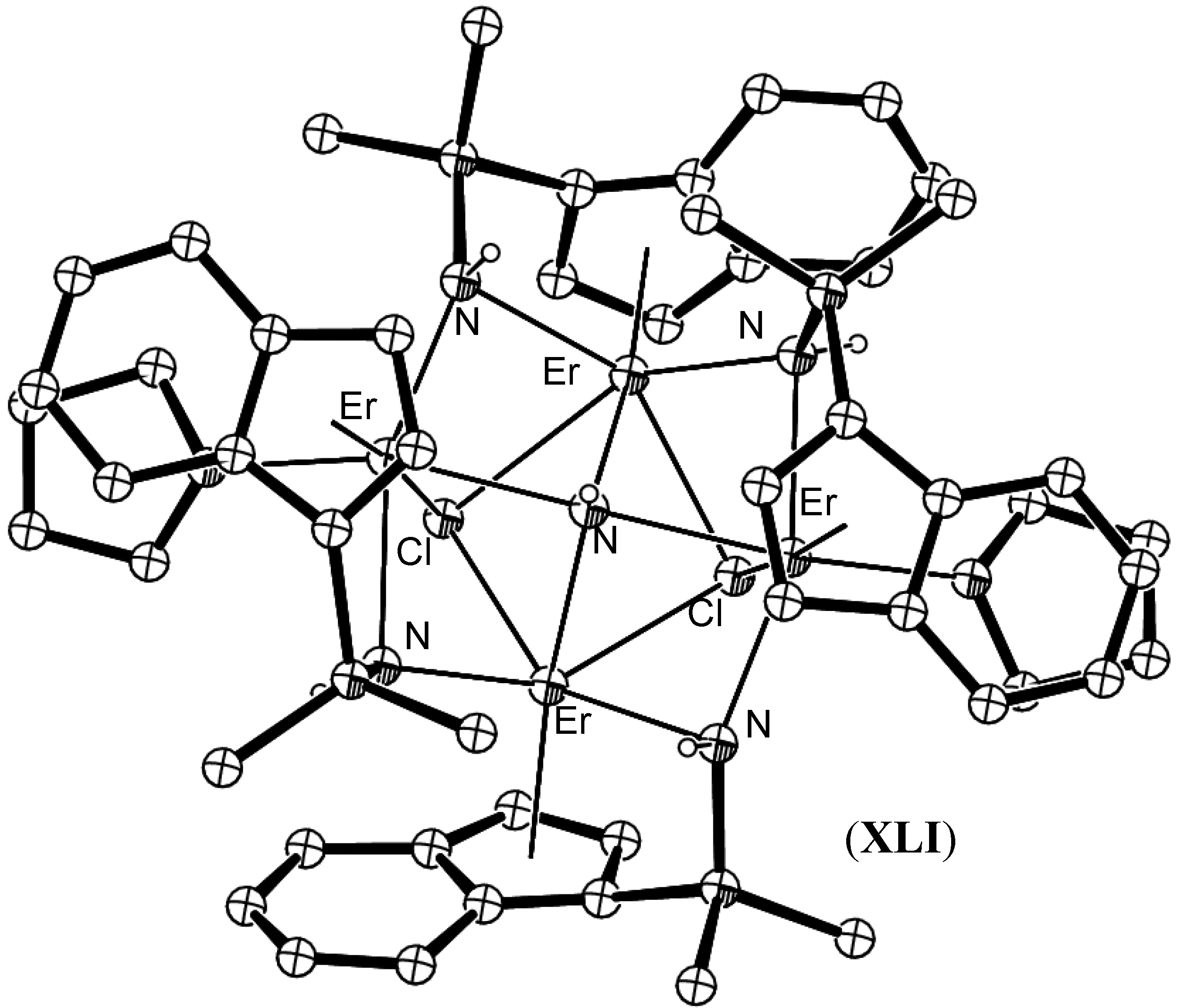
- Berthet, J.-C.; Thuéry, P.; Ephritikhine, M. Polyimido clusters of neodymium and uranium, including a cluster with an M6(μ3-N)8 core. Eur. J. Inorg. Chem. 2008, 5455–5459. [Google Scholar] [CrossRef]
- Grigsby, W.J.; Hascall, T.; Ellison, J.J.; Olmstead, M.M.; Power, P.P. Imide transfer properties and reactions of the magnesium imide ((THF)MgNPh)6: A versatile synthetic reagent. Inorg. Chem. 1996, 35, 3254–3261. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Gardiner, M.G. C-H Activation (γ-deprotonation) of a Sm(III) bis(trimethylsilyl)amide complex via macrocyclic stabilisation of the sodium counter ion. Chem. Commun. 2005, 1589–1591. [Google Scholar] [CrossRef]
- Stringer, D.N.; Gardiner, M.G. unpublished work.
- Giesbrecht, G.R.; Gordon, J.C. Lanthanide alkylidene and imido complexes. Dalton Trans. 2004, 16, 2387–2393. [Google Scholar] [CrossRef] [PubMed]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Gardiner, M.G.; Stringer, D.N. Dinitrogen and Related Chemistry of the Lanthanides: A Review of the Reductive Capture of Dinitrogen, As Well As Mono- and Di-aza Containing Ligand Chemistry of Relevance to Known and Postulated Metal Mediated Dinitrogen Derivatives. Materials 2010, 3, 841-862. https://doi.org/10.3390/ma3020841
Gardiner MG, Stringer DN. Dinitrogen and Related Chemistry of the Lanthanides: A Review of the Reductive Capture of Dinitrogen, As Well As Mono- and Di-aza Containing Ligand Chemistry of Relevance to Known and Postulated Metal Mediated Dinitrogen Derivatives. Materials. 2010; 3(2):841-862. https://doi.org/10.3390/ma3020841
Chicago/Turabian StyleGardiner, Michael G., and Damien N. Stringer. 2010. "Dinitrogen and Related Chemistry of the Lanthanides: A Review of the Reductive Capture of Dinitrogen, As Well As Mono- and Di-aza Containing Ligand Chemistry of Relevance to Known and Postulated Metal Mediated Dinitrogen Derivatives" Materials 3, no. 2: 841-862. https://doi.org/10.3390/ma3020841
APA StyleGardiner, M. G., & Stringer, D. N. (2010). Dinitrogen and Related Chemistry of the Lanthanides: A Review of the Reductive Capture of Dinitrogen, As Well As Mono- and Di-aza Containing Ligand Chemistry of Relevance to Known and Postulated Metal Mediated Dinitrogen Derivatives. Materials, 3(2), 841-862. https://doi.org/10.3390/ma3020841