Macroporous Semiconductors


Abstract
:Table of Contents
| 1. Introduction | 3007 |
| 1.1. Scope of the Paper | 3007 |
| 1.2. Some Pictures and Definitions | 3007 |
| 2. Pore Etching Experiments | 3012 |
| 2.1. Etching System and Degrees of Freedom | 3012 |
| 2.2. Current and Dissolution | 3016 |
| 2.3. First Insight into Pore Formation | 3017 |
| 3. Some General Aspects of Pores in Semiconductors | 3022 |
| 3.1. Major Differences and Common Features | 3022 |
| 3.2. Self Organization and Pore Growth Mode Transitions | 3025 |
| 3.3. Pore Geometry | 3030 |
| 3.4. A Closer Look at Self-Organization Issues | 3033 |
| 3.5. A Closer Look at Growing Deep Pores | 3036 |
| 4. Multi-Mode-In-Situ FFT Impedance Spectroscopy During Pore Etching | 3038 |
| 4.1. General Technique | 3038 |
| 4.2. Selected Results from Si Pore Etching | 3043 |
| 4.3. A Case Study for InP and GaAs | 3046 |
| 4.3.1. Studying Crysto and Curro Pores in InP | 3046 |
| 4.3.2. Crysto and Curro Pores in GaAs | 3054 |
| 5. Some Applications of Porous Semiconductors | 3056 |
| 5.1. General Remarks | 3056 |
| 5.2. Some New Applications | 3060 |
| 5.2.1. Sensors Exploiting the Piezoelectric Properties of Porous III-V | 3060 |
| 5.2.2. Single Holes with Large Aspect Ratios | 3060 |
| 5.2.3. Anodes for Li Ion Batteries | 3060 |
| 6. Summary and Conclusions | 3062 |
| Acknowledgements | 3063 |
| References and Notes | 3063 |
1. Introduction
1.1. Scope of the Paper
1.2. Some Pictures and Definitions



2. Pore Etching Experiments
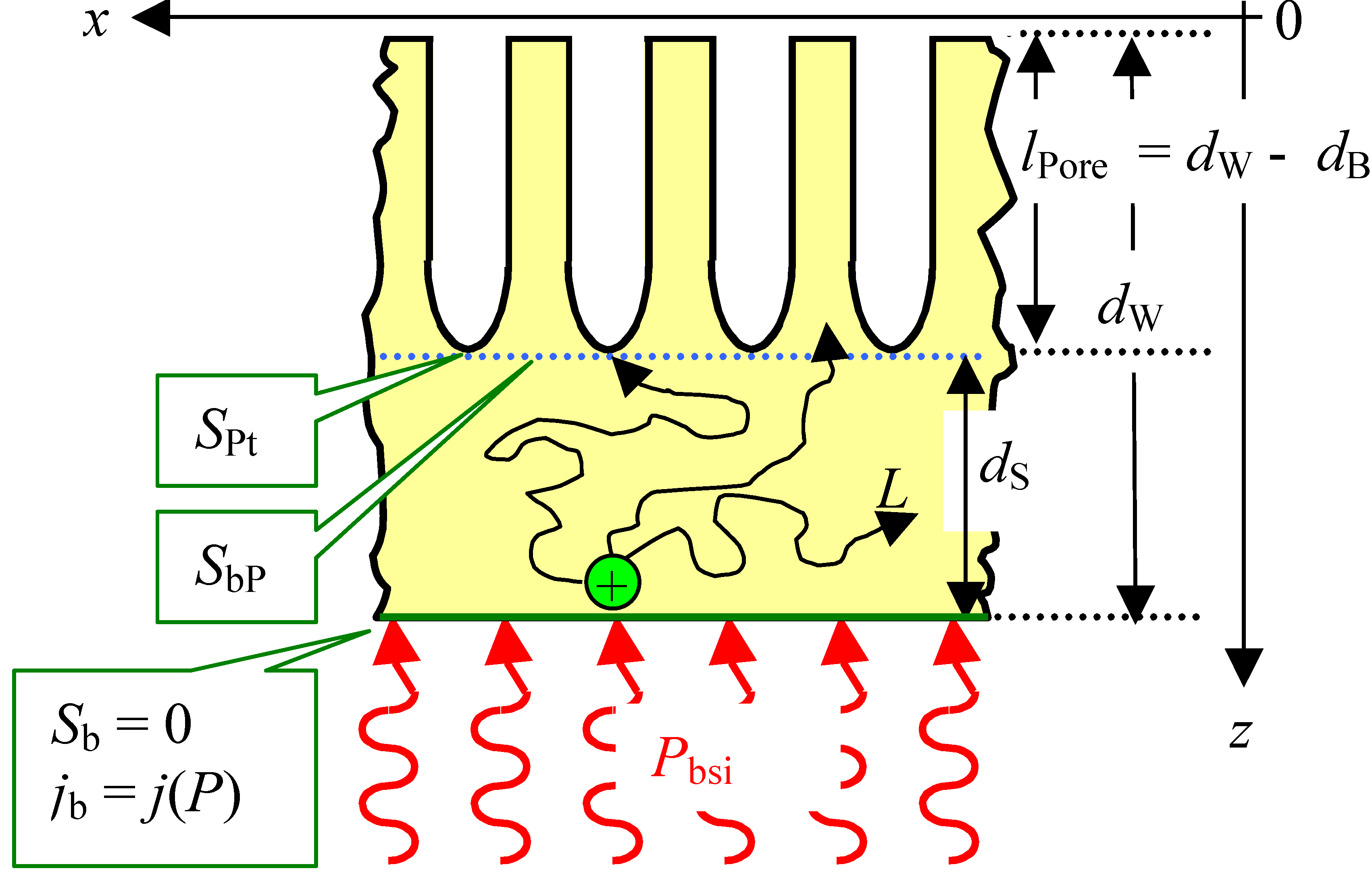
2.1. Etching System and Degrees of Freedom



2.2. Current and Dissolution
2.3. First Insight into Pore Formation
- 1)
- Direct dissolution and dissolution by oxide formation must occur “simultaneously” in a mix that produces a valence between 2 and 4.
- 2)
- This is, in a strict sense, impossible. One cannot have three different reactions taking place simultaneously, i.e., exactly at the same place at exactly the same time.
- 3)
- Holes and electrons are involved in the sum total of the dissolution reactions as given above. Holes, however, are the more important species since even for direct dissolution they are needed to initiate the reaction. This goes a long way to explain pore formation in n-type Si by some mechanism of local hole production. i.e., junction breakdown at high field strength.
- 4)
- On the other hand, it also implies that we will run into a problem understanding pore formation in p-type Si.
- 5)
- If the three reactions above do not have nearly identical time constants, the slowest one will dominate the reaction kinetics and thus the maximum local current density.
- Production of holes by illuminating the back side. Some of these holes will diffuse to the reaction front at the front side and will “automatically” end up at the pore tips (or tips initiated by lithography) because the concomitant bending of the space charge region cannot but focus holes at the tip. This SCR or “Lehmann/Föll” model [66] was (and still is) the basic model for the formation of the n-Si-macro(bsi, aqu, litho) pores. It was proposed to understand what we now would call n-Si-macro(fsi, aqu) pores, and the discovery that the first experiments immediately confirmed the prediction of the SCR model for n-Si-macro(bsi, aqu, litho) pores was seen as sufficient proof for the validity of the model for this mode of pore etching. Meanwhile, however, it has been shown that n-Si-macro(bsi/fsi, aq, litho) pore etching is more complex and not completely explained by the SCR model [11]. The SCR model is also restricted to semiconductors with a very large diffusion length of the minority carriers—in practice to very good single crystals of Si an Ge. However, in the case of Ge, back side illumination does not appear to influence pore formation very much [52], again questioning the general validity of the SCR model.
- Production of holes by electrical breakdown (tunneling or avalanche type) at points of high field strength = tips of pores (breakdown model, cf. [19]). Local hole production by breakdown is certainly a good reason to form pores in n-type semiconductors of all kinds, and there is no doubt that the breakdown model works, cf. for example, [19].

3. Some General Aspects of Pores in Semiconductors
3.1. Major Differences and Common Features

Major pore growth direction


3.2. Self Organization and Pore Growth Mode Transitions





3.3. Pore Geometry

3.4. A Closer Look at Self-Organization Issues

- 1).
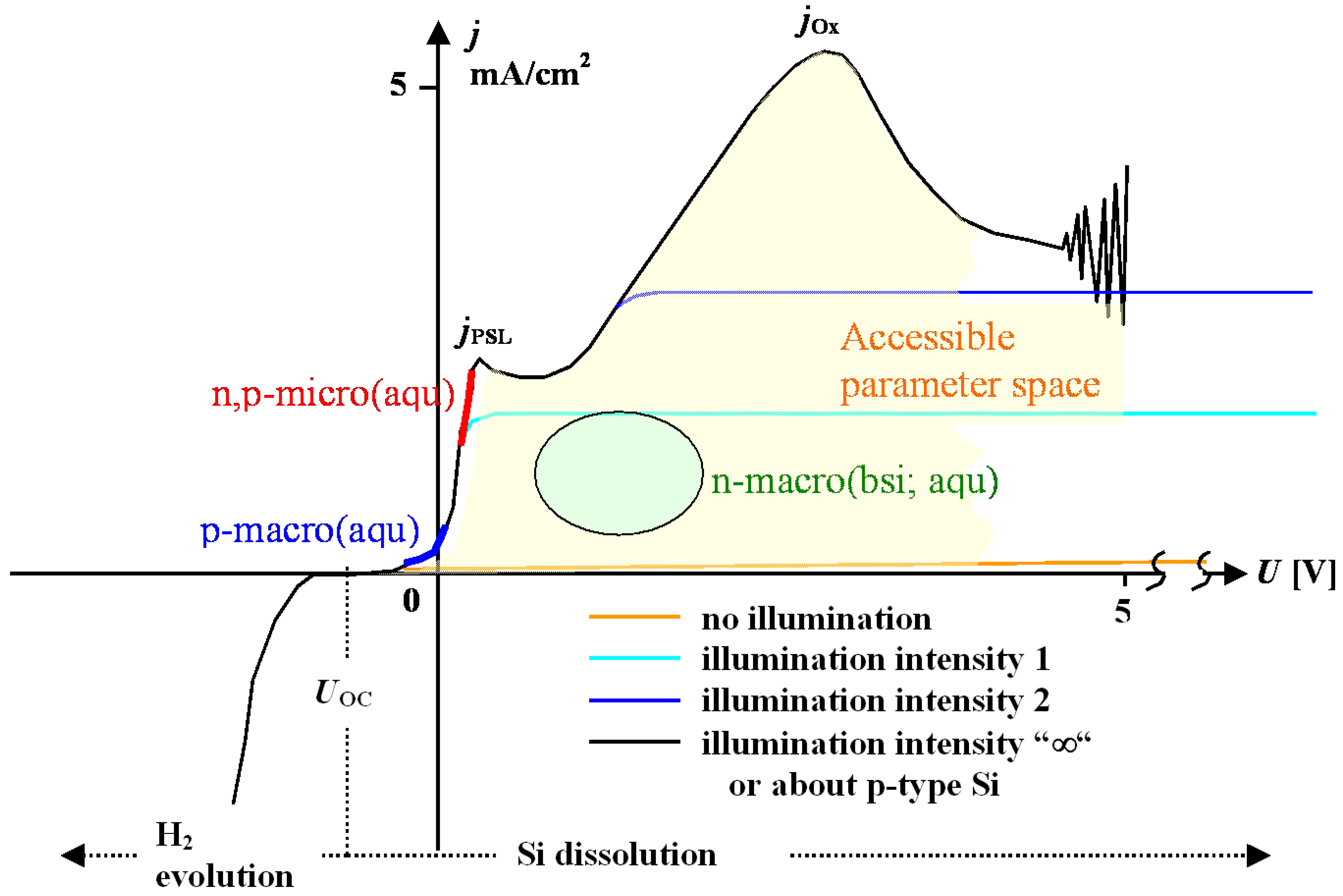
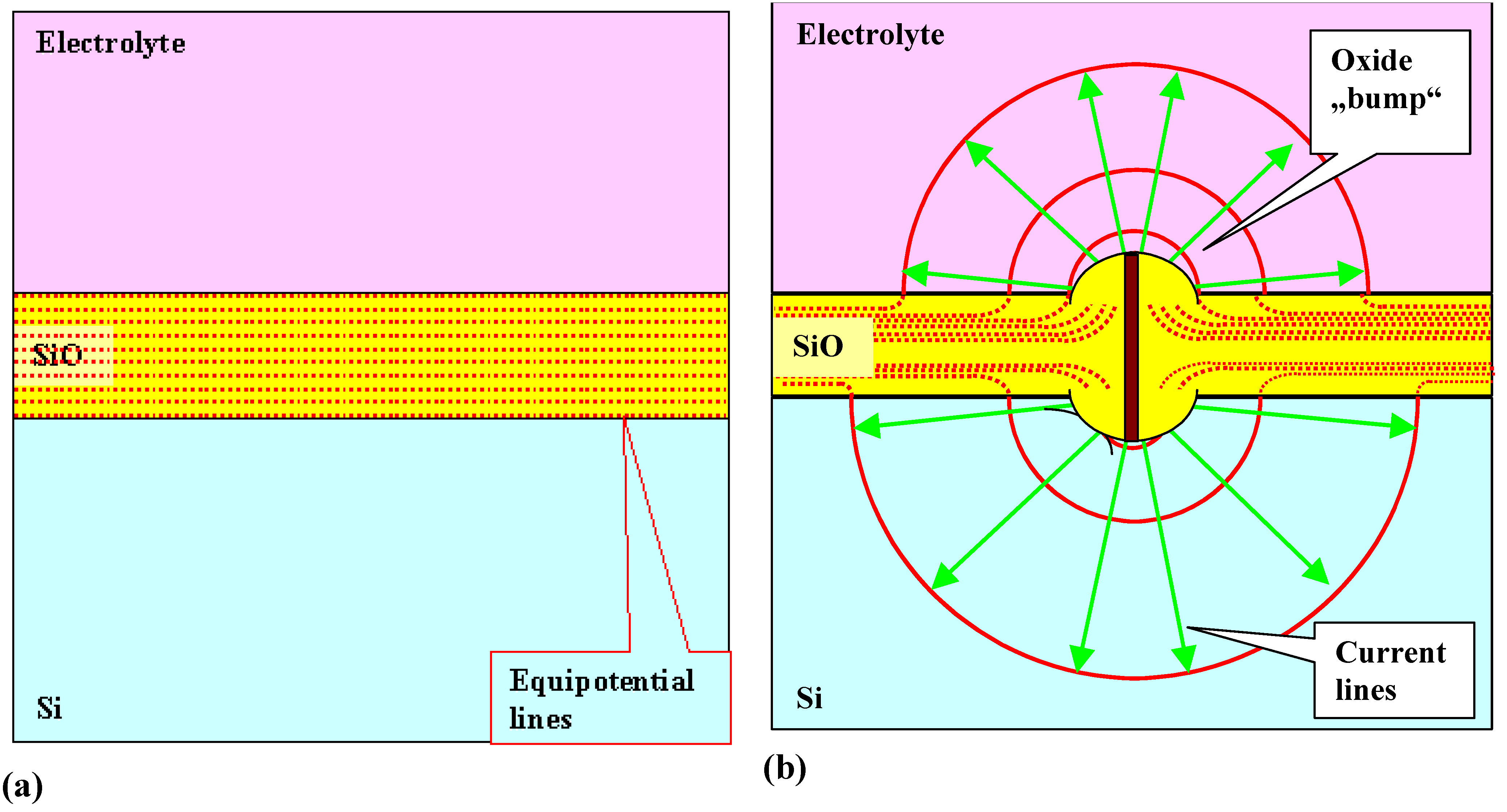
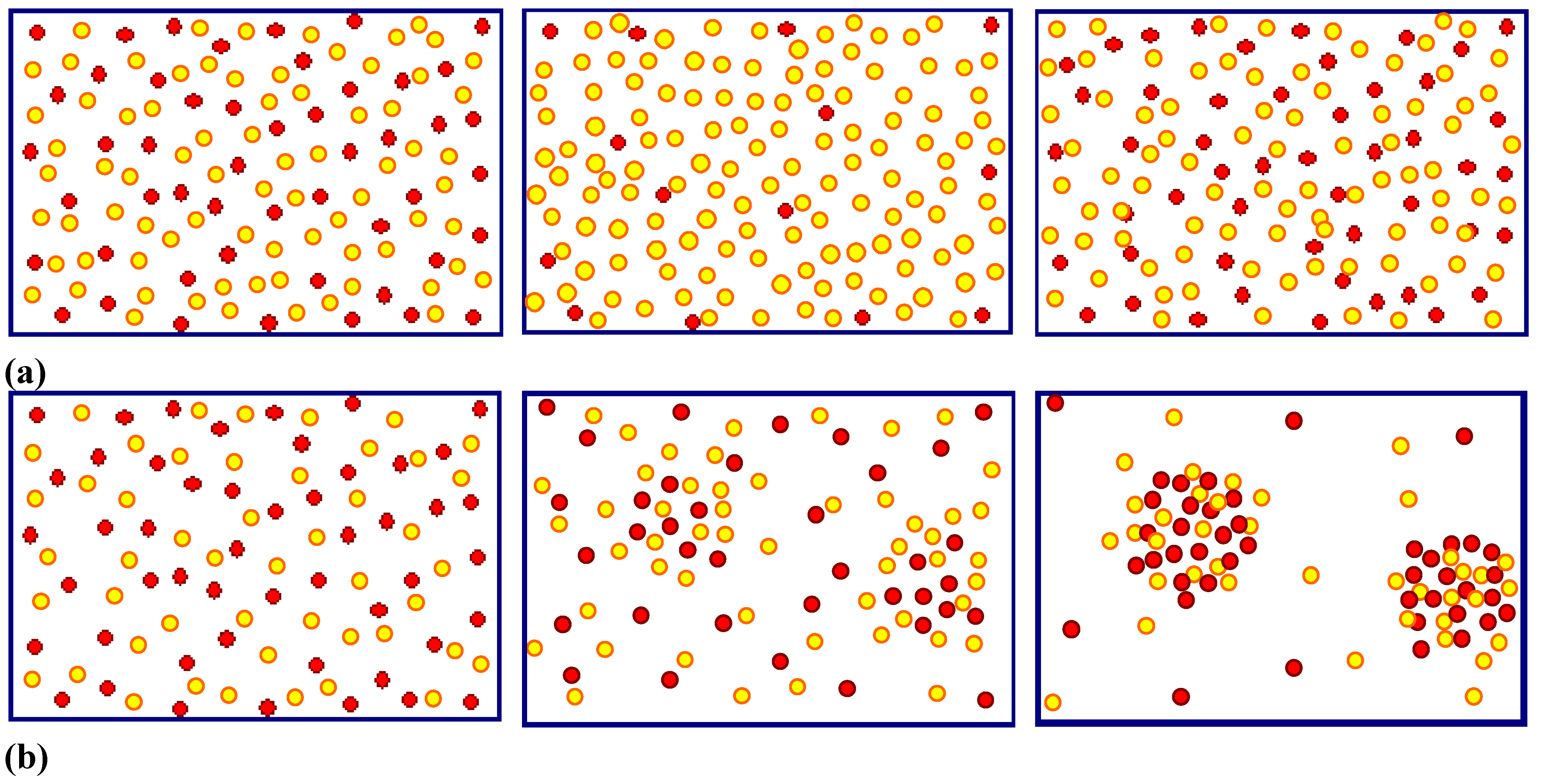
- Under conditions where direct dissolution prevails and the current is relatively large (i.e., in the micropore region of Figure 5), each individual current burst generates a nm-sized pore, “dug” by direct “divalent” dissolution and eventually closed by a small oxide lump. All that is required now to form sponge-like microporous Si is what one could call an “anti-correlation” in time: wherever a current burst has stopped, it is less likely that a new one will nucleate a short time after. This is likely to happen if there is a need to keep many current bursts “burning” all the time, and that is the case if the galvanostatically enforced current is close to a limiting value, here jPSL (cf. Figure 5). There is not enough time to wait for a new current burst until the oxide lump produced by the first one is sufficiently dissolved and a new current burst nucleates somewhere else—to the left or right of the oxide lump. The growing nanopore must dig into the Si in a kind of random walk (it cannot get too close to other pores, however), and a sponge-like network of nano-sized pores must result. We have not only “explained ”micropore formation in this way, but also the value of the first current peak at jPSL: it is simply the maximum current observable if the entire surface is covered with current bursts all the time.
- 2).
- Under conditions where direct dissolution prevails and the current is relatively small (i.e., in the macropore region of Figure 5), the situation is reversed. Current bursts correlate positively in time, i.e., it is more likely to nucleate a new one in places where an old one just expired. That appears contradictory to the first case but simply states that the nucleation probability as a function of time first is low (the pore tip is still covered with an oxide bump) but then increases (the oxide becomes thinner or disappears) and then decreases again (the surface starts to passivate with hydrogen). If a positive correlation prevails, the current bursts will start to cluster as shown in Figure 17(b) and macropore formation is inevitable

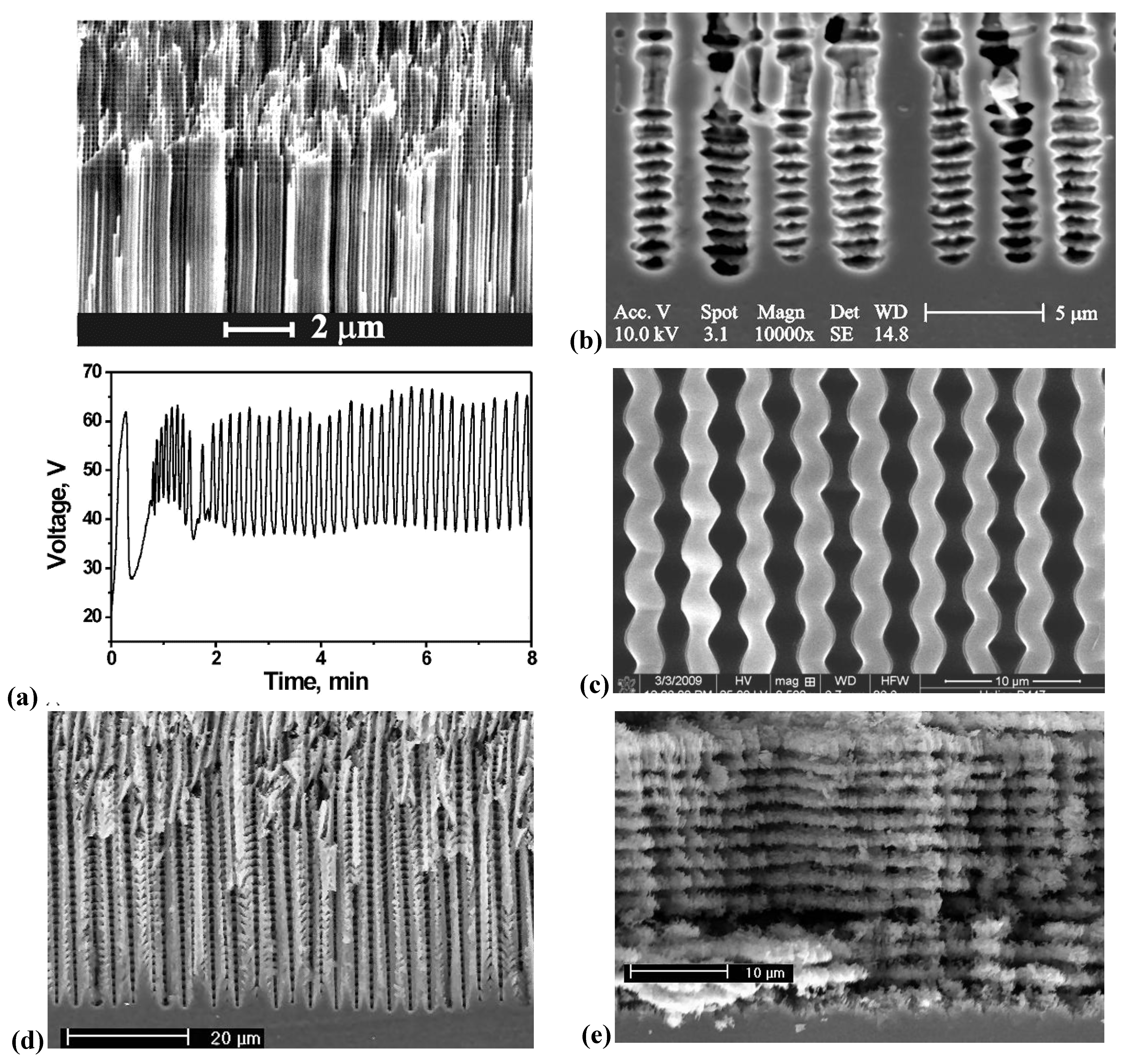
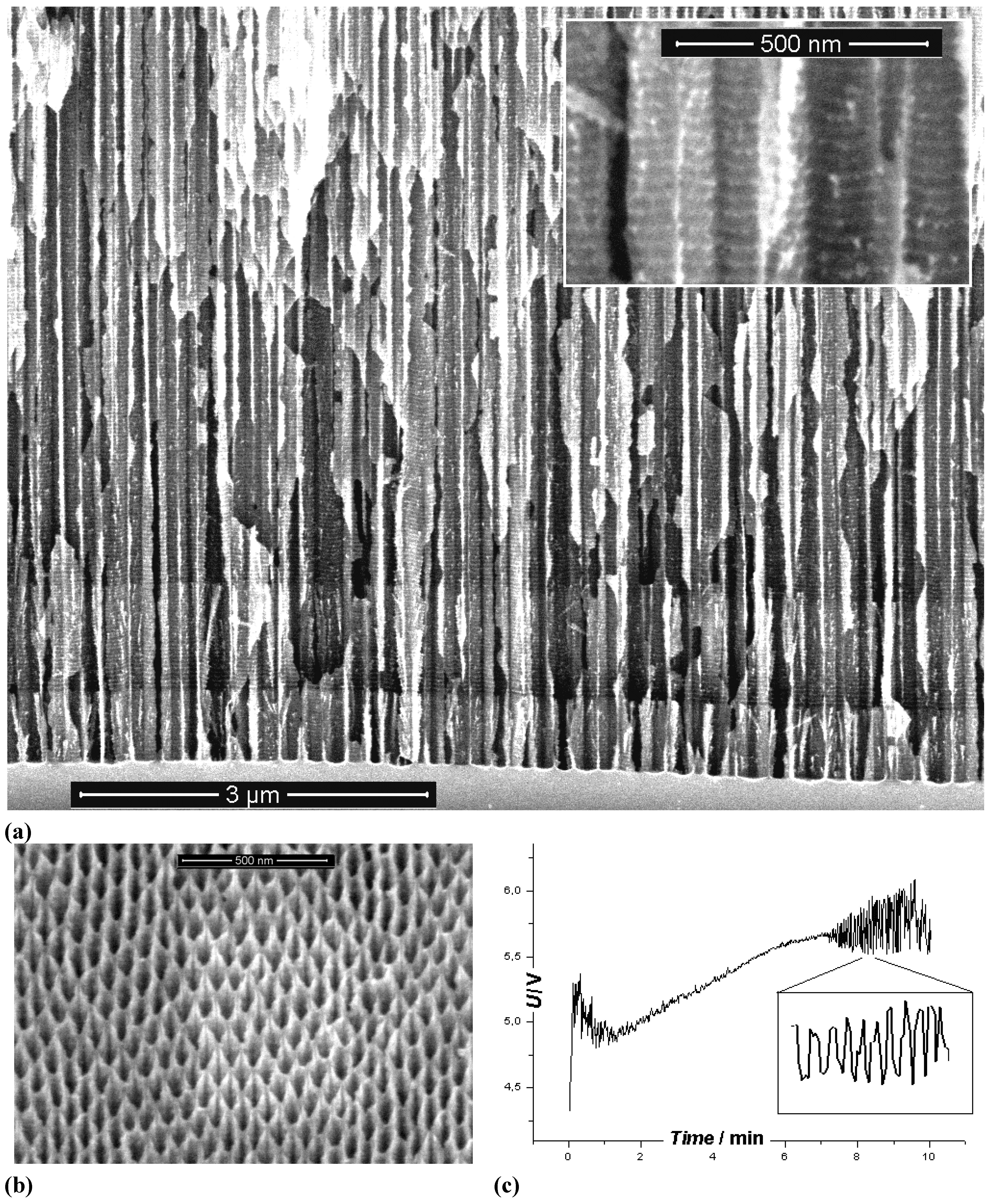
3.5. A Closer Look at Growing Deep Pores
4. Multi-Mode-In-Situ FFT Impedance Spectroscopy During Pore Etching
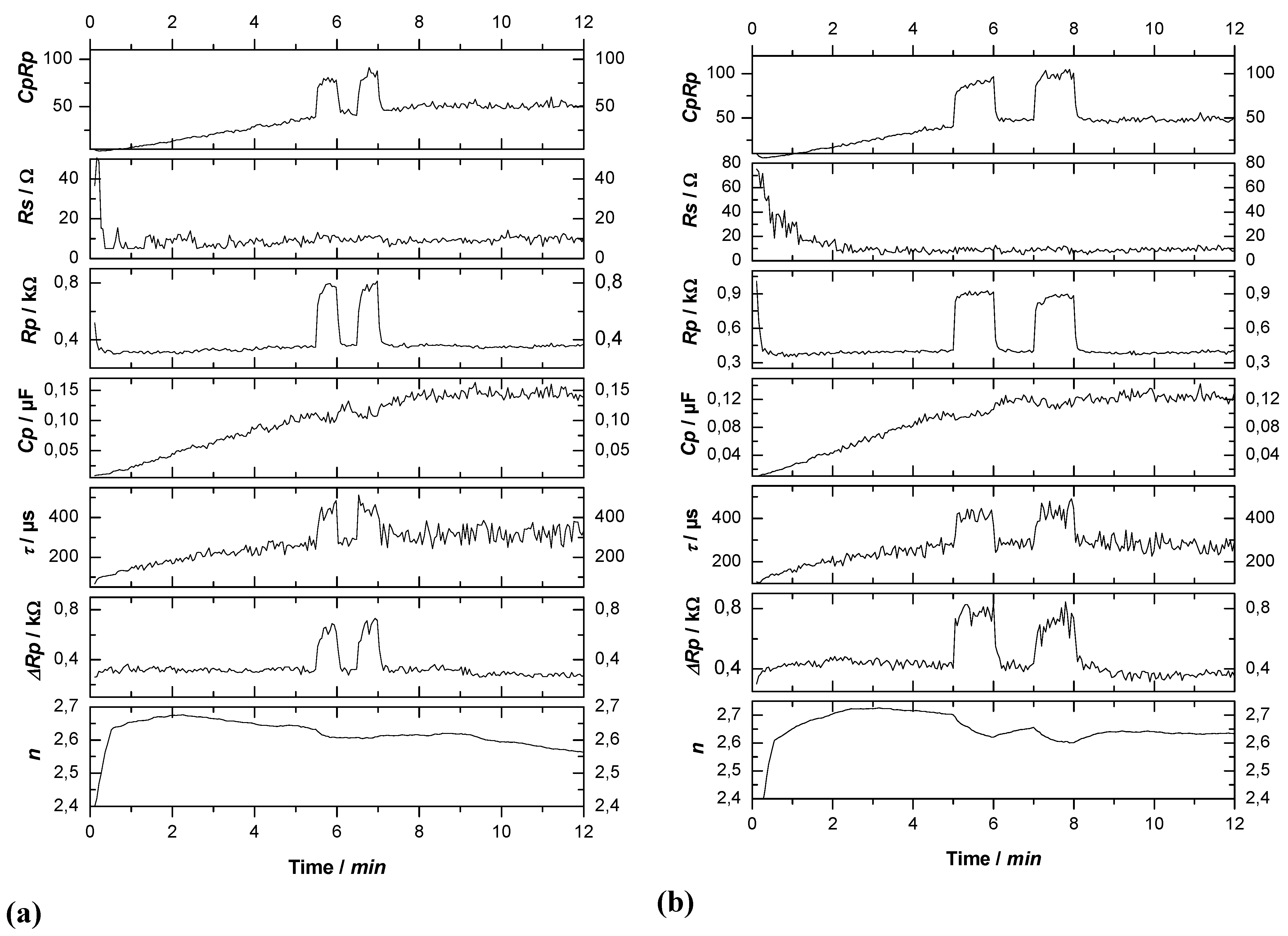
4.1. General Technique


4.2. Selected Results from Si Pore Etching



4.3. A Case Study for InP and GaAs
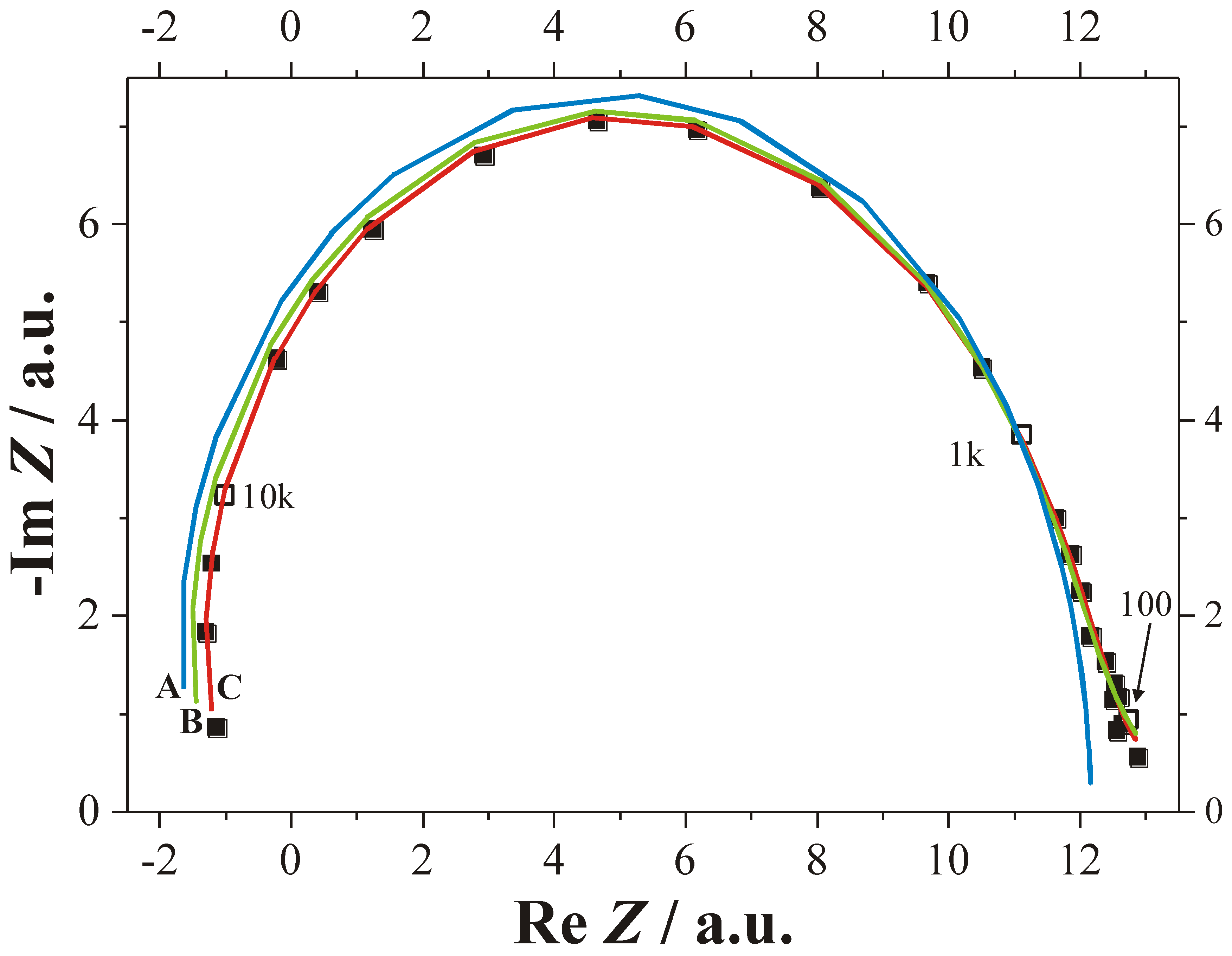
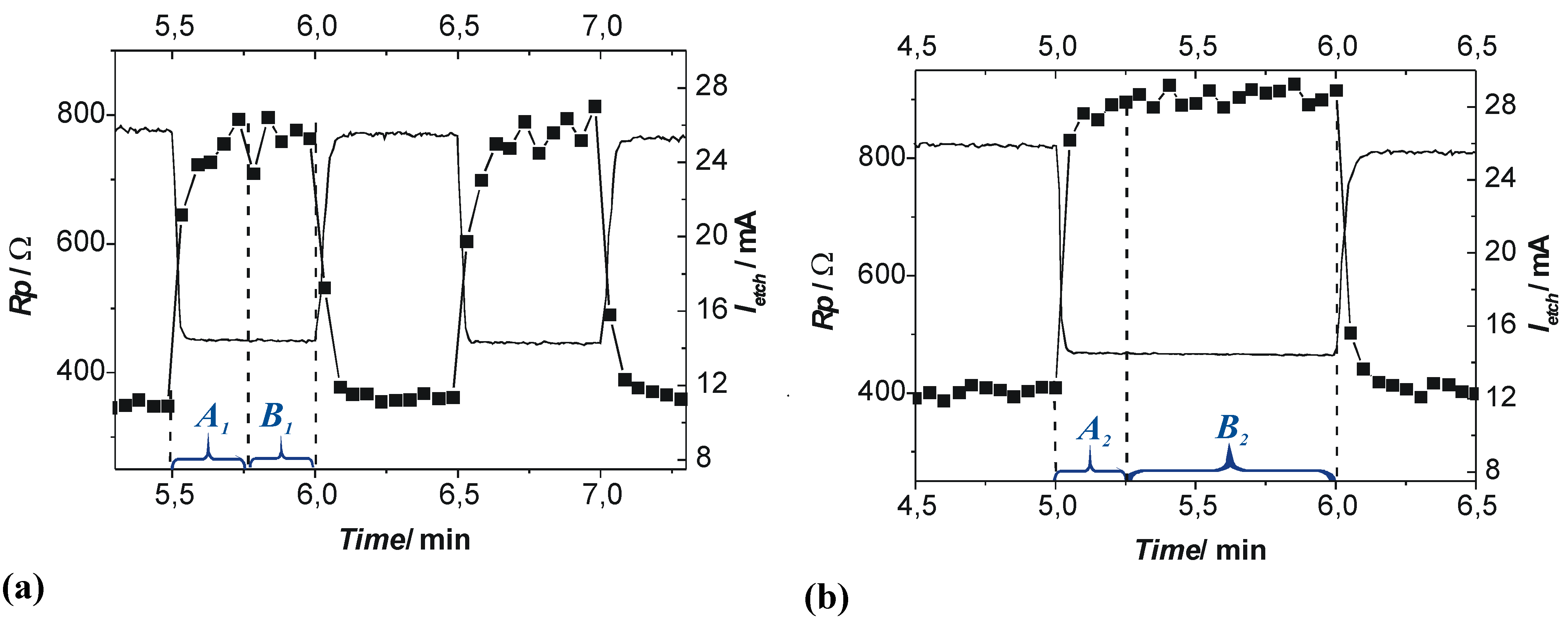
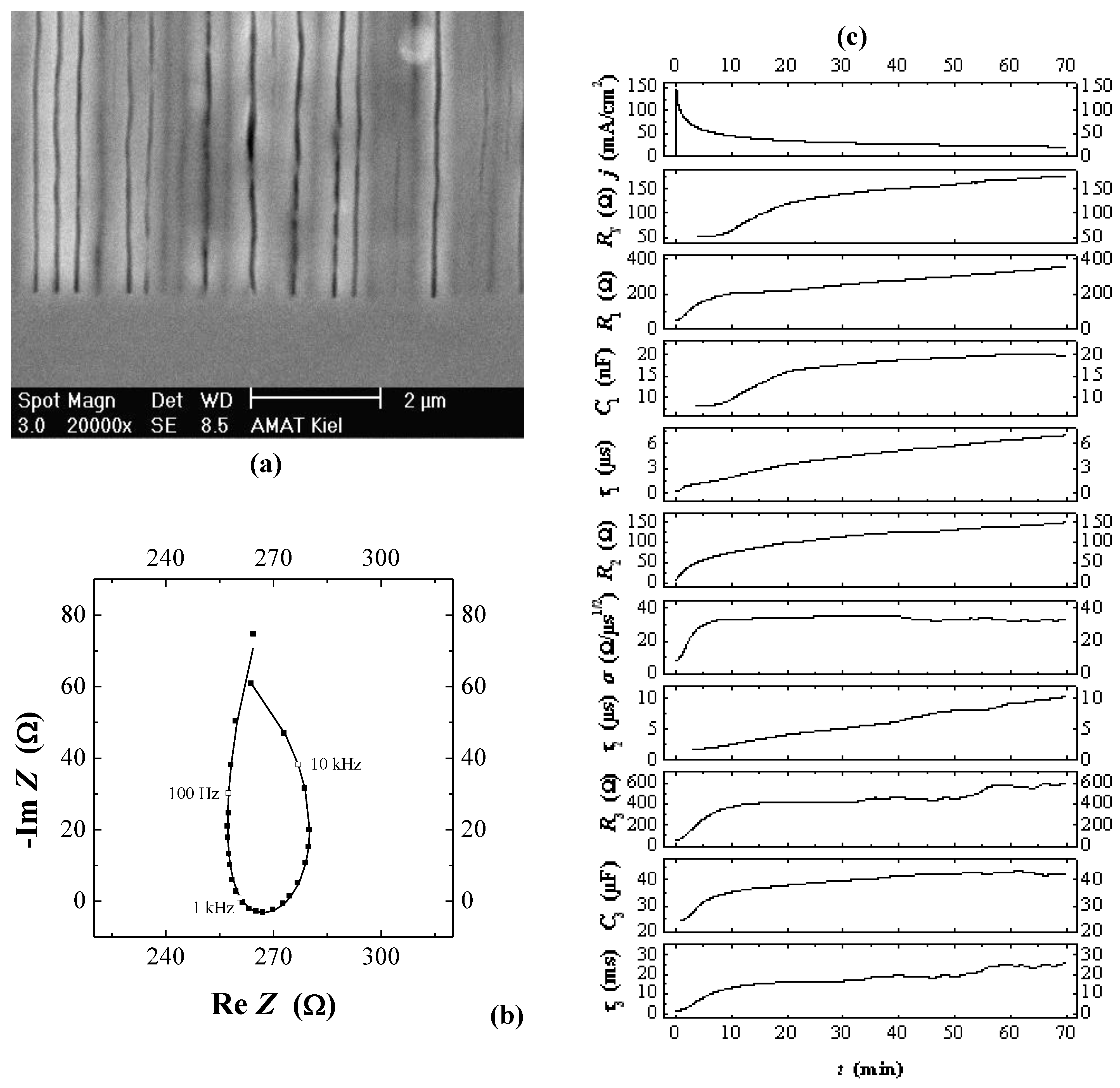
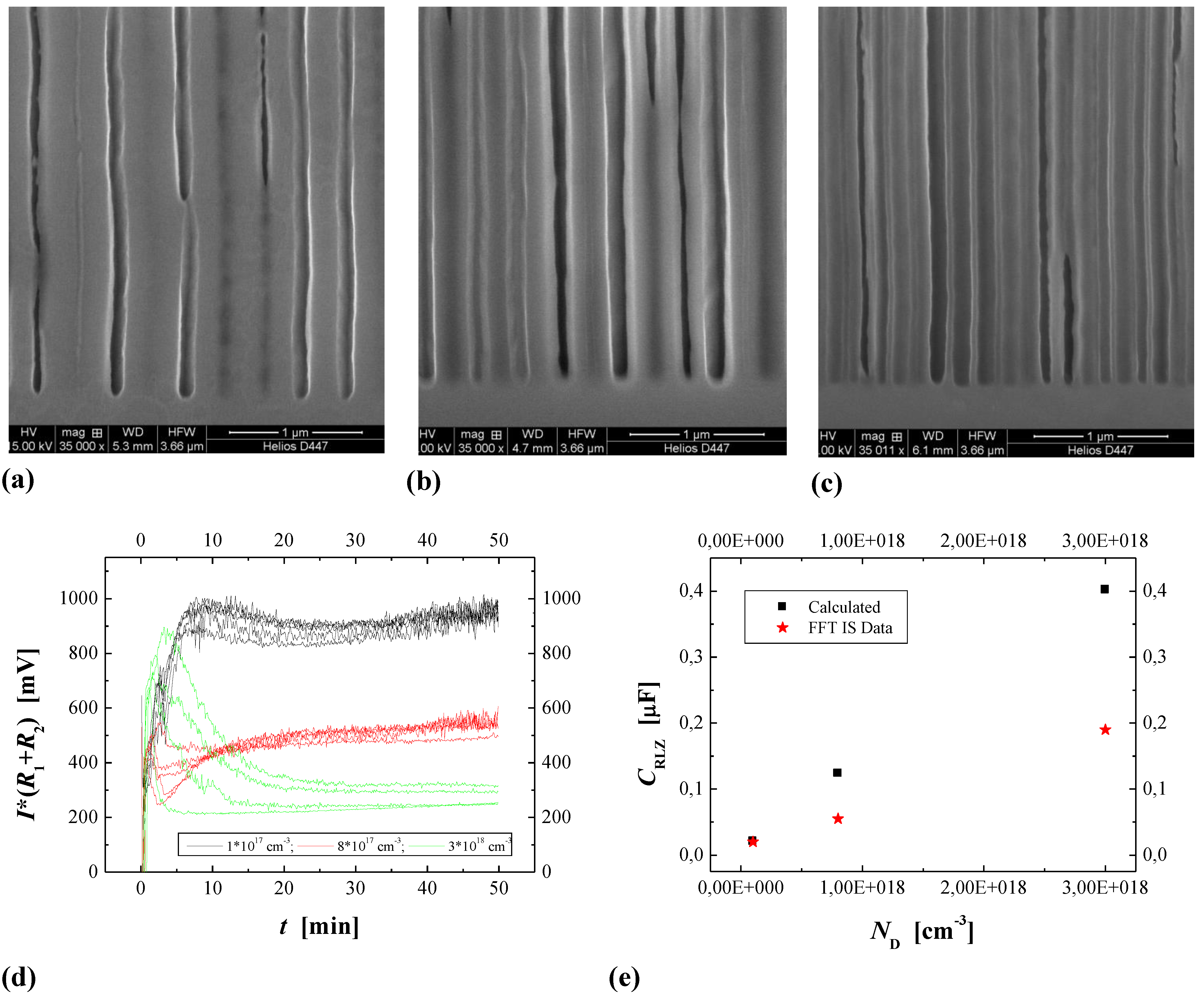
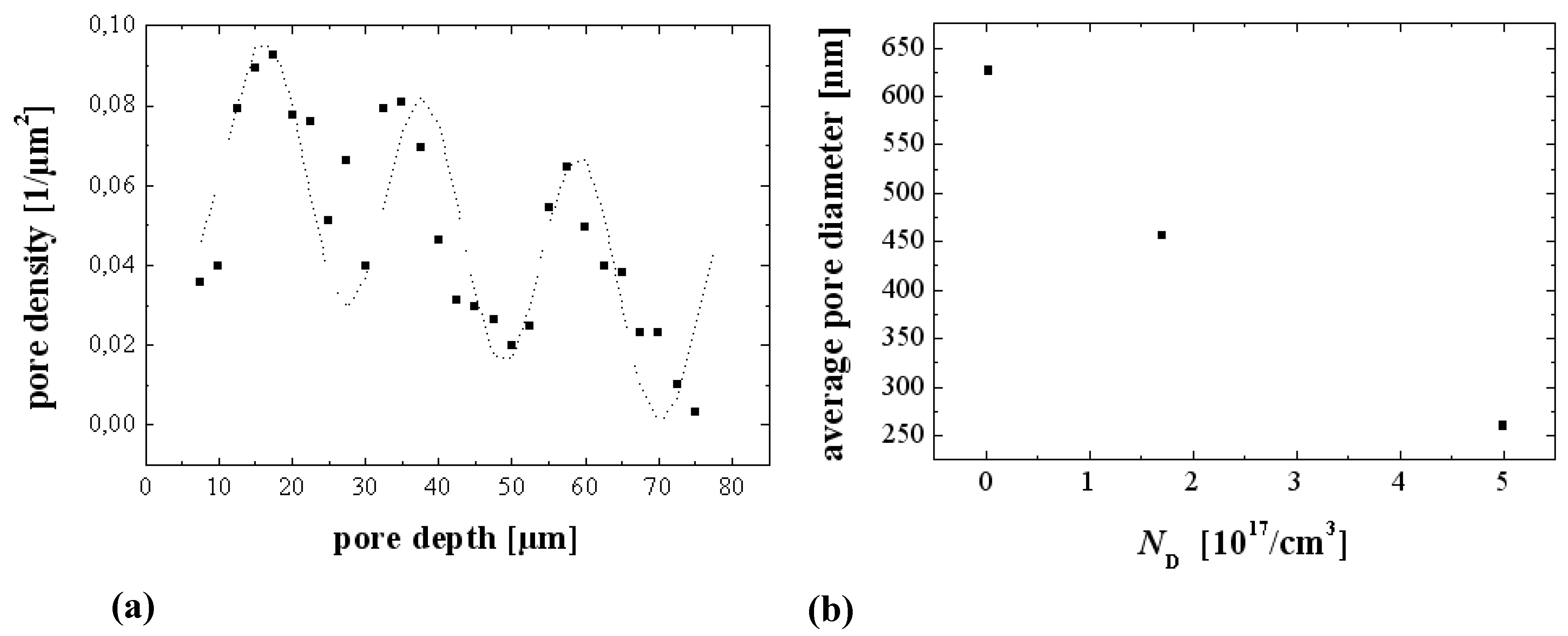
4.3.1. Studying Crysto and Curro Pores in InP






- 1)
- With the potential Uint the thickness of the space charge region dSCR can be calculated for the specific pore front geometry encountered (cf. [156]). The resulting values match the measured values (half of the pore wall thickness) very well.
- 2)
- With the potential Uint the capacitance of the space charge region CSCR can be calculated for the specific pore front geometry encountered assuming semi-spherical pore tips. The result matches well with the values for C1 as directly measured by FFT IS; this is illustrated in Figure 29(e). The somewhat higher values of the calculation for higher doping concentrations might be caused by significant deviations of the pore tip shape from an ideal semi-sphere. This changes the active area and thus C1.
- 3)
- With the potential Uint the breakdown field strength of the avalanche process can be calculated, again with the same geometrical considerations as for C1. A comparison with theoretical values [157] for solid pn-junctions shows that the values obtained in this work are somewhat smaller as one would expect and show the correct dependence on the doping concentration ND.

4.3.2. Crysto and Curro Pores in GaAs


5. Some Applications of Porous Semiconductors
5.1. General Remarks
- i)
- There are many potential applications where it does not matter that the porous material is actually a semiconductor. In other words, there are many applications outside the typical semiconductor domains of electronics and optics or optoelectronics.
- ii)
- iii)
- Some proposed applications, while perfectly viable, are rather impractical. For example, mono crystalline or multi crystalline Si solar cells come off the production line at a rate of about 1 (large) solar cell per second, leaving 1 second time for a single wafer process like pore etching. Producing many wafers in parallel would of course solve this problem but likely at prohibitive costs. Envisioning standard pore etching for some solar cell processes as proposed, e.g., in [178,179], is simply not practical. This is not to say that electrochemical processing is never useful in solar cell production; there are, in fact, considerable efforts under way to use a so-called layer transfer process involving (quick) pore etching for the production of cheap but highly efficient solar cells [180], but simply to say that economic or cost considerations cannot be neglected if one has a commercial product in mind.
- 1)
- High explosives from mesoporous Si. Two laboratories “discovered” the explosive properties of micro/mesoporous Si containing an oxidizing specimen more or less by accident (and fortunately without injuries to persons) cf. [181,182]. The mechanism is clear. The reaction Si + O2 ➔ SiO2 generates substantially more energy per kg than the explosion of TNT, and in a structure where a large percentage of all Si atoms are surface atoms the reaction can proceed rather fast. A sizeable project was undertaken to use “explosive Si” as a fuse for setting off airbags in cars [183], since it proceeds so fast that considerably more (valuable) time is available to process the sensory information, that might lead to an air bag deployment. The project did succeed in making a very reliable and easy-to-produce high explosive (we do not give the recipe for obvious reasons) but did not yet mature to production. There are also other efforts to use this effect [184]. This example provides perhaps the least intuitive application of porous Si and serves to illustrate that applications of porous Si may go far beyond the confines of traditional semiconductor uses.
- 2)
- Filters that pass X-rays in the direction perpendicular to the filter axis but not at some inclination (collimators) have been produced by filling n-Si-macro(bsi, lith, aqu) pores with Pb [185]. Filters like that are very useful for medical X-ray applications, in particular if soft tissue is to be investigated, e.g., in mammography. Working filters have been demonstrated, but no production has yet been started. We mention this old example, because it provides the by far easiest and possibly oldest technique for filling Si pores with a metal: just “dip” the porous substrate in the molten metal! Figure 32 gives an illustration. Note that other uses of porous Si for X-ray imaging are also possible by filling pores with a scintillator material [186,187].
- 3)
- In non-cubic single crystals the index of refraction is not a single number but a tensor, producing effects like birefringence [188]. Presently, advanced optics (including non-linear optics) exploiting this anisotropy must rely on what nature provides in the form of natural or laboratory-grown crystals. The effects are thus limited to “naturally occurring” numerical values of the tensor components and tensor symmetries that are always tied to the crystal symmetry. Pores in crystals break the symmetry. For example, cubic semiconductors like GaAs that contain several sets of pores [e.g., crysto pores in a {100} crystal that run along two <111> directions; cf. Figure 3, Figure 30(b), and Figure 25(a)], have a non-cubic symmetry from the viewpoint of light with a wavelength far smaller than the pore geometry. If crystals with an asymmetric pore set are produced, e.g., by cutting the specimen with an orientation somewhat off {100}, more symmetry breaking occurs and optical tensors not possible in any bulk crystals result [11]. The effect is beyond doubt, but needs yet to be demonstrated. Moreover, porous III-V crystals like GaP (and possibly also II-VI crystals) may also have very unusual non-linear properties as demonstrated in [176,177] for porous GaP, where a more than 100 fold increase of the second harmonic production was observed. This example serves to point out that so-called meta-materials can be made with porous semiconductors having properties that are so unusual that it is not surprising that they have not yet been exploited.Figure 32. (a) Part of a 150 mm diameter X-ray filter with partially etched back Si, exposing the Pb needles filling the n-Si-macro(bsi, aqu, litho) pores [185]. (b) n-Si-macro (bsi, aqu viscous, litho) pores coated with TiO2 intended to be used as an UV switchable membrane for biological filtering.Figure 32. (a) Part of a 150 mm diameter X-ray filter with partially etched back Si, exposing the Pb needles filling the n-Si-macro(bsi, aqu, litho) pores [185]. (b) n-Si-macro (bsi, aqu viscous, litho) pores coated with TiO2 intended to be used as an UV switchable membrane for biological filtering.
![Materials 03 03006 g032]()
- 4)
- Porous semiconductors (like other porous materials) lose whatever thermal conductivity they have, as soon as the distance between pores approaches the mean free path length of phonons. Microcrystalline porous Si is an almost perfect thermal insulator as has been demonstrated in [189,190,191]. This effect has been used for unusual kinds of loudspeakers [192] and may find future use for thermoelectric devices converting heat directly to electricity. A figure of merit for such a device is the ratio of thermal conductivity to electrical conductivity and numbers surpassing the present state of the art by a large margin should be possible with porous semiconductors. Efforts to exploit this feature are under way [193,194,195].
- 5)
- A membrane made of macroporous Si is a direct mechanical high-precision filter for particle sizes in the µm regime [196,197]. If one can induce the diameter to decrease locally to even smaller values, a sub-µm filter results, cf. [172,198] Figure 33(b). Coating the filter with e.g., TiO2 [cf. Figure 32(b)] adds “bio” possibilities—the filter, for example, “kills” bacteria if exposed to (ultraviolet) light [199]. Filters of this type might even be useful for brewing beer [200].
- 6)
- Biological applications of (micro and meso) porous Si have been pursued for several years by now. Not only is porous Si a very good biocompatible substrate (cell cultures thrive on it, in contrast to bulk Si [201,202,203]), porous Si implants that have been loaded with some drug before they are administered, might provide reliable long-term drug delivery without severe side effects [204,205,206].
- 7)
- Last, it is illuminating to mention the two major porous Si products that actually were on the market some years ago but discontinued at present. First, there was V. Lehmann’s DNA chip, based on n-Si-macro(bsi, aqu, litho) pores [207]. A pilot production was running successfully for several years but could not sustain its just tolerated “niche” status in a major micro-electronics production line. Second, the so-called ELTRAN™ (Epitaxial layer TRANsfer) process from CANON™ [208], also abandoned by now, employed Si mesopores as a “zipper” layer not unlike the layer transfer process for solar cells mentioned above [180].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
- 1)
- It seems to be no problem to process a 300 mm Si wafer with breathtaking complexity and precision (the cm2 of a 1 Gb memory chip, for example, sells for < $ 1) but very few people (if any) have anodically etched (deep) pores into Si wafers with the precision and reproducibility required for production. If that feat would be possible at all, the exceedingly large amount of money and time required to do so is simply prohibitive in laboratory work.
- 2)
- The pore etching process (and its in situ control) is still not understood to the extent needed in a production environment. To give an example: what do you have to specify in terms of the carrier life time if you were to etch n-Si-macro(bsi, aqu, litho) pores? It should be large, of course, but how large and with what kind of global tolerances? What is the lateral uniformity needed? If your specification is very tight, are you willing or able to pay the (possible large) additional price for those wafers? What do you have to specify in terms of the carrier life time if you were to etch p-Si-macro(org) pores? There are no data at present about the possible importance in this case. If it would be important, the question is: why?
5.2. Some New Applications
5.2.1. Sensors Exploiting the Piezoelectric Properties of Porous III-V’s

5.2.2. Single Holes with Large Aspect Ratios
5.2.3. Anodes for Li Ion Batteries

6. Summary and Conclusions
Acknowledgements
References and Notes\reflist
- Zhang, X.G. Electrochemistry of Silicon and Its Oxide; Kluwer Academic-Plenum Publishers: New York, NY, USA, 2001. [Google Scholar]
- Lehmann, V. Electrochemistry of Silicon; Wiley-VCH: Weinheim, Germany, 2002. [Google Scholar]
- Canham, L.T. (Ed.) Proceedings of the 2nd International Conference on Porous Semiconductors-Science and Technology, Madrid, Spain, 12–17 March 2000. Phys. Stat. Sol. (a) 2000, 182.
- Canham, L.T.; Nassiopoulou, A.; Parkhutik, V. (Eds.) Proceedings of 3rd International Conference, Porous Semiconductors - Science and Technology, Tenerife, Spain, 10-15 March 2002. Phys. Stat. Sol. (a) 2003, 197.
- Canham, L.T.; Nassiopoulou, A.; Parkhutik, V. (Eds.) Proceedings of 4th International Conference, Porous Semiconductors - Science and Technology, Valencia, Spain, 14-19 March 2004. Phys. Stat. Sol. (a) 2005, 202.
- Parkhutik, V.; Nassiopoulou, A.; Sailor, M.; Canham, L.T. (Eds.) Proceedings of 5th International Conference, Porous Semiconductors - Science and Technology, Sitges, Spain, 12-17 March 2006. Phys. Stat. Sol. (a) 2007, 204.
- Nassiopoulou, A.; Sailor, M.; Canham, L.; Schmuki, P. (Eds.) Proceedings of 6th International Conference, Porous Semiconductors - Science and Technology, Sa Coma, Mallorca, Spain, 10-14 March 2008. Phys. Stat. Sol. (a) 2009, 206.
- Chou, L.; Buckley, D.; Chang, P.; Etcheberry, A.; O'Dwyer, C.; Overberg, M.; Yoshimoto, M. (Eds.) State-of-the-art program on compound semiconductors 46 (SOTAPOCS 46) and processes at the semiconductor/solution interface 2. ECS Trans. 2007, 6. number 2.
- Schmuki, P.; Föll, H.; Goesele, U.; Kelly, J.J.; Lockwood, D.J.; Ogata, Y.H. (Eds.) Porous semiconductors: A Symposium Held in Memory of Vitali Parkhutik and Volker Lehmann. ECS Trans. 2008, 25. number 3.
- Baca, A.; O'Dwyer, C.; Brown, J.; Buckley, D.; Nam, P.; Etcheberry, A. (Eds.) State-of-the-art program on compound semiconductors 50 (SOTAPOCS 50) and processes at the semiconductor solution interface 3. ECS Trans. 2009, 19. number 3.
- Kochergin, V.; Föll, H. Porous Semiconductors: Optical Properties and Applications; Springer: London, UK, 2009. [Google Scholar]
- Notten, P.H.L.; van den Meerakker, J.E.A.M.; Kelly, J.J. Etching of III-V Semiconductors: An Electrochemical Approach; Elsevier Science Publishers Ltd.: Oxford, UK, 1991. [Google Scholar]
- Föll, H.; Christophersen, M.; Carstensen, J.; Hasse, G. Formation and application of porous silicon. Mat. Sci. Eng. R 2002, 39, 93–141. [Google Scholar] [CrossRef]
- Föll, H.; Langa, S.; Carstensen, J.; Lölkes, S.; Christophersen, M.; Tiginyanu, I.M. Review: Pores in III-V Semiconductors. Adv. Mater. 2003, 15, 183–198. [Google Scholar] [CrossRef]
- Chazalviel, J.-N.; Ozanam, F. Macropores in p-Type Silicon. In Ordered Porous Nanostructures and Applications; Wehrspohn, R.B., Ed.; Springer: Padeborn, Germany, 2005. [Google Scholar]
- Smith, R.L.; Collins, S.D. Porous silicon formation mechanisms. J. Appl. Phys. 1992, 71, R1–R22. [Google Scholar] [CrossRef]
- Bisi, O.; Ossicini, S.; Pavesi, L. Porous silicon: a quantum sponge structure for silicon based optoelectronics. Surf. Sci. Rep. 2000, 38, 1–126. [Google Scholar] [CrossRef]
- Ossicini, S.; Pavesi, L.; Priolo, F. Light Emitting Silicon for Microphotonics; Springer: Berlin, Germany, 2003. [Google Scholar]
- Lehmann, V.; Stengl, S.; Luigart, A. On the morphology and the electrochemical formation mechanism of mesoporous silicon. Mat. Sci. Eng. B 2000, 11, 69–70. [Google Scholar]
- Chazalviel, J.-N.; Wehrspohn, R.; Ozanam, F. Electrochemical preparation of porous semiconductors: from phenomenology to understanding. Mat. Sci. Eng. B 2000, 1, 69–70. [Google Scholar]
- Barillaro, G.; Bruschi, P.; Diligenti, A.; Nannini, A. Fabrication of regular silicon microstructures by photo-electrochemical etching of silicon. Phys. Stat. Sol. (c) 2005, 2, 3198–3202. [Google Scholar] [CrossRef]
- John, G.C.; Singh, V.A. Porous silicon: Theoretical Studies. Phys. Rep. 1995, 263, 93–101. [Google Scholar] [CrossRef]
- Langner, A.; Müller, F.; Gösele, U. Macroporous Silicon. In Molecular- and Nano-Tubes; Hayden, O., Nielsch, K., Wang, D., Eds.; Springer: Padeborn, Germany, 2008. [Google Scholar]
- Föll, H.; Leisner, M.; Cojocaru, A.; Carstensen, J. Self-organization phenomena at semiconductor electrodes. Electrochim. Acta 2009, 55, 327–339. [Google Scholar] [CrossRef]
- Sa´ar, A. Photoluminescence from silicon nanostructures: The Mutual Role of Quantum Confinement and Surface Chemistry. J. Nanophoton. 2009, 3, 032501. [Google Scholar] [CrossRef]
- Lharch, M.; Aggour, M.; Chazalviel, J.-N.; Ozanam, F. Anodic dissolution and electrolumin-escence of p-Si at high potentials in fluoride media. J. Electrochem. Soc. 2002, 149, C250–C255. [Google Scholar] [CrossRef]
- Cullis, A.G.; Canham, L.T.; Calcott, P.D.J. The structural and luminescence properties of porous silicon. J. Appl. Phys. 1997, 82, 909–966. [Google Scholar] [CrossRef]
- Carstensen, J.; Foca, E.; Keipert, S.; Föll, H.; Leisner, M.; Cojocaru, A. New modes of FFT impedance spectroscopy applied to semiconductor pore etching and materials characterization. Phys. Stat. Sol. (a) 2008, 205, 2485–2503. [Google Scholar] [CrossRef]
- Wloka, J.; Mueller, K.; Schmuki, P. Pore morphology and self-organization effects during etching of n-type GaP(100) in Bromide Solutions. Electrochem. Solid-State Lett. 2005, 8, B72–B75. [Google Scholar]
- Rojas, E.G.; Plagwitz, H.; Terheiden, B.; Hensen, J.; Baur, C.; La Roche, G.; Strobl, G.F.X.; Brendel, R. Mesoporous germanium formation by electrochemical etching. J. Electrochem. Soc. 2009, 156, D310–D313. [Google Scholar] [CrossRef]
- Monaico, E.; Tiginyanu, I.M.; Ursaki, V.V.; Saruna, A.; Kuball, M.; Nedeoglo, D.D.; Sirkeli, V.P. Photoluminescence and vibrational properties of nanostructured ZnSe templates. Semicond. Sci. Technol. 2007, 22, 1115–1121. [Google Scholar] [CrossRef]
- Irmer, G.; Monaico, E.; Tiginyanu, I.M.; Gärtner, G.; Ursaki, V.V.; Kolibaba, G.V.; Nedeoglo, D.D. Fröhlich vibrational modes in porous ZnSe studied by Raman scattering and Fourier transform infrared reflectance. J. Phys. D: Appl. Phys. 2009, 42, 045405. [Google Scholar]
- Sailor, M.J. Porous Silicon in Practice: Preparation, Characterization and Applications; Wiley-VCH: Hoboken, NJ, USA, 2010. [Google Scholar]
- Rouquerol, J.; Avnir, D.; Fairbridge, C.W.; Everett, D.H.; Haynes, J.H.; Pernicone, N.; Ramsay, J.D.F.; Sing, K.S.W.; Unger, K.K. Recommendations for the characterization of porous solids. Pure Appl. Chem. 1994, 66, 1739–1758. [Google Scholar]
- Christophersen, M.; Carstensen, J.; Feuerhake, A.; Föll, H. Crystal orientation and electrolyte dependence for macropore nucleation and stable growth on p-type Si. Mater. Sci. Eng. B 2000, 69–70, 194–198. [Google Scholar]
- Föll, H.; Carstensen, J.; Christophersen, M.; Hasse, G. A new view of silicon electrochemistry. Phys. Stat. Sol. (a) 2000, 182, 7–16. [Google Scholar] [CrossRef]
- Christophersen, M.; Carstensen, J.; Föll, H. Crystal orientation dependence of macropore formation in p-type Si using organic electrolytes. Phys. Stat. Sol. (a) 2000, 182, 103–107. [Google Scholar] [CrossRef]
- Christophersen, M.; Carstensen, J.; Föll, H. Crystal orientation dependence of macropore formation in n-type Si using organic electrolytes. Phys. Stat. Sol. (a) 2000, 182, 601–606. [Google Scholar] [CrossRef]
- Claussen, J.C.; Carstensen, J.; Christophersen, M.; Langa, S.; Föll, H. Self-organized pore formation and open-loop-control in semiconductor etching. Chaos 2003, 13, 217–224. [Google Scholar] [CrossRef]
- Carstensen, J.; Christophersen, M.; Lölkes, S.; Ossei-Wusu, E.; Bahr, J.; Langa, S.; Popkirov, G.; Föll, H. Large area etching of porous semiconductors. Phys. Stat. Sol. (c) 2005, 2, 3339–3343. [Google Scholar] [CrossRef]
- Shor, J.S.; Grimberg, I.; Weiss, B.-Z.; Kurtz, A.D. Direct observation of porous SiC formed by anodization in HF. Appl. Phys. Lett. 1993, 62, 2836–2837. [Google Scholar] [CrossRef]
- Parkhutik, V.P. Self-organization of pores in SiC/Si composite structure. J. Appl. Phys. 1998, 83, 4647–4651. [Google Scholar] [CrossRef]
- Zangooie, S.; Woolam, J.A.; Arwin, H. Self-organization in porous 6H-SiC. J. Mater. Res. 2000, 15, 1860–1864. [Google Scholar] [CrossRef]
- Zangooie, S.; Arwin, H. Porous Anodic 4H-SiC: Thickness Dependent Anisotropy in Pore Propagation and Ellipsometric Characterization. Phys. Stat. Sol. (a) 2000, 182, 213–221. [Google Scholar] [CrossRef]
- Sagar, A.; Lee, C.D.; Feenstra, P.M.; Inoki, C.K.; Kuan, T.S. Morphology and effect of hydrogen etching of porous SiC. J. Appl. Phys. 2002, 92, 4070–4075. [Google Scholar] [CrossRef]
- Zhuang, D.; Edgar, J.H. Wet etching of GaN, AlN, and SiC: A Review. Mat. Sci. Eng. 2005, 48, 1–46. [Google Scholar] [CrossRef]
- Sagüés, A.A.; Wolan, J.T.; De Fex, A.; Fawcett, T.J. Impedance behavior of nanoporous SiC. Electrochim. Acta 2006, 51, 1656–1663. [Google Scholar] [CrossRef]
- Langa, S.; Christophersen, M.; Carstensen, J.; Tiginyanu, I.M.; Föll, H. Electrochemical pore etching in Ge. Phys. Stat. Sol. (a) 2003, 195, R4–R11. [Google Scholar] [CrossRef]
- Langa, S.; Carstensen, J.; Christophersen, M.; Steen, K.; Frey, S.; Tiginyanu, I.M.; Föll, H. Uniform and nonuniform nucleation of pores during the anodization of Si, Ge, and III-V Semiconductors. J. Electrochem. Soc. 2005, 152, C525–C531. [Google Scholar] [CrossRef]
- Langa, S.; Carstensen, J.; Tiginyanu, I.M.; Föll, H. Nucleation and growth of macro pores on (100) n-type Ge. Phys. Stat. Sol. (c) 2005, 2, 3237–3242. [Google Scholar] [CrossRef]
- Cheng, F.; Carstensen, J.; Föll, H. Electrochemical pore etching in Ge. Mater. Sci. Semicond. Process. 2006, 9, 694–700. [Google Scholar] [CrossRef]
- Fang, C.; Föll, H.; Carstensen, J. Electrochemical pore etching in germanium. J. Electroanal. Chem. 2006, 589, 259–288. [Google Scholar] [CrossRef]
- Fang, C.; Carstensen, J.; Föll, H. Electrochemical pore etching in n- and p-type Ge. Sol. St. Phen. 2007, 121–123, 37–40. [Google Scholar]
- Fang, C.; Föll, H.; Carstensen, J.; Langa, S. Electrochemical pore etching in Ge-An overview. Phys. Stat. Sol. (a) 2007, 204, 1292–1296. [Google Scholar] [CrossRef]
- Langa, S.; Tiginyanu, I.M.; Carstensen, J.; Christophersen, M.; Föll, H. Formation of porous layers with different morphologies during anodic etching of n-InP. Electrochem. Solid-State Lett. 2000, 3, 514–516. [Google Scholar]
- Stevens-Kalceff, M.A.; Tiginyanu, I.M.; Langa, S.; Föll, H. Correlation between morphology and cathodoluminescence in porous GaP. J. Appl. Phys. 2001, 89, 2560–2564. [Google Scholar] [CrossRef]
- Stevens-Kalceff, M.A.; Langa, S.; Tiginyanu, I.M.; Carstensen, J.; Christophersen, M.; Föll, H. Comparative SEM and Cathodoluminescence Microanalysis of Porous GaP Structures. In MRS Proceedings Fall Meeting: Microcrystalline and Nanocrystalline Semiconductors, Boston, MA, USA, 26-30 November 2001; Volume 638. Section 5.31.
- Langa, S.; Carstensen, J.; Christophersen, M.; Föll, H.; Tiginyanu, I.M. Observation of crossing pores in anodically etched n-GaAs. Appl. Phys. Lett. 2001, 78, 1074–1077. [Google Scholar] [CrossRef]
- Tiginyanu, I.M.; Langa, S.; Christophersen, M.; Carstensen, J.; Sergentu, V.; Foca, E.; Föll, H. Properties of 2D and 3D Dielectric Structures Fabricated by Electrochemical Dissolution of III-V Compounds. In MRS Proceedings Fall Meeting: Progress in Semiconductor Materials for Optoelectronic Applications, Boston, USA, 26-30 November 2002; Volume 692. K2.7.
- Föll, H.; Langa, S.; Carstensen, J.; Christophersen, M.; Tiginyanu, I.M.; Dichtel, K. Pore Etching in Compound Semiconductors for the Production of Photonic Crystals. In MRS Proceedings Spring Meeting: Materials and Devices for Optoelectronics and Microphotonics, Boston, USA, 26-30 November 2002. Section L6.4.
- Föll, H.; Carstensen, J.; Langa, S.; Christophersen, M.; Tiginyanu, I.M. Porous III-V compound semiconductors: Formation, properties, and comparison to silicon. Phys. Stat. Sol. (a) 2003, 197, 61–70. [Google Scholar] [CrossRef]
- Christophersen, M.; Langa, S.; Carstensen, J.; Tiginyanu, I.M.; Föll, H. A comparison of pores in silicon and pores in III-V compound materials. Phys. Stat. Sol. (a) 2003, 197, 197–203. [Google Scholar] [CrossRef]
- Langa, S.; Carstensen, J.; Tiginyanu, I.M.; Christophersen, M.; Föll, H. Selfordering in porous III-V compounds. In Ordered Porous Nanostructures and Applications; Wehrspohn, R.B., Ed.; Springer: Paderborn, Germany, 2005; p. 57. [Google Scholar]
- Rönnebeck, S.; Ottow, S.; Carstensen, J.; Föll, H. Crystal orientation dependence of macropore formation in n-Si with backside-illumination in HF-electrolyte. J. Porous Mat. 2000, 7, 353–357. [Google Scholar] [CrossRef]
- Cojocaru, A.; Carstensen, J.; Föll, H. Growth modes of macropores in n-type silicon. ECS Trans. 2008, 16, 157–172. [Google Scholar]
- Lehmann, V.; Föll, H. Formation mechanism and properties of electrochemically etched trenches in n-type silicon. J. Electrochem. Soc. 1990, 137, 653–659. [Google Scholar] [CrossRef]
- Canham, L.T. Silicon quantum wire array fabrication by electrochemical and chemical dissolution of wafers. Appl. Phys. Lett. 1990, 57, 1046–1049. [Google Scholar] [CrossRef]
- Lehmann, V.; Gösele, U. Porous silicon formation: A Quantum Wire Effect. Appl. Phys. Lett. 1991, 58, 856–559. [Google Scholar] [CrossRef]
- Etch, ET&TE; GmbH, Technology. Available online: http://www.et-te.com (Accessed on 31 March 2010).
- Semitool. Available online: http://www.semitool.com (Accessed on 31 March 2010).
- Langa, S.; Christophersen, M.; Carstensen, J.; Tiginyanu, I.M.; Föll, H. Single crystalline 2D porous arrays obtained by self-organization in n-InP. Phys. Stat. Sol. (a) 2003, 197, 77–82. [Google Scholar] [CrossRef]
- Carstensen, J.; Cojocaru, A.; Leisner, M.; Föll, H. In situ assessment of macropore growth in low-doped n-type silicon. ECS Trans. 2008, 16, 21–27. [Google Scholar]
- Cojocaru, A.; Carstensen, J.; Leisner, M.; Föll, H.; Tiginyanu, I.M. Self-induced oscillation of the macropore diameter in n-type silicon. Phys. Stat. Sol. (c) 2009, 206, 1533–1537. [Google Scholar] [CrossRef]
- Matsumura, M.; Morrison, S.R. Anodic properties of n-Si and n-Ge electrodes in HF solution under illumination and in the dark. J. Electroanal. Chem. 1983, 147, 157–166. [Google Scholar] [CrossRef]
- Santinacci, M.; Bouttemy, I.; Gerard, L.; Etcheberry, A. Unexpected Dissolution Process at Porous n-InP Electrodes. ECS Trans. 2009, 19, 313–320. [Google Scholar]
- Krischer, K. Principles of Temporal and spatial pattern formation in electrochemical systems. In Modern Aspects of Electrochemistry; Conway, B.E., Bockris, J.o'M., White, R.E., Eds.; Plenum Press: New York, NY, USA, 1999; Volume 32, p. 1. [Google Scholar]
- Krischer, K. Nonlinear dynamics in electrochemical systems. In Advances in Electrochemical Science and Engineering; Alkire, R.C., Kolb, D.M., Eds.; Wiley-VCH: Weinheim, Germany, 2003; Volume 8. [Google Scholar]
- Gerischer, H.; Allongue, P.; Kieling, V.C. The mechanism of the anodic oxidation of silicon in acidic fluoride solutions revisited. Ber. Bunsenges. Phys. Chem. 1993, 97, 753–756. [Google Scholar] [CrossRef]
- Gerischer, H.; Lübke, M. Electrolytic growth and dissolution of oxide layers on silicon in aqueous solutions of fluorides. Ber. Bunsenges. Phys. Chem. 1988, 92, 573–577. [Google Scholar] [CrossRef]
- Lockwood, D.J.; Schmuki, P.; Labbe, H.J.; Fraser, J.W. Optical properties of porous GaAs. Physica E 1999, 4, 102–106. [Google Scholar] [CrossRef]
- Chazalviel, J.-N.; Ozanam, F.; Gabouze, N.; Fellah, S.; Wehrspohn, R.B. Quantitative analysis of the morphology of macropores on low-doped p-Si. What is the minimum resistivity. J. Electrochem. Soc. 2002, 149, C511–C520. [Google Scholar] [CrossRef]
- Menezes, S.; Miller, B.; Bachmann, K.J. Electrodissolution and passivation phenomena in III-V Semiconducting compounds. J. Vac. Sci. Technol. B 1983, 1, 48–54. [Google Scholar] [CrossRef]
- Moslavac, K.; Lovrecek, B. The anodic passivation on silicon single crystals. Electrochim. Acta 1969, 14, 373–376. [Google Scholar] [CrossRef]
- Seidel, H.; Csepregi, L.; Heuberger, A.; Baumgärtel, H. Anisotropic etching of crystalline silicon in alkaline solutions. I. Orientation dependence and behavior of passivation layers. J. Electrochem. Soc. 1990, 137, 3612–3626. [Google Scholar]
- Smith, R.L.; Klöck, B.; Collins, S.D. Anodic Passivation of (111) Silicon in KOH. J. Electrochem. Soc. 1988, 135, 2001–2008. [Google Scholar] [CrossRef]
- Lublow, M.; Lewerenz, H.J. Electrochemical conditioning of fractal topographies at the silicon oxide/silicon interface. In Proceedings of Extended Abstracts-The 3rd international IEEE scientific Conference on Physics and Control, Potsdam, Germany; 2007; Volume 188. [Google Scholar]
- Chazalviel, J.N.; Ozanam, F. In situ infrared characterization of the silicon surface in hydrofluoric acid. J. Appl. Phys. 1997, 81, 7684–7687. [Google Scholar] [CrossRef]
- Chazalviel, J.-N.; da Fonseca, C.; Ozanam, F. In situ infrared study of the oscillating anodic dissolution of silicon in fluoride electrolytes. J. Electrochem. Soc. 1998, 145, 964–974. [Google Scholar] [CrossRef]
- Chazalviel, J.N.; Erne, B.H.; Maroun, F.; Ozanam, F. New directions and challenges in modern electrochemistry: in situ infrared spectroscopy of the semiconductor - electrolyte interface. J. Electroanal. Chem. 2001, 50, 180–190. [Google Scholar] [CrossRef]
- Chazalviel, J.-N.; Erne, B.H.; Maroun, F.; Ozanam, F. In situ infrared spectroscopy of the semiconductor - electrolyte interface. J. Electroanal. Chem. 2001, 509, 108–118. [Google Scholar] [CrossRef]
- Rappich, J.; Lewerenz, H.-J. In situ fourier transform infrared investigations on the electrolytic hydrogenation of n-silicon(111). J. Electrochem. Soc. 1995, 142, 1233–1237. [Google Scholar] [CrossRef]
- Nast, O.; Rauscher, S.; Jungblut, H.; Lewerenz, H.-J. Micromorphology changes of silicon oxide on Si(111) during current oscillations: a comparative in situ AFM and FTIR study. J. Electroanal. Chem. 1998, 442, 169–174. [Google Scholar] [CrossRef]
- Lewerenz, H.J.; Aggour, M.; Murrell, C.; Kanis, M.; Jungblut, H.; Jakubowicz, J.; Cox, P.A.; Campbell, S.A.; Hoffmann, P.; Schmeisserd, D. Initial stages of structure formation on silicon electrodes investigated by photoelectron spectroscopy using synchrotron radiation and in situ atomic force microscopy. J. Electrochem. Soc. 2003, 150, E185–E189. [Google Scholar] [CrossRef]
- Grzanna, J.; Jungblut, H.; Lewerenz, H.J. A model for electrochemical oscillations at the Si-electrolyte contact, Part I. Theoretical development. J. Electroanal. Chem. 2000, 486, 181–189. [Google Scholar] [CrossRef]
- Grzanna, J.; Jungblut, H.; Lewerenz, H.J. A model for electrochemical oscillations at the Si-electrolyte contact, Part II. Simulations and experimental results. J. Electroanal. Chem. 2000, 486, 190–203. [Google Scholar]
- Grzanna, J.; Jungblut, H.; Lewerenz, H.J. Nano- and macropores in the model for current oscillations at the Si/electrolyte contact. Phys. Stat. Sol. (a) 2007, 204, 1245–1249. [Google Scholar] [CrossRef]
- Grzanna, J.; Notz, T.; Lewerenz, H.J. Model for current oscillations at the Si/electrolyte contact: Extension to Spatial Resolution. ECS Trans. 2008, 16, 173–180. [Google Scholar]
- Chazalviel, J.-N.; Ozanam, F.; Etman, M.; Paolucci, F.; Peter, L.M.; Stumper, J. The p-Si/Fluoride interface in the anodic region: Damped and/or sustained oscillations. J. Electroanal. Chem. 1992, 327, 343–349. [Google Scholar] [CrossRef]
- Chazalviel, J.-N.; Ozanam, F. A theory for resonant response of an electrochemical system: Self-Oscillating Domains, Hidden Oscillation and Synchronization Impedance. J. Electrochem. Soc. 1992, 139, 2501–2508. [Google Scholar] [CrossRef]
- Ozanam, F.; Blanchard, N.; Chazalviel, J.-N. Microscopic, self-oscillating domains at the silicon surface during its anodic dissolution in a fluoride electrolyte. Electrochim. Acta 1993, 38, 1627–1630. [Google Scholar] [CrossRef]
- Krischer, K.; Varela, H. Oscillations and other dynamic instabilities. In Handbook of Fuel Cells; Vielstich, W., Lamm, A., Gasteiger, H., Eds.; John Wiley & Sons, Ltd: Hoboken, NJ, USA, 2003; Volume 2, p. 679. [Google Scholar]
- Föll, H.; Carstensen, J.; Christophersen, M.; Hasse, G. A stochastic model for current oscillations in space and time at the silicon electrode. In ECS Proceedings of Pits and Pores II: Formation Properties and Significance for Advanced Materials, New Jersey, NJ, USA; 2000; pp. 36–48. [Google Scholar]
- Foca, E.; Carstensen, J.; Föll, H. Monte Carlo simulation of electrochemical oscillations in the electropolishing regime. Phys. Stat. Sol. (a) 2005, 202, 1524–1528. [Google Scholar] [CrossRef]
- Foca, E.; Carstensen, J.; Föll, H. Modeling electrochemical current and potential oscillations at the Si electrode. J. Electroanal. Chem. 2007, 603, 175–202. [Google Scholar] [CrossRef]
- Föll, H.; Carstensen, J.; Foca, E. Electrochemical pore formation in semiconductors: Oscillations, Structure Formation and Control. In Proceedings of The 3rd international IEEE Scientific Conference on Physics and Control (PhysCon2007), Potsdam, Germany, 3-7 September 2007; p. 185.
- Langa, S.; Monaico, E.; Föll, H.; Tiginyanu, I.M. Porous morphologies in Si, III-V and II-VI compounds: A Comparative Study. In Proceedings of the 6th International Conference on Microelectronics and Computer Science, Chisinau, Moldova, 19-21 October 2009; p. 175.
- Claussen, J.C.; Carstensen, J.; Christophersen, M.; Langa, S.; Föll, H. Open-Loop-Control of pore formation in semiconductor etching. In Proceedings of Physics and Control, St. Petersburg, Russia, 20-22 August 2003; pp. 895–899.
- Christophersen, M.; Carstensen, J.; Föll, H. Macropore Formation on Highly Doped n-Type Silicon. Phys. Stat. Sol. (a) 2000, 182, 45–50. [Google Scholar] [CrossRef]
- Frey, S.; Kemell, M.; Carstensen, J.; Langa, S.; Föll, H. Fast pore etching. Phys. Stat. Sol. (a) 2005, 202, 1369–1373. [Google Scholar] [CrossRef]
- Rönnebeck, S.; Carstensen, J.; Ottow, S.; Föll, H. Crystal orientation dependence of macropore growth in n-type silicon. Electrochem. Solid-State Lett. 1999, 2, 126–128. [Google Scholar]
- Christophersen, M.; Carstensen, J.; Rönnebeck, S.; Jäger, C.; Jäger, W.; Föll, H. Crystal orientation dependance and anisotropic properties of macropore formation of p- and n-type silicon. J. Electrochem. Soc. 2001, 148, E267–E275. [Google Scholar] [CrossRef]
- Föll, H.; Carstensen, J.; Frey, S. Porous and nanoporous semiconductors and emerging applications. In Sensor, and Gas Separation Applications; Lu, S.W., Hahn, H., Weissmuller, J., Gole, J.L., Eds.; Curran Associates, Inc.: Warrendale, PA, USA, 2005; pp. R12.1–R12.1.13. [Google Scholar]
- Langa, S.; Frey, S.; Carstensen, J.; Föll, H.; Tiginyanu, I.M.; Hermann, M.; Böttger, G. Waveguide Structures Based on Porous Indium Phosphide. Electrochem. Sol. State Lett. 2005, 8, C30–C32. [Google Scholar] [CrossRef]
- Föll, H.; Leisner, M.; Carstensen, J.; Schauer, P. Growth mode transition of crysto and curro pores in III-V semiconductors. ECS Trans. 2009, 19, 329–345. [Google Scholar]
- Zheng, J.; Christophersen, M.; Bergstrom, P.L. Thick macroporous membranes made of p-type silicon. Phys. Stat. Sol. (a) 2005, 202, 1402–1406. [Google Scholar] [CrossRef]
- Christophersen, M.; Carstensen, J.; Voigt, K.; Föll, H. Organic and aqueous electrolytes used for etching macro- and mesoporous silicon. Phys. Stat. Sol. (a) 2003, 197, 34–37. [Google Scholar] [CrossRef]
- Slimani, A.; Iratni, A.; Chazalviel, J.-N.; Gabouze, N.; Ozanam, F. Experimental study of macropore formation in p-type silicon in a fluoride solution and the transition between macropore formation and electropolishing. Electrochim. Acta 2009, 54, 3139–3144. [Google Scholar] [CrossRef]
- Chazalviel, J.-N.; Ozanam, F. Are the oscillations during the anodic dissolution of Si in dilute fluoride electrolyte damped or sustained? ECS Trans. 2008, 16, 189–194. [Google Scholar]
- Langa, S.; Tiginyanu, I.M.; Carstensen, J.; Christophersen, M.; Föll, H. Self-organized growth of single crystals of nanopores. Appl. Phys. Lett. 2003, 82, 278–289. [Google Scholar] [CrossRef]
- Tiginyanu, I.M.; Langa, S.; Sirbu, L.; Monaico, E.; Stevens-Kalceff, M.A.; Föll, H. Cathodoluminescence microanalysis of porous GaP and InP structures. Eur. Phys. J.: Appl. Phys. 2004, 27, 81–84. [Google Scholar]
- Langa, S.; Carstensen, J.; Tiginyanu, I.M.; Christophersen, M.; Föll, H. Formation of tetrahedron-like pores during anodic etching of (100)-oriented n-GaAs. Electrochem. Solid-State Lett. 2002, 5, C14–C17. [Google Scholar]
- Rumpf, K.; Granitzer, P.; Krenn, H. Transition metals specifically electrodeposited into porous silicon. Phys. Stat. Sol. 2009, 206, 1592–1595. [Google Scholar] [CrossRef]
- Bao, X.Q.; Jiao, J.W.; Zhou, J.; Wang, Y.L. Fast speed pore formation via strong oxidizers. Electrochim. Acta 2007, 52, 6728–6733. [Google Scholar] [CrossRef]
- Cojocaru, A.; Carstensen, J.; Ossei-Wusu, E.K.; Leisner, M.; Riemenschneider, O.; Föll, H. Fast macropore growth in n-type silicon. Phys. Stat. Sol. (c) 2009, 206, 1571–1574. [Google Scholar] [CrossRef]
- Tsuchiya, H.; Hueppe, M.; Djenizian, T.; Schmuki, P. Electrochemical formation of porous superlattices on n-type (100) InP. Surf. Sci. 2003, 547, 268–274. [Google Scholar] [CrossRef]
- Spiecker, E.; Rudel, M.; Jäger, W.; Leisner, M.; Föll, H. Morphology, interface polarity and branching of electrochemically etched pores in InP. Phys. Stat. Sol. (a) 2005, 202, 2950–2962. [Google Scholar] [CrossRef]
- Hejjo Al Rifai, M.; Christophersen, M.; Ottow, S.; Carstensen, J.; Föll, H. Dependence of macropore formation on n-Si on potential, temperature, and doping. J. Electrochem. Soc. 2000, 147, 627–635. [Google Scholar] [CrossRef]
- Sze, S.M. Physics of Semiconductor Devices; Wiley & Sons: New York, NY, USA, 1981. [Google Scholar]
- Lehmann, V.; Gösele, U. Porous silicon: quantum sponge structures grown via a self-adjusting etching process. Adv. Mater. 1992, 4, 114–116. [Google Scholar] [CrossRef]
- Carstensen, J.; Prange, R.; Popkirov, G.S.; Föll, H. A model for current oscillations in the Si-HF system based on a quantitative analysis of current transients. Appl. Phys. A 1998, 67, 459–467. [Google Scholar] [CrossRef]
- Carstensen, J.; Prange, R.; Föll, H. Percolation model for the current oscillation in the Si-HF system. In Proceedings of ECS' 193rd Meeting, San Diego, CA, USA; 1998; pp. 148–156. [Google Scholar]
- Carstensen, J.; Christophersen, M.; Hasse, G.; Föll, H. Parameter dependence of pore formation in silicon within the model of local current bursts. Phys. Stat. Sol. (a) 2000, 182, 63–69. [Google Scholar] [CrossRef]
- Salem, M.S.; Sailor, M.J.; Fukami, K.; Sakka, T.; Ogata, Y.H. Preparation and optical properties of porous silicon rugate-type multilayers with different pore sizes. Phys. Stat. Sol. (c) 2009, 6, 1620–1623. [Google Scholar] [CrossRef]
- Bao, X.Q.; Lin, J.L.; Jiao, J.W.; Wang, Y.L. Macropore density as a function of HF-concentration and bias. Electrochim. Acta 2007, 53, 823–828. [Google Scholar] [CrossRef]
- Leisner, M.; Carstensen, J.; Föll, H. FFT impedance spectroscopy analysis of the growth of anodic oxides on (100) p-Si for various solvents. J. Electroanal. Chem. 2008, 615, 124–134. [Google Scholar] [CrossRef]
- Leisner, M.; Carstensen, J.; Föll, H. Simulating crystallographic pore growth on III-V semiconductors. ECS Trans. 2009, 19, 321–328. [Google Scholar]
- Beale, M.I.J.; Benjamin, J.D.; Uren, M.J.; Chew, N.G.; Cullis, A.G. The formation of porous silicon by chemical stain etches. Surf. Sci. 1986, 75, 408–414. [Google Scholar]
- Cheggoua, R.; Kadouna, A.; Gabouzeb, N.; Ozanam, F.; Chazalviel, J.-N. Theoretical modelling of the I–V characteristics of p-type silicon in fluoride electrolyte in the first electropolishing plateau. Electrochim. Acta 2009, 54, 3053–3058. [Google Scholar] [CrossRef]
- Barillaro, G.; Pieri, F. A self-consistent theoretical model for macropore growth in n-type silicon. J. Appl. Phys. 2005, 97, 116105. [Google Scholar] [CrossRef]
- Ossei-Wusu, E.K.; Cojocaru, A.; Carstensen, J.; Leisner, M.; Föll, H. Etching deep macropores in n-type silicon in short times. ECS Trans. 2008, 16, 109–123. [Google Scholar]
- Macak, J.M.; Schmuki, P. Anodic growth of self-organized anodic TiO2 nanotubes in viscous electrolytes. Electrochim. Acta 2006, 52, 1258–1264. [Google Scholar] [CrossRef]
- Chazalviel, J.-N. Impedance studies at semiconductor electrodes: clasical and more exotic techniques. Electrochim. Acta 1990, 35, 1545–1552. [Google Scholar] [CrossRef]
- Searson, P.C.; Zhang, X.G. The anodic dissolution of silicon in HF solutions. J. Electrochem. Soc. 1990, 137, 2539–2548. [Google Scholar] [CrossRef]
- Ozanam, F.; Chazalviel, J.-N.; Radi, A.; Etman, M. Resonant and nonresonant behavior of the anodic dissolution of silicon in fluoride media: An impedance study. J. Electrochem. Soc. 1992, 139, 2491–2501. [Google Scholar] [CrossRef]
- Vanmaekelbergh, D.; Searson, P.C. On the electrical impedance due to the anodic dissolution of silicon in HF solutions. J. Electrochem. Soc. 1994, 141, 697–702. [Google Scholar] [CrossRef]
- Shen, W.M.; Tomkiewicz, M.; Levy-Clement, C. Impedance of porous Si. J. Appl. Phys. 1994, 76, 3635–3639. [Google Scholar] [CrossRef]
- Koshida, N.; Naggsu, M.; Echizenya, K.; Kiuchi, Y. Impendance spectra of p-type porous Si-electrolyte interfaces. J. Electrochem. Soc. 1986, 133, 2283–2291. [Google Scholar] [CrossRef]
- Frateur, I.; Cattarin, S.; Musiani, M.; Tribollet, B. Electrodissolution of Ti and p-Si in acidic fluoride media: formation ratio of oxide layers from electrochemical impedance spectroscopy. J. Electroanal. Chem. 2000, 482, 202–210. [Google Scholar] [CrossRef]
- MacDonald, J.R. Impedance Spectroscopy; John Wiley & Sons: Hoboken, NJ, USA, 1987. [Google Scholar]
- Orazem, M.E.; Tribollet, B. Electrochemical Impedance Spectroscopy; Wiley-VCH: Hoboken, NJ, USA, 2008. [Google Scholar]
- Carstensen, J.; Schütt, A.; Föll, H. CELLO measurements with FFT impedance analysis: Drastic Increase of Measurement Speed for Analysis of Local Solar Cell Defects. In Proceedings of the 23rd European Photovoltaic Solar Energy Conference, Valencia, Spain, 1-5 September 2008. 1AO.6.1.
- Carstensen, J.; Schütt, A.; Föll, H. CELLO FFT impedance analysis as a routine tool for identifying various defect types on crystalline silicon solar cells. In Proceedings of the 24th European Photovoltaic Solar Energy Conference, Hamburg, Germany, 21-25 September 2009. 1AO.4.5.
- Leisner, M.; Carstensen, J.; Cojocaru, A.; Föll, H. Pore growth on n-InP investigated by in situ FFT impedance spectroscopy. Phys. Stat. Sol. (c) 2009, 206, 1566–1571. [Google Scholar] [CrossRef]
- Leisner, M.; Carstensen, J.; Cojocaru, A.; Föll, H. In situ FFT impedance spectroscopy during the growth of crystallographically oriented pores in InP. ECS Trans. 2008, 16, 133–142. [Google Scholar]
- Carstensen, J.; Cojocaru, A.; Leisner, M.; Föll, H. Dynamics of macropore growth in n-type silicon investigated by FFT in situ impedance analysis. ECS Trans. 2009, 19, 355–361. [Google Scholar]
- Zhang, X.G. Mechanism of pore formation on n-type silicon. J. Electrochem. Soc. 1991, 138, 3750–3756. [Google Scholar] [CrossRef]
- Hauser, J.R. Avalanche breakdown voltage for III-V semiconductors. Appl. Phys. Lett. 1978, 33, 351–356. [Google Scholar] [CrossRef]
- Canham, L.T. Gaining light from silicon. Nature 2000, 408, 411–412. [Google Scholar] [CrossRef]
- Propst, E.; Kohl, P.A. The photoelectrochemical oxidation of n-Si in anhydrous HF-Acetonitrile. J. Electrochem. Soc. 1993, 140, L78–L80. [Google Scholar] [CrossRef]
- Imai, K.; Unno, H. FIPOS (Full Isolation by Porous Oxidized. Silicon) Technology and Its Application to LSIs. IEEE Trans. Electron Devices. 1984, 31, 297–302. [Google Scholar]
- Föll, H. Anodic etching of defects in p-type silicon. J. Electrochem. Soc. 1980, 127, 1925–1931. [Google Scholar] [CrossRef]
- Föll, H. Anodic etching of p-type silicon as a method for discriminating electrically active and inactive defects. Appl. Phys. Lett. 1980, 37, 316–318. [Google Scholar] [CrossRef]
- Gösele, U.; Föll, H. Volker Lehmann: An unconventional scientist. ECS Trans. 2008, 16, 7–20. [Google Scholar]
- Carstensen, J.; Lippik, W.; Liebert, S.; Köster, S.; Föll, H. ELYMAT technique on multicrystalline silicon for solar cell application. In Conference proceedings of the 13th European Photovoltaic Solar Energy Conference, Nice, France, 23-27 October 1995; p. 1344.
- Carstensen, J.; Lippik, W.; Föll, H. Mapping of defect related bulk and surface properties with the ELYMAT technique. In Semiconductor Silicon; Huff, H.R., Bergholz, W., Sumino, K., Eds.; Electrochemical Society: San Francisco, CA, USA, 1994; p. 1105. [Google Scholar]
- Carstensen, J.; Lippik, W.; Föll, H. Mapping of defect related silicon properties with the ELYMAT technique in three dimensions. Mater. Sci. Forum 1995, 173, 159–164. [Google Scholar] [CrossRef]
- Föll, H.; Carstensen, J.; Frey, S. Porous and nanoporous semiconductors and emerging applications. J. Nanomater 2006. Article ID 91635. [Google Scholar]
- Stewart, M.P.; Buriak, J.M. Chemical and biological applications of porous silicon technology. Adv. Mat. 2000, 12, 859–869. [Google Scholar] [CrossRef]
- Müller, F.; Birner, A.; Gösele, U.; Lehmann, V.; Ottow, S.; Föll, H. Structuring of macroporous silicon for applications as photonic crystals. J. Por. Mat. 2000, 7, 201–204. [Google Scholar] [CrossRef]
- Kochergin, V.; Sanghavi, M.; Swinehart, P.R. Porous silicon filters for low-temperature far IR applications. Proc. SPIE 2005, 5883, 184–191. [Google Scholar]
- Langa, S.; Lölkes, S.; Carstensen, J.; Hermann, M.; Böttger, G.; Tiginyanu, I.M.; Föll, H. Engineering the morphology of porous InP for waveguide applications. Phys. Stat. Sol. (c) 2005, 2, 3253–3257. [Google Scholar] [CrossRef]
- Kochergin, V.; Föll, H. Commercial applications of porous Si: Optical Filters And Components. Phys. Stat. Sol. (c) 2007, 4, 1933–1940. [Google Scholar] [CrossRef]
- Wehrspohn, R.B.; Schilling, J. Electrochemically prepared pore arrays for photonic crystal applications. MRS Bull. 2001, 26, 623–626. [Google Scholar] [CrossRef]
- Wehrspohn, R.B.; Schweizer, S.L.; Schilling, J.; Geppert, T.; Jamois, C.; Glatthaar, R. Application of Photonic Crystals for Gas Detection and Sensing. In Photonic Crystals: Advances in Design, Fabrication, and Characterization; Busch, K., Lölkes, S., Wehrspohn, R., Föll, H., Eds.; Wiley-VCH: Weinheim, Germany, 2004. [Google Scholar]
- Akkari, E.; Benachour, Z.; Aouida, S.; Touayar, O.; Bessais, B.; Benbrahim, J. Study and characterization of porous germanium for radiometric measurements. Phys. Stat. Sol. (c) 2009, 6, 1685–1688. [Google Scholar] [CrossRef]
- Tiginyanu, I.M.; Langa, S.; Hjort, K.; Monecke, J.; Hartnagel, H.L. Nanotexturization of III-V compounds for THz-wave generation. In Proceedings of 8th International Conference on Terahertz Electronics, Darmstadt, Germany, 28–29 September 2000; p. 275.
- Melnikov, V.A.; Golovan, L.A.; Konorov, S.O.; Fedotov, A.B.; Petrov, G.I.; Li, L.; Yakovlev, V.V.; Gavrilov, S.A.; Zheltikov, A.M.; Timoshenko, V.Yu.; Kashkarov, P.K. Porous gallium phosphide: Challenging Material for Nonlinear-optical Applications. Phys. Stat. Sol. C 2005, 2, 3248–3252. [Google Scholar] [CrossRef]
- Bastide, S.; Quang, N.L.; Monna, R.; Levy-Clément, C. Chemical etching of Si by Ag nanocatalysts in HF-H2O2: Application to Multicrystalline Si Solar Cell Texturisation. Phys. Stat. Sol. (c) 2009, 206, 1536–1540. [Google Scholar] [CrossRef]
- Yae, S.; Tanaka, H.; Kobayashi, T.; Fukumuro, N.; Matsuda, H. Porous silicon formation by HF chemical etching for antireflection of solar cells. Phys. Stat. Sol. (c) 2005, 2, 3476–3480. [Google Scholar] [CrossRef]
- Brendel, R. Review of layer transfer processes for crystalline thin-film silicon solar cells. Jpn. J. Appl. Phys. Part 1 2001, 40, 4431–4439. [Google Scholar] [CrossRef]
- Kovalev, D.; Timoshenko, V.Yu.; Künzner, N.; Gross, E.; Koch, F. Strong explosive interaction of hydrogenated porous silicon with oxygen at cryogenic temperatures. Phys. Rev. Lett. 2001, 87, 068301. [Google Scholar] [CrossRef]
- Mikulec, F.V.; Kirtland, J.D.; Sailor, M.J. Explosive nanocrystalline porous silicon and its use in atomic emission spectroscopy. Adv. Mater. 2002, 14, 38–41. [Google Scholar] [CrossRef]
- Völlmeke, S.; Bartuch, H.; Laucht, H.; Kovalev, D. Poröses Silizium - Basiswerkstoff für pyroltechnische Bauelemente. In Proceedings of 51th International Scientific Colloquium, Ilmenau, Germany, 11-15 September 2006.
- Plessis, M. Investgating nanoporous silicon explosive devices. Phys. Stat. Sol. (c) 2009, 206, 1763. [Google Scholar] [CrossRef]
- Lehmann, V.; Rönnebeck, S. MEMS techniques applied to the fabrication of anti-scatter grids for X-ray imaging. Sens. Actuator. A 2001, 95, 202–207. [Google Scholar]
- Badel, X.; Galeckas, A.; Linnros, J.; Kleimann, P.; Fröjdh, C.; Petersson, C.S. Improvement of an X-ray imaging detector based on a scintillating guides screen. Nucl. Instrum. Methods Phys. Res. Sect. A 2002, 487, 129–135. [Google Scholar] [CrossRef]
- Badel, X.; Linnros, J.; Kleimann, P.; Norlin, B.; Koskiahde, E.; Valpas, K.; Nenonen, S.; Petersson, C.S.; Fröjdh, C. Metallized and oxidized silicon macropore arrays filled with a scintillator for CCD-based X-ray imaging detectors. IEEE Trans. Nucl. Sci. 2004, 51, 1001–1005. [Google Scholar] [CrossRef]
- Bonder, Y.; Wang, C. Ab initio study of birefringent porous silicon. Phys. Stat. Sol. (a) 2005, 202, 1552–1556. [Google Scholar] [CrossRef]
- Kaltsas, G.; Nassiopoulou, A.G. Novel C-MOS compatible monolithic silicon gas flow sensor with porous silicon thermal isolation. Sens. Actuat. A 1999, 76, 133–138. [Google Scholar] [CrossRef]
- Roussel, Ph.; Lysenko, V.; Remaki, B.; Delhomme, G.; Dittmar, A.; Barbier, D. Thick oxidised porous silicon layers for the design of a biomedical thermal conductivity microsensor. Sens. Actuat. 1999, 74, 100–103. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, J.; Xu, S.; Wang, L.; Cao, Z.; Zhan, P.; Wang, Z. 1D partially oxidized porous silicon photonic crystal reflector for mid-infrared application. J. Phys. D: Appl. Phys. 2007, 40, 4482–4484. [Google Scholar]
- Shinoda, H.; Nakajima, T.; Ueno, K.; Koshida, N. Thermally induced ultrasonic emission from porous silicon. Nature 1999, 400, 853–855. [Google Scholar] [CrossRef]
- Joshi, G.; Lee, H.; Lan, Y.; Wang, X.; Zhu, G.; Wang, D.; Gould, R.W.; Cuff, D.C.; Tang, M.Y.; Dresselhaus, M.S.; Chen, G.; Ren, Z. Enhanced Thermoelectric Figure-of-Merit in Nanostructured p-type Silicon Germanium Bulk Alloys. Nano Lett. 2008, 8, 4670–4674. [Google Scholar] [CrossRef]
- Lee, J.; Galli, G.A.; Grossman, J.C. Nanoporous Si as an Efficient Thermoelectric Material. Nano Lett. 2008, 8, 3750–3754. [Google Scholar] [CrossRef]
- Goldsmid, H.J. Porous Thermoelectric Materials. Materials 2009, 2, 903–910. [Google Scholar] [CrossRef]
- Müller, F.; Birner, A.; Schilling, J.; Gösele, U.; Kettner, C.; Hänggi, P. Membranes for micropumps from macroporous silicon. Phys. Stat. Sol. (a) 2000, 182, 585–590. [Google Scholar] [CrossRef]
- Wallner, J.Z.; Bergstrom, P.L. A porous silicon based particle filter for microsystems. Phys. Stat. Sol. (a) 2007, 204, 1469–1473. [Google Scholar] [CrossRef]
- Campbell, J.; Corno, J.A.; Larsen, N.; Gole, J.L. Development of porous-silicon-based active microfilters. J. Electrochem. Soc. 2008, 155, D128–D132. [Google Scholar] [CrossRef]
- Gole, J.L.; Corno, J.; Ozdemir, S.; Prokes, S.; Shin, H.-C. Active microfiltered sensor interfaces, photocatalytic reactors, and microbatteries using combined micro/nanoporous interfaces. Phys. Stat. Sol. (c) 2009, 6, 1773–1776. [Google Scholar] [CrossRef]
- Vainrot, N.; Eisen, M.S.; Semiat, R. Membranes in Desalination and Water Treatment. MRS Bull. 2008, 33, 16–20. [Google Scholar] [CrossRef]
- Bengtsson, M.; Ekström, S.; Drott, J.; Collins, A.; Csöregi, E.; Marko-Varga, G.; Laurell, T. Applications of microstructured porous silicon as a biocatalytic surface. Phys. Stat. Sol. (a) 2000, 182, 495–504. [Google Scholar] [CrossRef]
- Mayne, A.H.; Bayliss, S.C.; Barr, P.; Tobin, M.; Backberry, L.D. Biologically interfaced porous Silicon devices. Phys. Stat. Sol. (a) 2000, 182, 505–513. [Google Scholar] [CrossRef]
- Chan, S.; Fauchet, P.M.; Li, Y.; Rothberg, L.J.; Miller, B.L. Porous silicon microcavities for biosensing applications. Phys. Stat. Sol. (a) 2000, 182, 541–546. [Google Scholar] [CrossRef]
- Stewart, M.P.; Robins, E.G.; Geders, T.W.; Allen, M.J.; Cheul Choi, H.; Buriak, J.M. Derivatization, stabilization and oxidation - three methods for stabilization and functionalization of porous silicon surfaces via hydrosilylation and electrografting reactions. Phys. Stat. Sol. (a) 2000, 182, 109–115. [Google Scholar] [CrossRef]
- Li, Y.Y.; Cunin, F.; Link, J.R.; Gao, T.; Betts, R.E.; Reiver, S.H.; Chin, V.; Bhatia, S.N.; Sailor, M.J. Polymer replicas of photonic porous silicon for sensing and drug delivery applications. Science 2003, 299, 2045–2047. [Google Scholar] [CrossRef]
- Pastor, E.; Salonen, J.; Lehto, V.; Matveeva, E. Electrochemically induced bioactivity of porous silicon functionalized by acetylene. Phys. Stat. Sol. (c) 2009, 206, 1333–1338. [Google Scholar] [CrossRef]
- Lehmann, V. Biosensors: Barcoded Molecules. Nat. Mater. 2002, 1, 12–13. [Google Scholar] [CrossRef]
- Yonehara, T.; Sakaguchi, K. ELTRAN; SOI-Epi wafer™ by epitaxial layer transfer from porous Si. In Proceedings of 2nd International Symposium on Pits and Pores: Formation, Properties, Significance for Advanced Materials, Phoenix, Arizona, 22-27 October 2000; Volume 198.
- Bell, T.E.; Gennissen, P.T.J.; DeMunter, D.; Kuhl, M. Porous silicon as a sacrificial material. J. Micromech. Microeng. 1996, 6, 361–369. [Google Scholar] [CrossRef]
- Rottner, K.; Helbig, R.; Müller, G. Piezoelectric constant of InP. Appl. Phys. Lett. 1993, 62, 352–353. [Google Scholar] [CrossRef]
- McKitterick, J.B. First-principles calculation of dielectric properties of GaAs: Dielectric Constant, Effective Charges, and Piezoelectric Constant. Phys. Rev. B 1983, 28, 7384–7386. [Google Scholar] [CrossRef]
- Bernardini, F.; Fiorentini, V.; Vanderbilt, D. Spontaneous polarization and piezoelectric constants of III-V nitrides. Phys. Rev. B 1997, 56, R10024–R10027. [Google Scholar] [CrossRef]
- Ramesh, R.; Spaldin, N.A. Multiferroics: Progress and Prospects in Thin Films. Nat. Mat. 2007, 6, 21–29. [Google Scholar] [CrossRef]
- Howorka, S.; Cheley, S.; Bayley, H. Sequence-specific detection of individual DNA strands using engineered nanopores. Nat. Biotechnol. 2001, 19, 636–639. [Google Scholar] [CrossRef]
- Braha, O.; Gu, L.-Q.; Zhou, L.; Lu, X.; Cheley, S.; Bayley, H. Simultaneous stochastic sensing of divalent metal ions. Nat. Biotechnol. 2000, 18, 1005–1007. [Google Scholar] [CrossRef]
- Li, J.; Stein, D.; McMullan, C.; Branton, D.; Aziz, M.J.; Golovchenko, J.A. Ion-beam sculpting at nanometre length scales. Nature 2001, 412, 166–169. [Google Scholar] [CrossRef]
- Deamer, D.W.; Branton, D. Characterization of nucleic acids by nanopore analysis. Nature 2002, 35, 817–825. [Google Scholar]
- Fertig, N.; Blick, R.H.; Behrends, J.C. Whole cell patch recording performed on a planar glass chip. Biophys. J. 2002, 82, 3056–3062. [Google Scholar] [CrossRef]
- Clayton, J. Go with the microflow. Nat. Methods 2005, 2, 621–627. [Google Scholar] [CrossRef]
- Föll, H.; Gerngroß, M.-D.; Cojocaru, A.; Leisner, M.; Bahr, J.; Carstensen, J. How to make single small holes with large aspect ratios. Phys. Stat. Sol. (RRL) 2009, 3, 55–59. [Google Scholar] [CrossRef]
- Boukamp, B.A.; Lesh, G.C.; Huggins, R.A. All-solid lithium electrodes with mixed-conductor matrix. J. Electrochem. Soc. 1981, 128, 725–729. [Google Scholar] [CrossRef]
- Chan, C.K.; Peng, H.; Liu, G.; McIlwrath, K.; Zhang, X.F.; Huggins, R.A.; Cui, Y. High-performance lithium battery anodes using silicon nanowires. Nat. Nanotechnol. 2008, 3, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Föll, H.; Hartz, H.; Ossei-Wusu, E.K.; Carstensen, J.; Riemenschneider, O. Si nanowires arrays as anodes in Li ion batteries. Phys. Stat. Sol. RRL 2010, 4, 4–8. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Föll, H.; Leisner, M.; Cojocaru, A.; Carstensen, J. Macroporous Semiconductors. Materials 2010, 3, 3006-3076. https://doi.org/10.3390/ma3053006
Föll H, Leisner M, Cojocaru A, Carstensen J. Macroporous Semiconductors. Materials. 2010; 3(5):3006-3076. https://doi.org/10.3390/ma3053006
Chicago/Turabian StyleFöll, Helmut, Malte Leisner, Ala Cojocaru, and Jürgen Carstensen. 2010. "Macroporous Semiconductors" Materials 3, no. 5: 3006-3076. https://doi.org/10.3390/ma3053006
APA StyleFöll, H., Leisner, M., Cojocaru, A., & Carstensen, J. (2010). Macroporous Semiconductors. Materials, 3(5), 3006-3076. https://doi.org/10.3390/ma3053006